
Unravelling Differential DNA Methylation Patterns in Genotype Dependent Manner under Salinity Stress Response in Chickpea

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
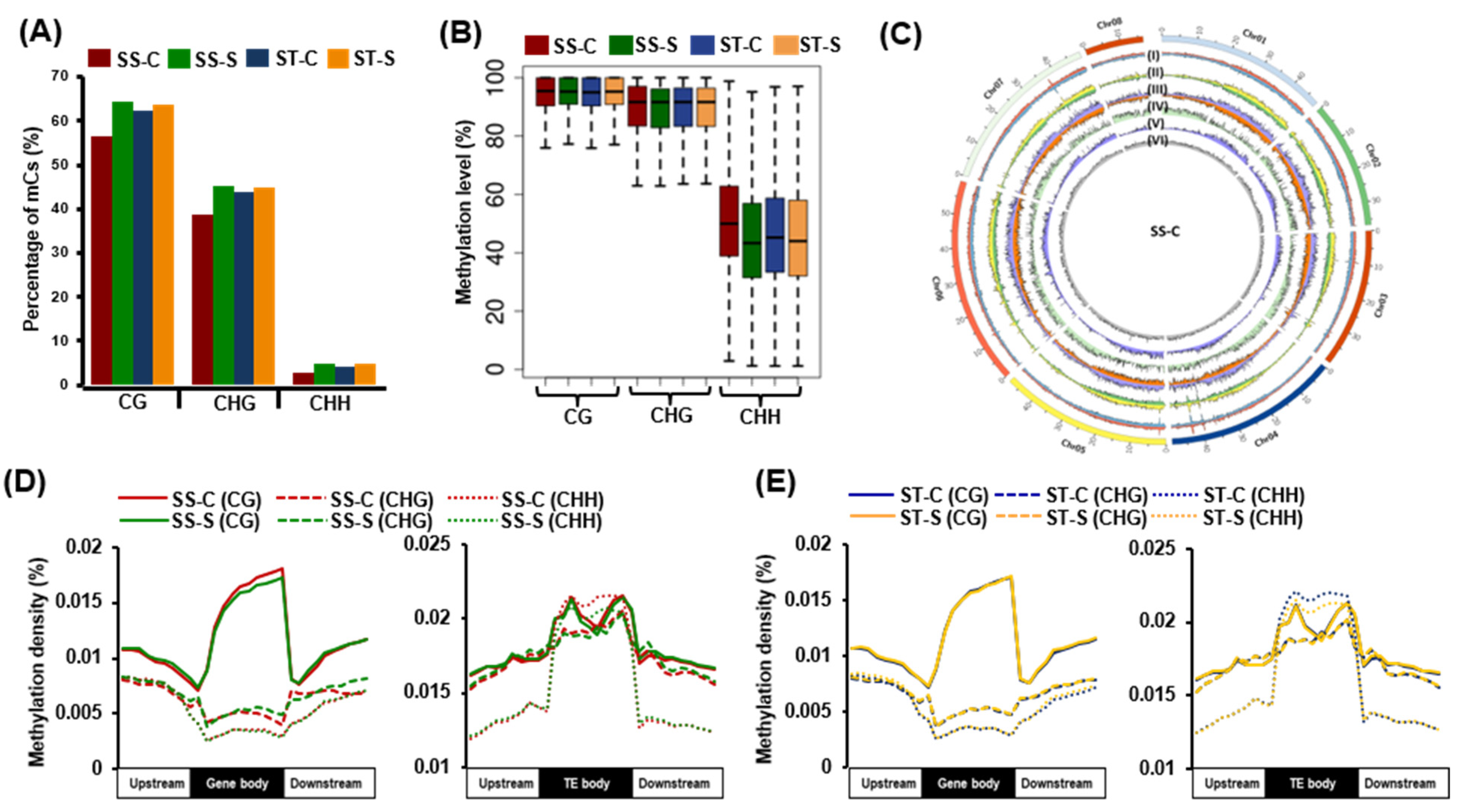
2.1. Genome-Wide DNA Methylation Landscape in Chickpea Genotypes
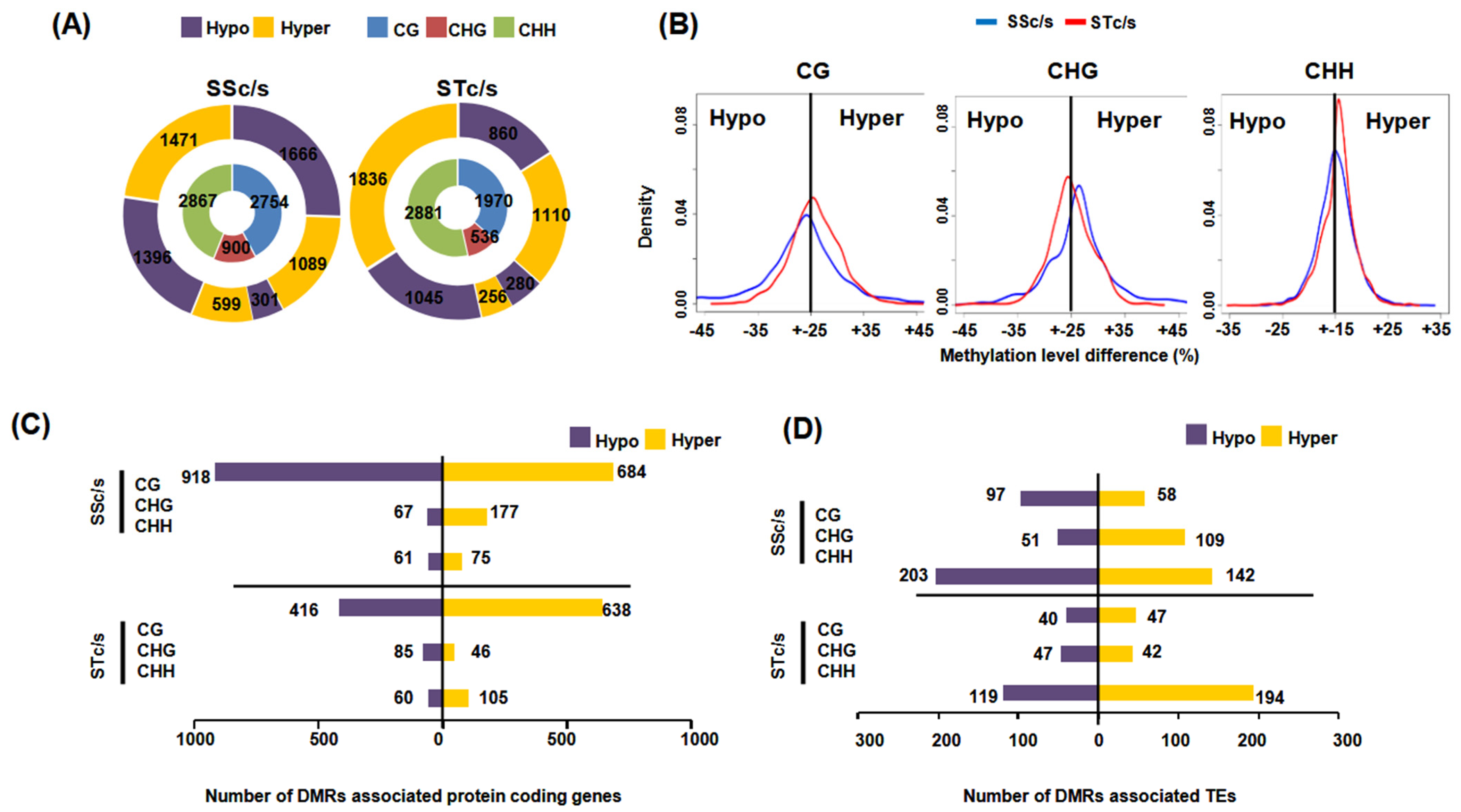
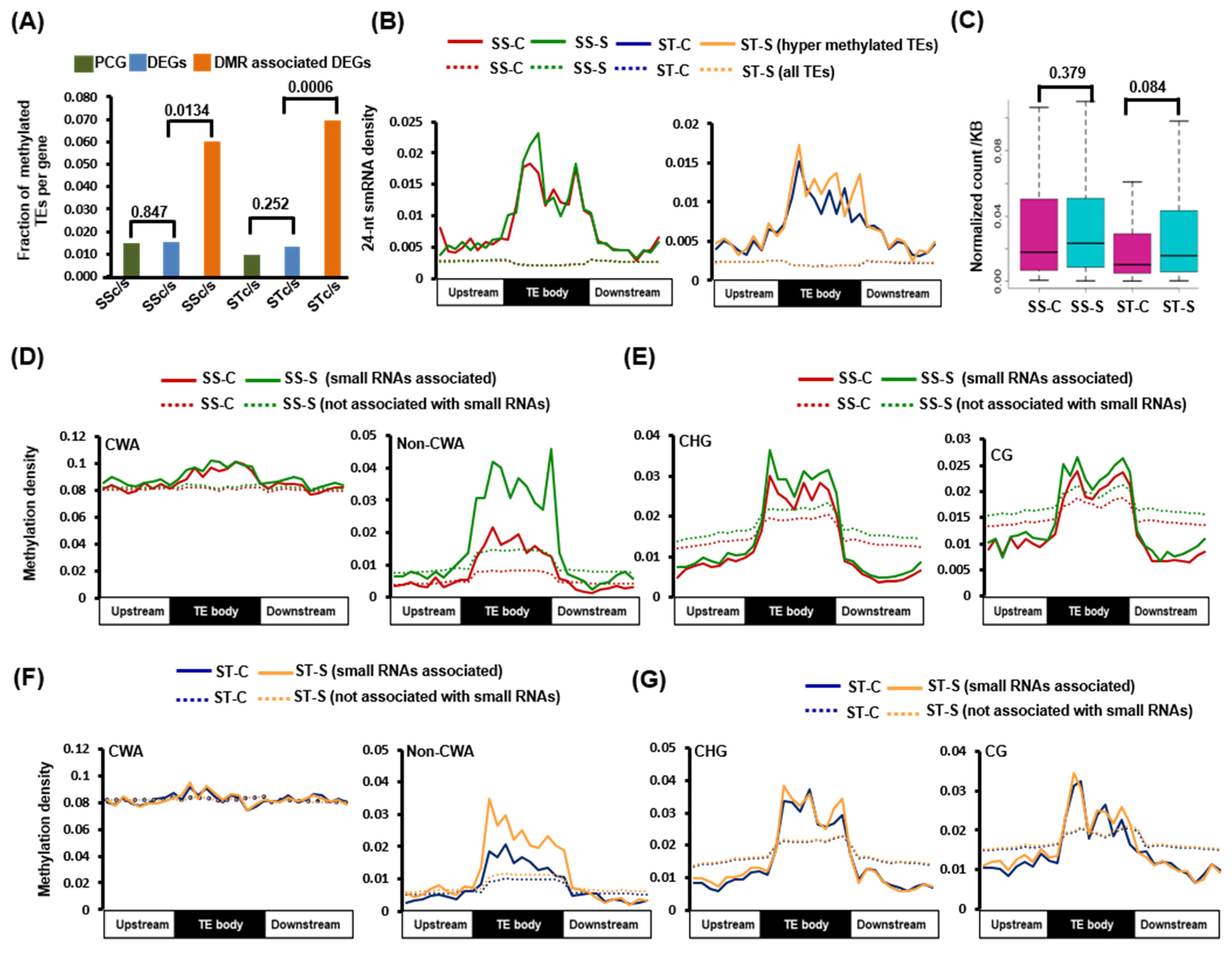
2.2. Identification of Differentially Methylated Regions in Protein-Coding Genes and TEs
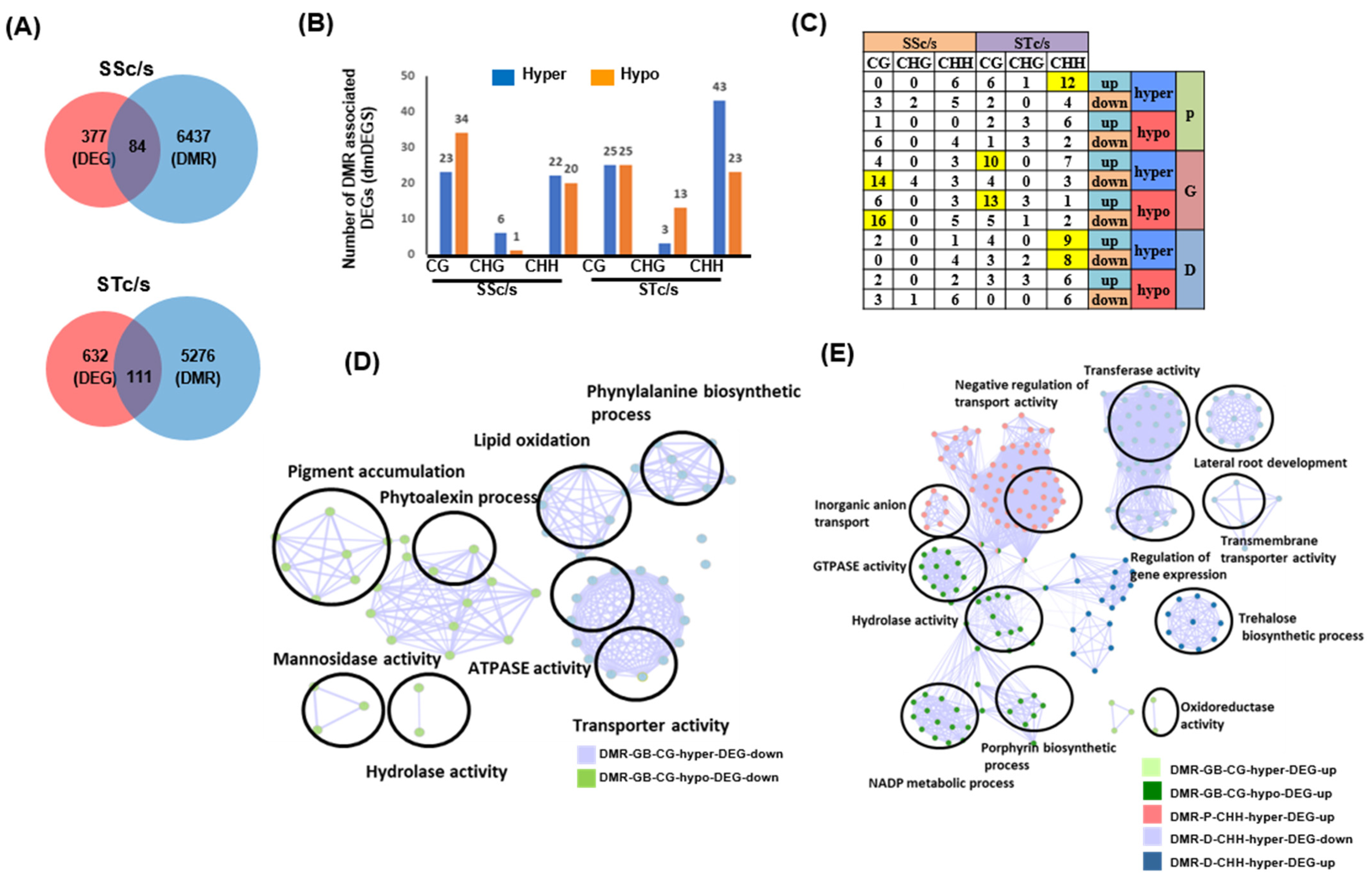
2.3. Differential Gene Expression under Salinity Stress in the Chickpea Genotypes
2.4. Influence of DNA Methylation on Differential Gene Expression and Co-Relation between Different Methylation Contexts
2.5. Small RNA-Mediated DNA Methylation Dynamics in TEs
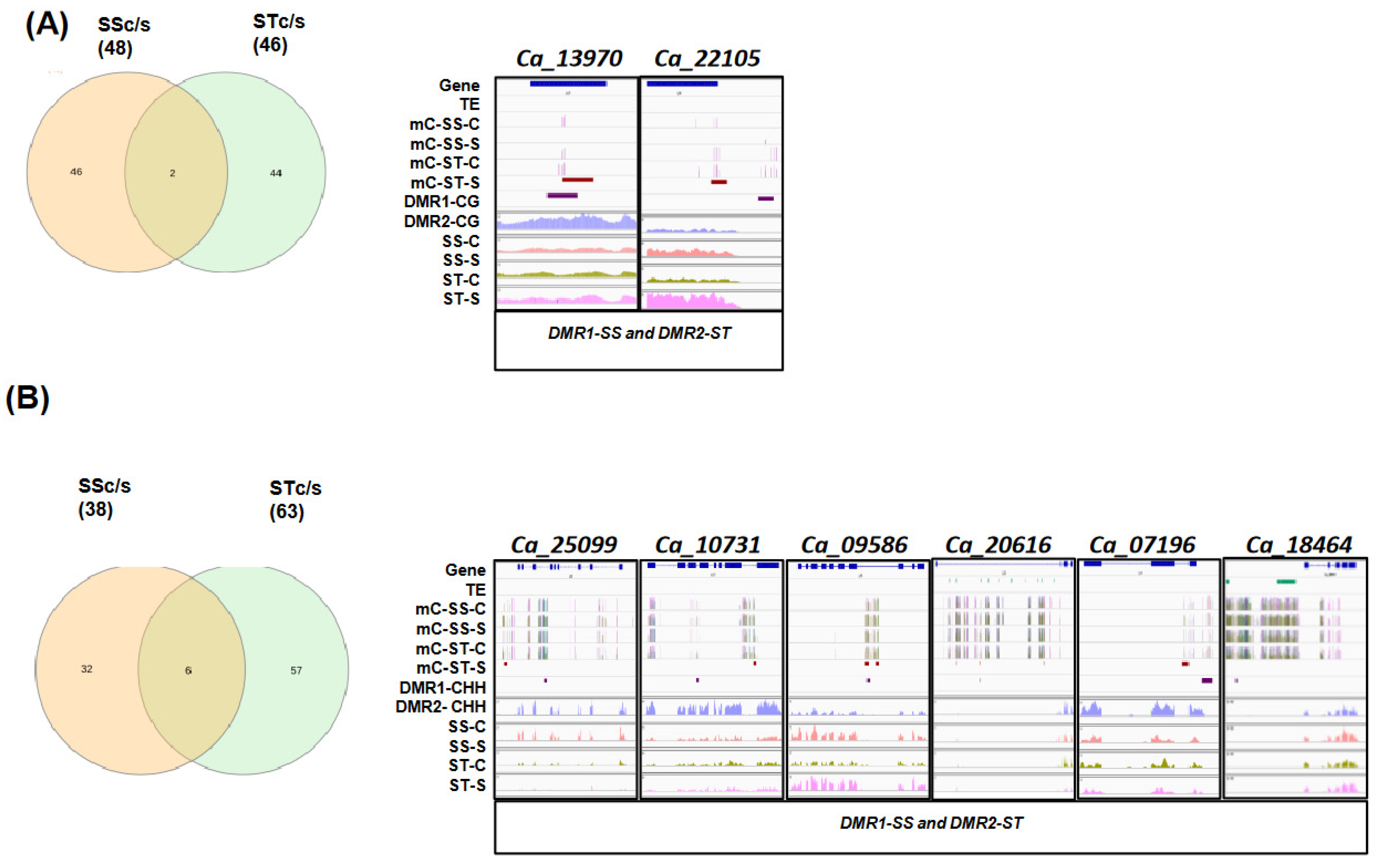
2.6. Identification of Conserved and Specific DMR-Associated DEGs (dmDEGs) between Genotypes
3. Discussion
4. Conclusions
5. Material and Methods
5.1. Plant Materials and Genomic DNA Isolation
5.2. Whole-Genome Bisulphite Sequencing and Data Analysis
5.3. RNA Sequencing and Data Analysis
5.4. Small RNA Sequencing and Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flowers, T.J.; Gaur, P.M.; Gowda, C.L.; Krishnamurthy, L.; Samineni, S.; Siddique, K.H.; Turner, N.C.; Vadez, V.; Varshney, R.K.; Colmer, T.D. Salt sensitivity in chickpea. Plant Cell Environ. 2010, 33, 490–509. [Google Scholar] [CrossRef]
- Gupta, B.; Huang, B. Mechanism of Salinity Tolerance in Plants: Physiological Biochemical and Molecular Characterization. Int. J. Genom. 2014, 2014, e701596. [Google Scholar] [CrossRef] [PubMed]
