
Molecular Determinants of Chronic Venous Disease: A Comprehensive Review

 , , , , and
, , , , and 
Abstract
1. Introduction
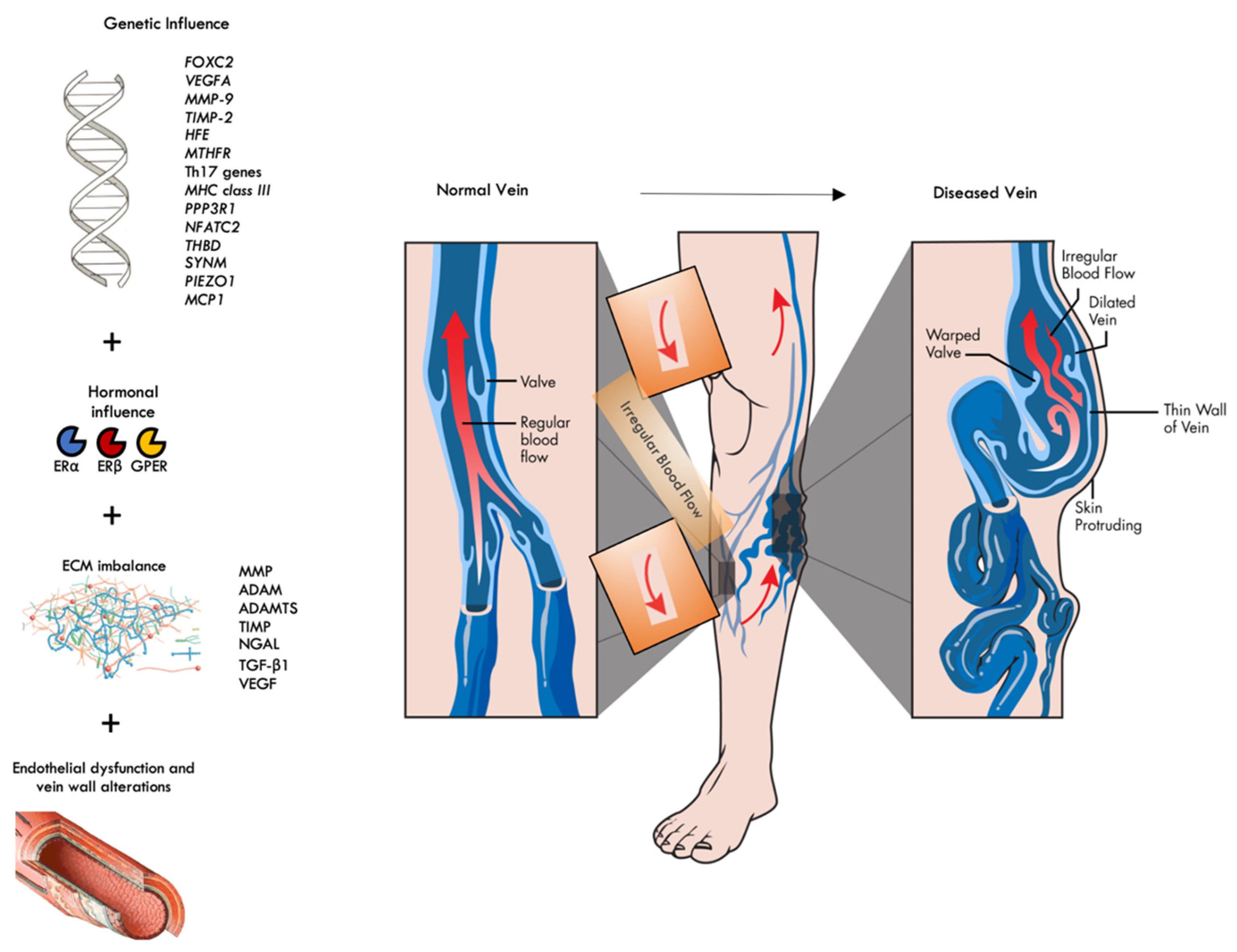
2. The Genetic Influence
3. The Hormonal Influence
4. ECM Imbalance and Histopathology of CVD
5. The Role of Endothelial Dysfunction in CVD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Serra, R.; Grande, R.; Butrico, L.; Fugetto, F.; de Franciscis, S. Epidemiology, diagnosis and treatment of chronic venous disease: A systematic review. Chirurgia 2016, 29, 34–45. [Google Scholar]
- Serra, R.; Butrico, L.; Ruggiero, M.; Rossi, A.; Buffone, G.; Fugetto, F.; De Caridi, G.; Massara, M.; Falasconi, C.; Rizzuto, A.; et al. Epidemiology, diagnosis and treatment of chronic leg ulcers: A systematic review. Acta Phlebolol. 2015, 16, 9–18. [Google Scholar]
- Lurie, F.; Passman, M.; Meisner, M.; Dalsing, M.; Masuda, E.; Welch, H.; Bush, R.L.; Blebea, J.; Carpentier, P.H.; De Maeseneer, M.; et al. The 2020 update of the CEAP classification system and reporting standards. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 342–352. [Google Scholar] [CrossRef]
- Raffetto, J.D. Pathophysiology of Chronic Venous Disease and Venous Ulcers. Surg. Clin. N. Am. 2018, 98, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Amato, B.; Butrico, L.; Barbetta, A.; De Caridi, G.; Massara, M.; Caliò, F.G.; Longo, C.; Dardano, G.; Cannistrà, M.; et al. Study on the efficacy of surgery of the superficial venous system and of compression therapy at early stages of chronic venous disease for the prevention of chronic venous ulceration. Int. Wound J. 2016, 13, 1385–1388. [Google Scholar] [CrossRef]
- Evans, C.J.; Fowkes, F.G.; Ruckley, C.V.; Lee, A.J. Prevalence of varicose veins and chronic venous insufficiency in men and women in the general population: Edinburgh Vein Study. J. Epidemiol. Community Health 1999, 53, 149–153. [Google Scholar] [CrossRef]
- O’Donnell, T.F.; Balk, E.M.; Dermody, M.; Tangney, E.; Iafrati, M.D. Recurrence of varicose veins after endovenous ablation of the great saphenous vein in randomized trials. J. Vasc. Surg. Venous Lymphat. Disord. 2016, 4, 97–105. [Google Scholar] [CrossRef]
- Ielapi, N.; Andreucci, M.; Licastro, N.; Faga, T.; Grande, R.; Buffone, G.; Mellace, S.; Sapienza, P.; Serra, R. Precision Medicine and Precision Nursing: The Era of Biomarkers and Precision Health. Int. J. Gen. Med. 2020, 13, 1705–1711. [Google Scholar] [CrossRef]
- Serra, R.; Ielapi, N.; Barbetta, A.; Buffone, G.; Bevacqua, E.; Andreucci, A.; de Franciscis, S.; Gasbarro, V. Biomarkers for precision medicine in phlebology and wound care: A systematic review. Acta Phlebol. 2017, 18, 52–56. [Google Scholar] [CrossRef]
- Ahmed, W.U.; Kleeman, S.; Ng, M.; Wang, W.; Auton, A.; Lee, R.; Handa, A.; Zondervan, K.T.; Wiberg, A.; Furniss, D. Genome-wide association analysis and replication in 810,625 individuals with varicose veins. Nat. Commun. 2022, 13, 3065. [Google Scholar] [CrossRef]
- Radhakrishnan, N. Chapter 5–The pathophysiology of varicose veins of the lower limb. In Genesis, Pathophysiology and Management of Venous and Lymphatic Disorders; Radhakrishnan, N., Ed.; Academic Press: London, UK, 2022; pp. 95–137. [Google Scholar]
- Cornu-Thenard, A.; Boivin, P.; Baud, J.-M.; DE Vincenzi, I.; Carpentier, P.H. Importance of the Familial Factor in Varicose Disease. J. Dermatol. Surg. Oncol. 1994, 20, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Krysa, J.; Jones, G.T.; Van Rij, A.M. Evidence for a genetic role in varicose veins and chronic venous insufficiency. Phlebology 2012, 27, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Smetanina, M.A.; Shevela, A.I.; Gavrilov, K.A.; Filipenko, M.L. The genetic constituent of varicose vein pathogenesis as a key for future treatment option development. Vessel Plus 2021, 5, 19. [Google Scholar] [CrossRef]