- Atieno, J.; Li, Y.; Langridge, P.; Dowling, K.; Brien, C.; Berger, B.; Varshney, R.K.; Sutton, T. Exploring genetic variation for salinity tolerance in chickpea using image-based phenotyping. Sci. Rep. 2017, 7, 1300. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, M.S.; Jain, M.; Garg, R. Discovery of DNA polymorphisms via whole genome resequencing and their functional relevance in salinity stress response in chickpea. Physiol. Plant. 2021, 173, 1573–1586. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.L.; Zhang, X.; Bernatavichute, Y.V.; Jacobsen, S.E. Two-Step Recruitment of RNA-Directed DNA Methylation to Tandem Repeats. PLOS Biol. 2006, 4, e363. [Google Scholar] [CrossRef]
- Miryeganeh, M. Plants’ Epigenetic Mechanisms and Abiotic Stress. Genes 2021, 12, 1106. [Google Scholar] [CrossRef]
- Ashapkin, V.V.; Kutueva, L.I.; Aleksandrushkina, N.I.; Vanyushin, B.F. Epigenetic Mechanisms of Plant Adaptation to Biotic and Abiotic Stresses. Int. J. Mol. Sci. 2020, 21, 7457. [Google Scholar] [CrossRef]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA Methylome Is Stable under Transgenerational Drought Stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef]
- Rajkumar, M.S.; Gupta, K.; Khemka, N.K.; Garg, R.; Jain, M. DNA methylation reprogramming during seed development and its functional relevance in seed size/weight determination in chickpea. Commun. Biol. 2020, 3, 340. [Google Scholar] [CrossRef]
- Domcke, S.; Bardet, A.F.; Ginno, P.A.; Hartl, D.; Burger, L.; Schübeler, D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 2015, 528, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, R.C.; Ecker, J.R. Epiallelic variation in Arabidopsis thaliana. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Sudan, J.; Raina, M.; Singh, R. Plant epigenetic mechanisms: Role in abiotic stress and their generational heritability. 3 Biotech 2018, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, A.; Becker, C.; Marconi, G.; Durr, J.; Price, J.; Hagmann, J.; Papareddy, R.; Putra, H.; Kageyama, J.; Becker, J.; et al. Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by DNA glycosylase activity. ELife 2016, 5, e13546. [Google Scholar] [CrossRef]
- Bilichak, A.; Kovalchuk, I. Transgenerational response to stress in plants and its application for breeding. J. Exp. Bot. 2016, 67, 2081–2092. [Google Scholar] [CrossRef]
- Garg, R.; Shankar, R.; Thakkar, B.; Kudapa, H.; Krishnamurthy, L.; Mantri, N.; Varshney, R.K.; Bhatia, S.; Jain, M. Transcriptome analyses reveal genotype- and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci. Rep. 2016, 6, 19228. [Google Scholar] [CrossRef]
- Vadez, V.; Krishnamurthy, L.; Thudi, M.; Thudi, C.; Colmer, T.D.; Turner, N.C.; Siddique, K.H.M.; Gaur, P.M.; Varshney, R.K. Assessment of ICCV 2× JG 62 chickpea progenies shows sensitivity of reproduction to salt stress and reveals QTLs for seed yield and seed number. Mol. Breed. 2012, 30, 9–21. [Google Scholar] [CrossRef]
- Zhong, X.; Hale, C.J.; Nguyen, M.; Ausin, I.; Groth, M.; Hetzel, J.; Vashisht, A.A.; Henderson, I.R.; Wohlschlegel, J.A.; Jacobsen, S.E. Domains rearranged methyltransferase3 controls DNA methylation and regulates RNA polymerase V transcript abundance in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 911–916. [Google Scholar] [CrossRef]
- Duan, C.G.; Zhang, H.; Tang, K.; Zhu, X.; Qian, W.; Hou, Y.J.; Wang, B.; Lang, Z.; Zhao, Y.; Wang, X.; et al. Specific but interdependent functions for Arabidopsis AGO4 and AGO6 in RNA-directed DNA methylation. EMBO J. 2015, 34, 581–592. [Google Scholar] [CrossRef]
- Wendte, J.M.; Zhang, Y.; Ji, L.; Shi, X.; Hazarika, R.R.; Shahryary, Y.; Johannes, F.; Schmitz, R.J. Epimutations are associated with CHROMOMETHYLASE 3-induced de novo DNA methylation. ELife 2019, 8, e47891. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Lee, S.H.; Cho, M.; Lee, O.R.; Yoo, H.; Cho, H.T. Differential auxin-transporting activities of PIN-FORMED proteins in Arabidopsis root hair cells. Plant Physiol. 2010, 153, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.L.; Zhang, G.; Tang, K.; Li, J.; Yang, L.; Huang, H.; Zhang, H.; Zhu, J.K. Dicer-independent RNA-directed DNA methylation in Arabidopsis. Cell Res. 2016, 26, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Gouil, Q.; Baulcombe, D.C. DNA Methylation Signatures of the Plant Chromomethyltransferases. PLoS Genet. 2016, 12, e1006526. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.D.; Zemach, A. Contiguous and stochastic CHH methylation patterns of plant DRM2 and CMT2 revealed by single-read methylome analysis. Genome Biol. 2020, 21, 194. [Google Scholar] [CrossRef]
- Morgado, L.; Jansen, R.C.; Johannes, F. Learning sequence patterns of AGO-sRNA affinity from high-throughput sequencing libraries to improve in silico functional small RNA detection and classification in plants. bioRxiv 2017, 1, 173575. [Google Scholar]