- Serra, R.; Ssempijja, L.; Provenzano, M.; Andreucci, M. Genetic biomarkers in chronic venous disease. Biomarkers Med. 2020, 14, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Davies, A.H. Pathogenesis of primary varicose veins. Br. J. Surg. 2009, 96, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Mellor, R.H.; Brice, G.; Stanton, A.W.; French, J.; Smith, A.; Jeffery, S.; Levick, J.R.; Burnand, K.G.; Mortimer, P.S. Mutations in FOXC2 Are Strongly Associated With Primary Valve Failure in Veins of the Lower Limb. Circulation 2007, 115, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Mangion, J.; Rahman, N.; Mansour, S.; Brice, G.; Rosbotham, J.; Child, A.; Murday, V.; Mortimer, P.; Barfoot, R.; Sigurdsson, A.; et al. A Gene for Lymphedema-Distichiasis Maps to 16q24.3. Am. J. Hum. Genet. 1999, 65, 427–432. [Google Scholar] [CrossRef]
- Petrova, T.V.; Karpanen, T.; Norrmén, C.; Mellor, R.; Tamakoshi, T.; Finegold, D.; Ferrell, R.; Kerjaschki, D.; Mortimer, P.; Ylä-Herttuala, S.; et al. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 2004, 10, 974–981. [Google Scholar] [CrossRef]
- Shimoda, H.; Bernas, M.J.; Witte, M.H. Dysmorphogenesis of lymph nodes in Foxc2 haploinsufficient mice. Histochem. Cell Biol. 2011, 135, 603–613. [Google Scholar] [CrossRef]
- Wu, X.; Liu, N.-F. FOXC2 transcription factor: A novel regulator of lymphangiogenesis. Lymphology 2011, 44, 35–41. [Google Scholar]
- Zhang, C.; Li, H.; Guo, X. FOXC2-AS1 regulates phenotypic transition, proliferation and migration of human great saphenous vein smooth muscle cells. Biol. Res. 2019, 52, 59. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kume, T. Foxc Transcription Factors Directly Regulate Dll4 and Hey2 Expression by Interacting with the VEGF-Notch Signaling Pathways in Endothelial Cells. PLoS ONE 2008, 3, e2401. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.; Ramegowda, K.S.; Suresh, A.; Raj, S.B.; Lakkappa, R.K.B.; Kamalapurkar, G.; Radhakrishnan, N.; Kartha, C.C. Arterialization and anomalous vein wall remodeling in varicose veins is associated with upregulated FoxC2-Dll4 pathway. Lab. Investig. 2016, 96, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kume, T. Foxc2 transcription factor as a regulator of angiogenesis via induction of integrin beta3 expression. Cell Adhes. Migr. 2009, 3, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kume, T. Forkhead transcription factors regulate expression of the chemokine receptor CXCR4 in endothelial cells and CXCL12-induced cell migration. Biochem. Biophys. Res. Commun. 2008, 367, 584–589. [Google Scholar] [CrossRef]
- Representing, T.; Andrew, T.; Spector, T.D.; Jeffery, S. Linkage to the FOXC2 region of chromosome 16 for varicose veins in otherwise healthy, unselected sibling pairs. J. Med. Genet. 2005, 42, 235–239. [Google Scholar] [CrossRef]
- Al-Batayneh, K.M.; Battah, R.M. Genetic variation in the proximal 5’UTR of FOXC2 gene in varicose veins and hemorrhoids patients. Int. J. Integr. Biol. 2008, 4, 79. [Google Scholar]
- Serra, R.; Buffone, G.; de Franciscis, A.; Mastrangelo, D.; Molinari, V.; Montemurro, R.; de Franciscis, S. A Genetic Study of Chronic Venous Insufficiency. Ann. Vasc. Surg. 2012, 26, 636–642. [Google Scholar] [CrossRef]