- Luhua, S.; Hegie, A.; Suzuki, N.; Shulaev, E.; Luo, X.; Cenariu, D.; Ma, V.; Kao, S.; Lim, J.; Gunay, M.B.; et al. Linking genes of unknown function with abiotic stress responses by high-throughput phenotype screening. Physiol. Plant. 2013, 148, 322–333. [Google Scholar] [CrossRef]
- Forsthoefel, N.R.; Klag, K.A.; McNichol, S.R.; Arnold, C.E.; Vernon, C.R.; Wood, W.W.; Vernon, D.M. Arabidopsis PIRL6 Is Essential for Male and Female Gametogenesis and Is Regulated by Alternative Splicing. Plant Physiol. 2018, 178, 1154–1169. [Google Scholar] [CrossRef]
- Patanun, O.; Ueda, M.; Itouga, M.; Kato, Y.; Utsumi, Y.; Matsui, A.; Tanaka, M.; Utsumi, C.; Sakakibara, H.; Yoshida, M.; et al. The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Alleviates Salinity Stress in Cassava. Front. Plant Sci. 2016, 7, 2039. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, N.; Li, Y.; Zhu, S.; Zhang, S.; Sun, Y.; Zhang, H.X.; Wang, L.; Su, H. Overexpression of PeMIPS1 confers tolerance to salt and copper stresses by scavenging reactive oxygen species in transgenic poplar. Tree Physiol. 2018, 38, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Domka, A.; Jędrzejczyk, R.; Ważny, R.; Gustab, M.; Kowalski, M.; Nosek, M.; Bizan, J.; Puschenreiter, M.; Vaculίk, M.; Kováč, J.; et al. Endophytic yeast protect plants against metal toxicity by inhibiting plant metal uptake through an ethylene-dependent mechanism. Plant Cell Env. 2023, 46, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Akhter, Z.; Bi, Z.; Ali, K.; Sun, C.; Fiaz, S.; Haider, F.U.; Bai, J. In Response to Abiotic Stress DNA Methylation Confers EpiGenetic Changes in Plants. Plants 2021, 10, 1096. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Misra, G.; Patel, R.K.; Priya, P.; Jhanwar, S.; Khan, A.W.; Shah, N.; Singh, V.K.; Garg, R.; Jeena, G.; et al. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 2013, 74, 715–729. [Google Scholar] [CrossRef]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef]