- Surendran, S.; Girijamma, A.; Nair, R.; Ramegowda, K.S.; Nair, D.H.; Thulaseedharan, J.V.; Lakkappa, R.B.; Kamalapurkar, G.; Kartha, C.C. Forkhead box C2 Promoter Variant c.-512C>T Is Associated with Increased Susceptibility to Chronic Venous Diseases. PLoS ONE 2014, 9, e90682. [Google Scholar] [CrossRef]
- Shadrina, A.S.; Smetanina, M.A.; Sokolova, E.A.; Sevost’Ianova, K.S.; Shevela, A.I.; Demekhova, M.Y.; Shonov, O.A.; Ilyukhin, E.A.; Voronina, E.N.; Zolotukhin, I.A.; et al. Association of polymorphisms near the FOXC2 gene with the risk of varicose veins in ethnic Russians. Phlebology 2016, 31, 640–648. [Google Scholar] [CrossRef]
- Ferrara, N. Molecular and biological properties of vascular endothelial growth factor. J. Mol. Med. 1999, 77, 527–543. [Google Scholar] [CrossRef]
- Kowalewski, R.; Małkowski, A.; Sobolewski, K.; Gacko, M. Vascular endothelial growth factor and its receptors in the varicose vein wall. Acta Angiol. 2011, 17, 141–149. [Google Scholar]
- Kunt, A.T.; Isbir, S.; Görmüş, U.; Kahraman, O.T.; Arsan, S.; Yılmaz, S.G.; Isbir, T. Polymorphisms of MMP9 and TIMP2 in Patients with Varicose Veins. Vivo 2015, 29, 461–465. [Google Scholar]
- Xu, H.-M.; Zhao, Y.; Zhang, X.-M.; Zhu, T.; Fu, W.-G. Polymorphisms in MMP-9 and TIMP-2 in Chinese Patients with Varicose Veins. J. Surg. Res. 2011, 168, e143–e148. [Google Scholar] [CrossRef]
- Zamboni, P.; Tognazzo, S.; Izzo, M.; Pancaldi, F.; Scapoli, G.L.; Liboni, A.; Gemmati, D. Hemochromatosis C282Y gene mutation increases the risk of venous leg ulceration. J. Vasc. Surg. 2005, 42, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Budzyń, M.; Iskra, M.; Krasiński, Z.; Dzieciuchowicz, Ł.; Kasprzak, M.; Gryszczyńska, B. Serum iron concentration and plasma oxidant-antioxidant balance in patients with chronic venous insufficency. Experiment 2011, 17, CR719–CR727. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, Z.; Seidenbaum, M.; Loewenthal, E.; Rubinow, A. Overload of iron in the skin of patients with varicose ulcers. Possible contributing role of iron accumulation in progression of the disease. Arch. Dermatol. 1988, 124, 1376–1378. [Google Scholar] [CrossRef]
- Sokolova, E.A.; Shadrina, A.; Sevost’Ianova, K.S.; Shevela, A.I.; Soldatsky, E.Y.; Seliverstov, E.I.; Demekhova, M.; Shonov, O.A.; Ilyukhin, E.; Smetanina, M.; et al. HFE p.C282Y gene variant is associated with varicose veins in Russian population. Clin. Exp. Med. 2016, 16, 463–470. [Google Scholar] [CrossRef]
- Zamboni, P.; Scapoli, G.; Lanzara, V.; Izzo, M.; Fortini, P.; Legnaro, R.; Palazzo, A.; Tognazzo, S.; Gemmati, D. Serum iron and matrix metalloproteinase-9 variations in limbs affected by chronic venous disease and venous leg ulcers. Dermatol. Surg. 2005, 31, 644–649. [Google Scholar] [CrossRef]
- Wilmanns, C.; Cooper, A.; Wockner, L.; Katsandris, S.; Glaser, N.; Meyer, A.; Bartsch, O.; Binder, H.; Walter, P.K.; Zechner, U. Morphology and Progression in Primary Varicose Vein Disorder Due to 677C>T and 1298A>C Variants of MTHFR. Ebiomedicine 2015, 2, 158–164. [Google Scholar] [CrossRef]
- Amato, R.; Dattilo, V.; Brescia, C.; D’Antona, L.; Iuliano, R.; Trapasso, F.; Perrotti, N.; Costa, D.; Ielapi, N.; Aiello, F.; et al. Th17-Gene Expression Profile in Patients with Chronic Venous Disease and Venous Ulcers: Genetic Modulations and Preliminary Clinical Evidence. Biomolecules 2022, 12, 902. [Google Scholar] [CrossRef]