- Jain, M.; Bansal, J.; Rajkumar, M.S.; Garg, R. An integrated transcriptome mapping the regulatory network of coding and long non-coding RNAs provides a genomics resource in chickpea. Commun. Biol. 2022, 5, 1106. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Chevala, V.V.; Garg, R. Genome-wide discovery and differential regulation of conserved and novel microRNAs in chickpea via deep sequencing. J. Exp. Bot. 2014, 65, 5945–5958. [Google Scholar] [CrossRef]
- Garg, R.; Chevala, V.N.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 14922. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.K.; Colot, V. Gene body DNA methylation in plants: A means to an end or an end to a means? EMBO J. 2009, 28, 997–998. [Google Scholar] [CrossRef]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef]
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally induced phenotypes and DNA methylation: How to deal with unpredictable conditions until the next generation and after. Mol. Ecol. 2010, 19, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qin, L.; Xie, C.; Li, W.; Yuan, J.; Kong, L.; Yu, W.; Xia, G.; Liu, S. Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol. 2014, 55, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Lee, C.; Hwang, S.G.; Park, Y.C.; Lim, H.L.; Jang, C.S. Overexpression of the OsChI1 gene encoding a putative laccase precursor increases tolerance to drought and salinity stress in transgenic Arabidopsis. Gene 