- Bell, R.K.; Durand, E.Y.; McLean, C.Y.; Tung, J.; Hinds, D. A Large Scale Genome Wide Association Study of Varicose Veins in the 23andMe Cohort. In Proceedings of the 64th Annual Meeting of the American Society of Human Genetics, San Diego, CA, USA, 18–22 October 2014; p. 487. Available online: https://blog23andme.wpengine.com/wp-content/uploads/2014/10/Bell_ASHG2014_varicose.pdf (accessed on 13 December 2022).
- Ellinghaus, E.; Ellinghaus, D.; Krusche, P.; Greiner, A.; Schreiber, C.; Nikolaus, S.; Gieger, C.; Strauch, K.; Lieb, W.; Rosenstiel, P.; et al. Genome-wide association analysis for chronic venous disease identifies EFEMP1 and KCNH8 as susceptibility loci. Sci. Rep. 2017, 7, 45652. [Google Scholar] [CrossRef] [PubMed]
- Shadrina, A.; Tsepilov, Y.; Sokolova, E.; Smetanina, M.; Voronina, E.; Pakhomov, E.; Sevost’Ianova, K.; Shevela, A.; Ilyukhin, E.; Seliverstov, E.; et al. Genome-wide association study in ethnic Russians suggests an association of the MHC class III genomic region with the risk of primary varicose veins. Gene 2018, 659, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, E.; Flores, A.M.; Lindholm, D.; Gustafsson, S.; Zanetti, D.; Ingelsson, E.; Leeper, N.J. Clinical and Genetic Determinants of Varicose Veins. Circulation 2018, 138, 2869–2880. [Google Scholar] [CrossRef]
- Shadrina, A.S.; Sharapov, S.Z.; Shashkova, T.I.; Tsepilov, Y.A. Varicose veins of lower extremities: Insights from the first large-scale genetic study. PLoS Genet. 2019, 15, e1008110. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-L.; Liang, C.; Chuang, C.-H.; Lee, P.-S.; Chen, T.-H.; Sun, S.; Liao, K.-W.; Huang, H.-D. A genome-wide association study for varicose veins. Phlebology 2022, 37, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Boisseau, M. Chronic venous disease and the genetic influence. Phlebolymphology 2014, 21, 100–111. [Google Scholar]
- Shadrina, A.S.; Smetanina, M.A.; Sevost’Ianova, K.S.; Seliverstov, E.I.; Ilyukhin, E.A.; Voronina, E.N.; Zolotukhin, I.; Filipenko, M.L. Functional polymorphism rs1024611 in the MCP1 gene is associated with the risk of varicose veins of lower extremities. J. Vasc. Surgery: Venous Lymphat. Disord. 2017, 5, 561–566. [Google Scholar] [CrossRef]
- de Franciscis, S.; Metzinger, L.; Serra, R. The Discovery of Novel Genomic, Transcriptomic, and Proteomic Biomarkers in Cardiovascular and Peripheral Vascular Disease: The State of the Art. BioMed Res. Int. 2016, 2016, 7829174. [Google Scholar] [CrossRef]
- Wei, J.; Zhu, H.; Zhang, Q.; Zhang, Q. Prediction of Functional Genes in Primary Varicose Great Saphenous Veins Using the lncRNA-miRNA-mRNA Network. Comput. Math. Methods Med. 2022, 2022, 4722483. [Google Scholar] [CrossRef]
- Cao, Y.; Cao, Z.; Wang, W.; Jie, X.; Li, L. MicroRNA-199a-5p regulates FOXC2 to control human vascular smooth muscle cell phenotypic switch. Mol. Med. Rep. 2021, 24, 627. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, Z.; Shen, L.; Jin, Y.; Xu, G.; Zhang, Z.; Fang, C.; Guan, W.; Liu, C. Augmentation of miR-202 in varicose veins modulates phenotypic transition of vascular smooth muscle cells by targeting proliferator-activated receptor-γ coactivator-1α. J. Cell. Biochem. 2019, 120, 10031–10042. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, J.; Liu, Y.; Yang, S.; Wang, Q. microRNA-301a-3p is a potential biomarker in venous ulcers vein and gets involved in endothelial cell dysfunction. Bioengineered 2022, 13, 14138–14158. [Google Scholar] [CrossRef] [PubMed]