2014, 552, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Kim, J.K.; Jia, C.; Yin, F.; Kim, H.J.; Akram, W.; Hu, X.; Li, X. Comparative Transcriptome and Metabolic Profiling Analysis of Buckwheat (Fagopyrum Tataricum (L.) Gaertn,) under Salinity Stress. Metabolites 2019, 9, 225. [Google Scholar] [CrossRef]
- Yang, Z.; Li, J.L.; Liu, L.N.; Xie, Q.; Sui, N. Photosynthetic Regulation Under Salt Stress and Salt-Tolerance Mechanism of Sweet Sorghum. Front. Plant Sci. 2019, 10, 1722. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, M.S.; Shankar, R.; Garg, R.; Jain, M. Bisulphite sequencing reveals dynamic DNA methylation under desiccation and salinity stresses in rice cultivars. Genomics 2020, 112, 3537–3548. [Google Scholar] [CrossRef]
- Wang, W.; Huang, F.; Qin, Q.; Zhao, X.; Li, Z.; Fu, B. Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery. Biochem. Biophys. Res. Commun. 2015, 465, 790–796. [Google Scholar] [CrossRef]
- Takanashi, K.; Shitan, N.; Yazaki, K. The multidrug and toxic compound extrusion (MATE) family in plants. Plant Biotechnol. 2014, 31, 417–430. [Google Scholar] [CrossRef]
- Sui, W.; Guo, K.; Li, L.; Liu, S.; Takano, T.; Zhang, X. Arabidopsis Ca2+-dependent nuclease AtCaN2 plays a negative role in plant responses to salt stress. Plant Sci. Int. J. Exp. Plant Biol. 2019, 281, 213–222. [Google Scholar] [CrossRef]
- Jiang, Y.; Deyholos, M.K. Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 2009, 69, 91–105. [Google Scholar] [CrossRef]