- Biranvand, A.S.; Khosravi, M.; Esfandiari, G.; Poursaleh, A.; Hosseini-Fard, S.R.; Amirfarhangi, A.; Najafi, M. Associations between miR-661, miR-1202, lncRNA-HOTAIR, lncRNA-GAS5 and MMP9 in differentiated M2-macrophages of patients with varicose veins. Int. Angiol. A J. Int. Union Angiol. 2018, 37, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, D.P.; Ruszel, K.P.; Stępniewski, A.; Gałkowski, D.; Bogucki, J.; Komsta, Ł.; Kołodziej, P.; Chmiel, P.; Zubilewicz, T.; Feldo, M.; et al. Dysregulations of MicroRNA and Gene Expression in Chronic Venous Disease. J. Clin. Med. 2020, 9, 1251. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Gallelli, L.; Perri, P.; De Francesco, E.M.; Rigiracciolo, D.C.; Mastroroberto, P.; Maggiolini, M.; de Franciscis, S. Estrogen Receptors and Chronic Venous Disease. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2016, 52, 114–118. [Google Scholar] [CrossRef]
- Honduvilla, N.G.; Asúnsolo, Á.; Ortega, M.A.; Sainz, F.; Leal, J.; Lopez-Hervas, P.; Pascual, G.; Buján, J. Increase and Redistribution of Sex Hormone Receptors in Premenopausal Women Are Associated with Varicose Vein Remodelling. Oxidative Med. Cell. Longev. 2018, 2018, 3974026. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Qiao, X.; Beauregard, K.G.; Khalil, R.A. Estrogen receptor-mediated enhancement of venous relaxation in female rat: Implications in sex-related differences in varicose veins. J. Vasc. Surg. 2010, 51, 972–981. [Google Scholar] [CrossRef]
- Ropacka-Lesiak, M.; Bręborowicz, G.H.; Kasperczak, J. Risk factors for the development of venous insufficiency of the lower limbs during pregnancy--part 1. Ginekol. Pol. 2012, 83, 939–942. [Google Scholar]
- Kendler, M.; Makrantonaki, E.; Kratzsch, J.; Anderegg, U.; Wetzig, T.; Zouboulis, C.; Simon, J.C. Elevated sex steroid hormones in great saphenous veins in men. J. Vasc. Surg. 2010, 51, 639–646. [Google Scholar] [CrossRef]
- Zhao, M.-Y.; Zhao, T.; Meng, Q.-Y.; Zhao, L.; Li, X.-C. Estrogen and estrogen receptor affects MMP2 and MMP9 expression through classical ER pathway and promotes migration of lower venous vascular smooth muscle cells. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1460–1467. [Google Scholar] [PubMed]
- Ismail, L.; Normahani, P.; Standfield, N.J.; Jaffer, U. A systematic review and meta-analysis of the risk for development of varicose veins in women with a history of pregnancy. J. Vasc. Surg. Venous Lymphat. Disord. 2016, 4, 518–524.e1. [Google Scholar] [CrossRef]
- Serra, R.; Gallelli, L.; Butrico, L.; Buffone, G.; Caliò, F.G.; De Caridi, G.; Massara, M.; Barbetta, A.; Amato, B.; Labonia, M.; et al. From varices to venous ulceration: The story of chronic venous disease described by metalloproteinases. Int. Wound J. 2017, 14, 233–240. [Google Scholar] [CrossRef]
- Serra, R.; Buffone, G.; Falcone, D.; Molinari, V.; Scaramuzzino, M.; Gallelli, L.; De Franciscis, S. Chronic venous leg ulcers are associated with high levels of metalloproteinases-9 and neutrophil gelatinase-associated lipocalin. Wound Repair Regen. 2013, 21, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Horecka, A.; Hordyjewska, A.; Biernacka, J.; Dąbrowski, W.; Zubilewicz, T.; Malec, A.; Musik, I.; Kurzepa, J. Intense remodeling of extracellular matrix within the varicose vein: The role of gelatinases and vascular endothelial growth factor. Ir. J. Med. Sci. 2021, 190, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, W.; Raffetto, J.D.; Khalil, R.A. Matrix Metalloproteinases in Remodeling of Lower Extremity Veins and Chronic Venous Disease. Prog. Mol. Biol. Transl. Sci. 2017, 147, 267–299. [Google Scholar] [CrossRef]
- Busceti, M.T.; Grande, R.; Amato, B.; Gasbarro, V.; Buffone, G.; Amato, M.; Gallelli, L.; Serra, R.; de Franciscis, S. Pulmonary embolism, metalloproteinsases and neutrophil gelatinase associated lipocalin. Acta Phlebol. 2013, 14, 115–121. [Google Scholar]
- Raffetto, J.D.; Ross, R.L.; Khalil, R.A. Matrix metalloproteinase 2–induced venous dilation via hyperpolarization and activation of K+ channels: Relevance to varicose vein formation. J. Vasc. Surg. 2007, 45, 373–380. [Google Scholar] [CrossRef]
- Serra, R.; Gallelli, L.; Buffone, G.; Molinari, V.; Stillitano, D.M.; Palmieri, C.; De Franciscis, S. Doxycycline speeds up healing of chronic venous ulcers. Int. Wound J. 2013, 12, 179–184. [Google Scholar] [CrossRef]
- Serra, R.; Grande, R.; Buffone, G.; Gallelli, L.; de Franciscis, S. The effects of minocycline on extracellular matrix in patients with chronic venous leg ulcers. Acta Phlebol. 2013, 14, 99–107. [Google Scholar]
- Wali, M.A.; Dewan, M.; Eid, R.A. Histopathological changes in the wall of varicose veins. Int. Angiol. 2003, 22, 188–193. [Google Scholar] [PubMed]
- Ducasse, E.; Giannakakis, K.; Speziale, F.; Midy, D.; Sbarigia, E.; Baste, J.C.; Faraggiana, T. Association of primary varicose veins with dysregulated vein wall apoptosis. Eur. J. Vasc. Endovasc. Surg. 2008, 35, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Ielapi, N.; Barbetta, A.; Gallelli, L.; Michael, A.; Gasbarro, V.; Andreucci, M.; de Francisicis, S. Chronic leg ulcers: The role of fibrosis, stem cells, and tissue regeneration. Acta Phlebol. 2019, 20, 61–66. [Google Scholar] [CrossRef]
- Serra, R.; Bracale, U.M.; Chilà, C.; Renne, M.; Mignogna, C.; Ielapi, N.; Ciranni, S.; Torcia, G.; Bevacqua, E.; Di Taranto, M.D.; et al. Clinical and Pathological Correlations in Chronic Venous Disease. Ann. Vasc. Surg. 2021, 78, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Ghaderian, S.M.H.; Lindsey, N.J.; Graham, A.M.; Homer-Vanniasinkam, S.; Najar, R.A. Pathogenic mechanisms in varicose vein disease: The role of hypoxia and inflammation. Pathology 2010, 42, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Buffone, G.; Costanzo, G.; Montemurro, R.; Perri, P.; Damiano, R.; de Franciscis, S. Varicocele in Younger as Risk Factor for Inguinal Hernia and for Chronic Venous Disease in Older: Preliminary Results of a Prospective Cohort Study. Ann. Vasc. Surg. 2013, 27, 329–331. [Google Scholar] [CrossRef]
- Serra, R.; Buffone, G.; Costanzo, G.; Montemurro, R.; Scarcello, E.; Stillitano, D.M.; Damiano, R.; de Franciscis, S. Altered Metalloproteinase-9 Expression as Least Common Denominator between Varicocele, Inguinal Hernia, and Chronic Venous Disorders. Ann. Vasc. Surg. 2014, 28, 705–709. [Google Scholar] [CrossRef]
- Castro-Ferreira, R.; Cardoso, R.; Leite-Moreira, A.; Mansilha, A. The Role of Endothelial Dysfunction and Inflammation in Chronic Venous Disease. Ann. Vasc. Surg. 2018, 46, 380–393. [Google Scholar] [CrossRef]
- Iring, A.; Jin, Y.-J.; Albarrán-Juárez, J.; Siragusa, M.; Wang, S.; Dancs, P.T.; Nakayama, A.; Tonack, S.; Chen, M.; Künne, C.; et al. Shear stress–induced endothelial adrenomedullin signaling regulates vascular tone and blood pressure. J. Clin. Investig. 2019, 129, 2775–2791. [Google Scholar] [CrossRef]
- Hogan, B.; Shen, Z.; Zhang, H.; Misbah, C.; Barakat, A.I. Shear stress in the microvasculature: Influence of red blood cell morphology and endothelial wall undulation. Biomech. Model. Mechanobiol. 2019, 18, 1095–1109. [Google Scholar] [CrossRef]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C., IV; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jin, T.; Weng, J. Endothelial Cells as a Key Cell Type for Innate Immunity: A Focused Review on RIG-I Signaling Pathway. Front. Immunol. 2022, 13, 95161. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Villalba, N.; Baby, S.; Yuan, S.Y. The Endothelial Glycocalyx as a Double-Edged Sword in Microvascular Homeostasis and Pathogenesis. Front. Cell Dev. Biol. 2021, 9, 711003. [Google Scholar] [CrossRef]