- Rietz, S.; Dermendjiev, G.; Oppermann, E.; Tafesse, F.G.; Effendi, Y.; Holk, A.; Parker, J.E.; Teige, M.; Scherer, G.F.E. Roles of Arabidopsis patatin-related phospholipases a in root development are related to auxin responses and phosphate deficiency. Mol. Plant 2010, 3, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, V.K.; Jain, A.; Poling, M.D.; Lewis, A.J.; Raghothama, K.G.; Smith, A.P. Arabidopsis Pht1;5 mobilizes phosphate between source and sink organs and influences the interaction between phosphate homeostasis and ethylene signaling. Plant Physiol. 2011, 156, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, H.; Khemka, N.; Jain, M.; Garg, R. Genome-wide bisulphite-sequencing reveals organ-specific methylation patterns in chickpea. Sci. Rep. 2018, 8, 9704. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, K.; Garg, R. Unravelling Differential DNA Methylation Patterns in Genotype Dependent Manner under Salinity Stress Response in Chickpea. Int. J. Mol. Sci. 2023, 24, 1863. https://doi.org/10.3390/ijms24031863
Gupta K, Garg R. Unravelling Differential DNA Methylation Patterns in Genotype Dependent Manner under Salinity Stress Response in Chickpea. International Journal of Molecular Sciences. 2023; 24(3):1863. https://doi.org/10.3390/ijms24031863
Chicago/Turabian StyleGupta, Khushboo, and Rohini Garg. 2023. "Unravelling Differential DNA Methylation Patterns in Genotype Dependent Manner under Salinity Stress Response in Chickpea" International Journal of Molecular Sciences 24, no. 3: 1863. https://doi.org/10.3390/ijms24031863
APA StyleGupta, K., & Garg, R. (2023). Unravelling Differential DNA Methylation Patterns in Genotype Dependent Manner under Salinity Stress Response in Chickpea. International Journal of Molecular Sciences, 24(3), 1863. https://doi.org/10.3390/ijms24031863