- Jedlicka, J.; Becker, B.F.; Chappell, D. Endothelial Glycocalyx. Crit. Care Clin. 2020, 36, 217–232. [Google Scholar] [CrossRef]
- Moore, K.H.; Murphy, H.A.; George, E.M. The glycocalyx: A central regulator of vascular function. Am. J. Physiol. Integr. Comp. Physiol. 2021, 320, R508–R518. [Google Scholar] [CrossRef]
- Foote, C.A.; Soares, R.N.; Ramirez-Perez, F.I.; Ghiarone, T.; Aroor, A.; Manrique-Acevedo, C.; Padilla, J.; Martinez-Lemus, L. Endothelial Glycocalyx. Compr. Physiol. 2022, 12, 3781–3811. [Google Scholar]
- Birdina, J.; Pilmane, M.; Ligers, A. The Morphofunctional Changes in the Wall of Varicose Veins. Ann. Vasc. Surg. 2017, 42, 274–284. [Google Scholar] [CrossRef]
- Saharay, M.; Shields, D.A.; Georgiannos, S.N.; Porter, J.B.; Scurr, J.H.; Coleridge Smith, P.D. Endothelial activation in patients with chronic venous disease. Eur. J. Vasc. Endovasc. Surg. 1998, 15, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Khalil, R.A. Mechanisms of lower extremity vein dysfunction in chronic venous disease and implications in management of varicose veins. Vessel. Plus 2021, 5, 36. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed]
- Chandran Latha, K.; Sreekumar, A.; Beena, V.; SS, B.R.; Lakkappa, R.B.; Kalyani, R.; Nair, R.; Kalpana, S.R.; Kartha, C.C.; Surendran, S. Shear Stress Alterations Activate BMP4/pSMAD5 Signaling and Induce Endothelial Mesenchymal Transition in Varicose Veins. Cells 2021, 10, 3563. [Google Scholar] [CrossRef] [PubMed]
- Saberianpour, S.; Modaghegh, M.H.S.; Rahimi, H.; Kamyar, M.M. Role of mechanosignaling on pathology of varicose vein. Biophys. Rev. 2021, 13, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2021, 41, 2357–2369. [Google Scholar] [CrossRef]
- Yuan, C.; Ni, L.; Zhang, C.; Hu, X.; Wu, X. Vascular calcification: New insights into endothelial cells. Microvasc. Res. 2021, 134, 104105. [Google Scholar] [CrossRef]
- Tseng, C.-N.; Chang, Y.-T.; Yen, C.-Y.; Lengquist, M.; Kronqvist, M.; Eriksson, E.E.; Hedin, U. Early Inhibition of P-Selectin/P-Selectin Glycoprotein Ligand-1 Reduces Intimal Hyperplasia in Murine Vein Grafts through Platelet Adhesion. Thromb. Haemost. 2019, 119, 2014–2024. [Google Scholar] [CrossRef]
- Ortega, M.; Fraile-Martínez, O.; García-Montero, C.; Álvarez-Mon, M.; Chaowen, C.; Ruiz-Grande, F.; Pekarek, L.; Monserrat, J.; Asúnsolo, A.; García-Honduvilla, N.; et al. Understanding Chronic Venous Disease: A Critical Overview of Its Pathophysiology and Medical Management. J. Clin. Med. 2021, 10, 3239. [Google Scholar] [CrossRef]
- del Rio Solá, L.; Aceves, M.; Dueñas, A.I.; González-Fajardo, J.A.; Vaquero, C.; Crespo, M.S.; Garcia-Rodriguez, C. Varicose veins show enhanced chemokine expression. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 635–641. [Google Scholar] [CrossRef]
- Lattimer, C.R.; Kalodiki, E.; Geroulakos, G.; Hoppensteadt, D.; Fareed, J. Are Inflammatory Biomarkers Increased in Varicose Vein Blood? Clin. Appl. Thromb. Hemost. 2016, 22, 656–664. [Google Scholar] [CrossRef]
- Bilancini, S.; Lucchi, M.; Ciacciarelli, M. Stasis microangiopathy: From pathogenesis to treatment. Vessel Plus 2021, 5, 39. [Google Scholar] [CrossRef]
- Carroll, B.J.; Piazza, G.; Goldhaber, S.Z. Sulodexide in venous disease. J. Thromb. Haemost. 2019, 17, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Shayo, S.C.; Kawade, S.; Ogiso, K.; Yoshihiko, N. Strategies to ameliorate endothelial dysfunction associated with metabolic syndrome, where are we? Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 2164–2169. [Google Scholar] [CrossRef] [PubMed]
- De Franciscis, S.; Butrico, L.; Settimio, U.F.; Grande, R.; Serra, R. The endothelial dysfunction in chronic venous disease: A systematic review. Acta Phebol. 2015, 16, 69–76. [Google Scholar]
- Eschrich, J.; Meyer, R.; Kuk, H.; Wagner, A.H.; Noppeney, T.; Debus, S.; Hecker, M.; Korff, T. Varicose Remodeling of Veins Is Suppressed by 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitors. J. Am. Heart Assoc. 2016, 5, e002405. [Google Scholar] [CrossRef] [PubMed]
- Arase, H.; Sugasawa, N.; Kawatani, Y.; Sugano, M.; Kurobe, H.; Fujimoto, E.; Kitaichi, T.; Kitagawa, T. Appropriate Surgical Treatment of Symptomatic Primary Varicose Veins Decreases Systemic Inflammatory Biomarkers. Ann. Vasc. Dis. 2019, 12, 367–371. [Google Scholar] [CrossRef]
- Tisato, V.; Zauli, G.; Gianesini, S.; Menegatti, E.; Brunelli, L.; Manfredini, R.; Zamboni, P.; Secchiero, P. Modulation of Circulating Cytokine-Chemokine Profile in Patients Affected by Chronic Venous Insufficiency Undergoing Surgical Hemodynamic Correction. J. Immunol. Res. 2014, 2014, 473765. [Google Scholar] [CrossRef]
- Zamboni, P.; Spath, P.; Tisato, V.; Tessari, M.; Caneva, P.D.; Menegatti, E.; Occhionorelli, S.; Gianesini, S.; Secchiero, P. Oscillatory flow suppression improves inflammation in chronic venous disease. J. Surg. Res. 2016, 205, 238–245. [Google Scholar] [CrossRef]
- Zolotukhin, I.; Golovanova, O.; Efremova, O.; Golovina, V.; Seliverstov, E. Monocyte chemoattractant protein 1 plasma concentration in blood from varicose veins decreases under venoactive drug treatment. Int. Angiol. J. Int. Union Angiol. 2022. [Google Scholar] [CrossRef]


{kind=link}
| Gene Alteration or Polymorphism | Implication in Vascular Development and Angiogenesis | Implication in Vein Wall Integrity and Function | Implication in Chronic Inflammation |
|---|---|---|---|
| FOXC2 | X | X | |
| VEGFA | X | X | X |
| MMP9 | X | X | |
| TIMP2 | X | X | |
| HFE | X | X | |
| MTHFR | X | X | |
| Th17 genes | X | ||
| MHC class III | X | ||
| PPP3R1 | X | ||
| NFATC2 | X | ||
| THBD | X | ||
| SYNM | X | ||
| PIEZO1 | X | X | |
| MCP1 | X | X |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, D.; Andreucci, M.; Ielapi, N.; Serraino, G.F.; Mastroroberto, P.; Bracale, U.M.; Serra, R. Molecular Determinants of Chronic Venous Disease: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 1928. https://doi.org/10.3390/ijms24031928
Costa D, Andreucci M, Ielapi N, Serraino GF, Mastroroberto P, Bracale UM, Serra R. Molecular Determinants of Chronic Venous Disease: A Comprehensive Review. International Journal of Molecular Sciences. 2023; 24(3):1928. https://doi.org/10.3390/ijms24031928
Chicago/Turabian StyleCosta, Davide, Michele Andreucci, Nicola Ielapi, Giuseppe Filiberto Serraino, Pasquale Mastroroberto, Umberto Marcello Bracale, and Raffaele Serra. 2023. "Molecular Determinants of Chronic Venous Disease: A Comprehensive Review" International Journal of Molecular Sciences 24, no. 3: 1928. https://doi.org/10.3390/ijms24031928
APA StyleCosta, D., Andreucci, M., Ielapi, N., Serraino, G. F., Mastroroberto, P., Bracale, U. M., & Serra, R. (2023). Molecular Determinants of Chronic Venous Disease: A Comprehensive Review. International Journal of Molecular Sciences, 24(3), 1928. https://doi.org/10.3390/ijms24031928