
Development of a Versatile Method to Construct Direct Electron Transfer-Type Enzyme Complexes Employing SpyCatcher/SpyTag System

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
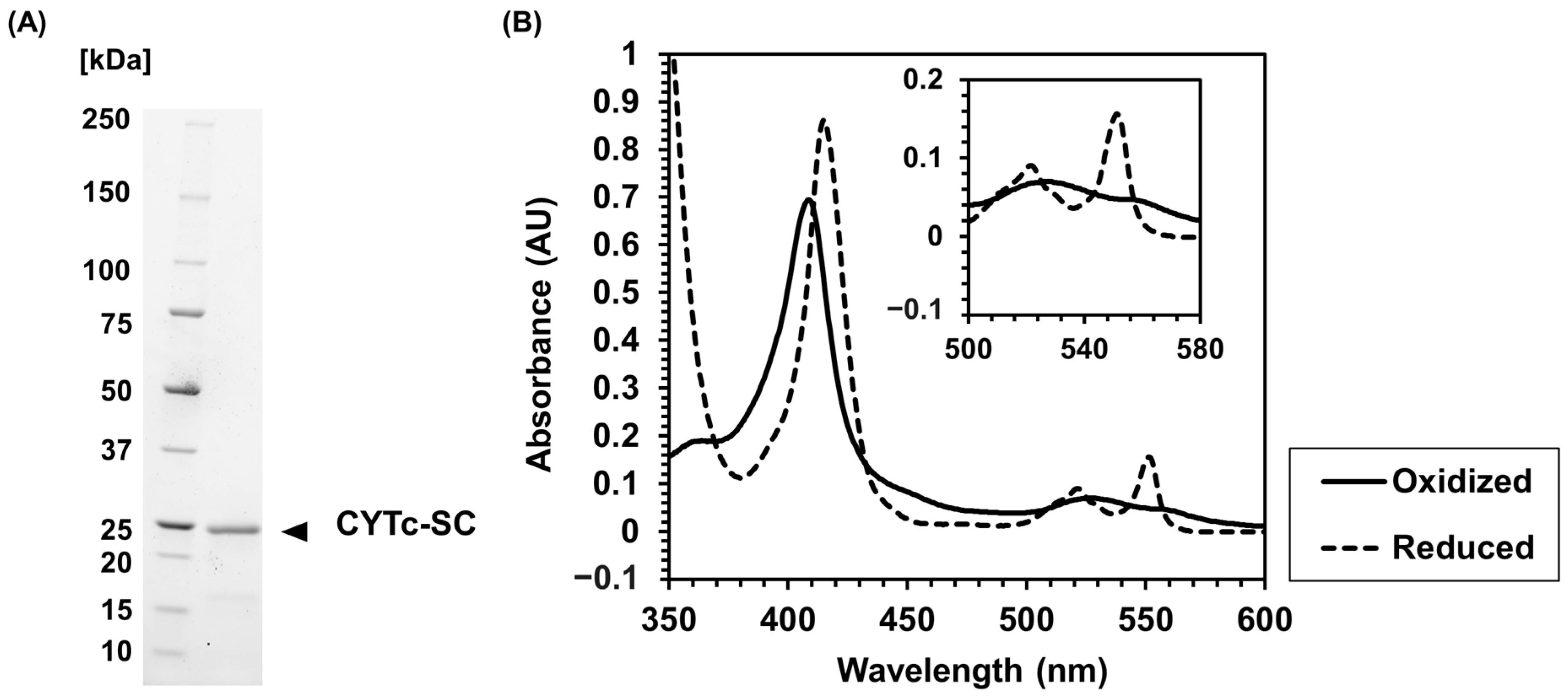
2.1. Recombinant Expression and Characterization of CYTc-SC
2.2. Complex Formation between CYTc-SC and ST-Enzymes
2.3. Investigation of Intramolecular Electron Transfer in CYTc-SC/ST-Enzyme Complexes
2.4. Electrochemical Evaluation of CYTc-SC/ST-Enzyme Complexes
3. Discussion
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Construction of the Expression Vectors
4.3. Recombinant Expression
4.4. Preparation of Oxidized or Reduced Sample of CYTc-SC
4.5. Formation of CYTc-SpyCatcher/SpyTag-Enzyme
4.6. Enzyme Activity and Absorption Spectrum Analysis of the Complexes
4.7. Electrochemical Measurement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ferri, S.; Kojima, K.; Sode, K. Review of Glucose Oxidases and Glucose Dehydrogenases: A Bird’s Eye View of Glucose Sensing Enzymes. J. Diabetes Sci. Technol. 2011, 5, 1068–1076. [Google Scholar] [CrossRef]
- Yamashita, Y.; Lee, I.; Loew, N.; Sode, K. Direct Electron Transfer (DET) Mechanism of FAD Dependent Dehydrogenase Complexes ∼from the Elucidation of Intra- and Inter-Molecular Electron Transfer Pathway to the Construction of Engineered DET Enzyme Complexes∼. Curr. Opin. Electrochem. 2018, 12, 92–100. [Google Scholar] [CrossRef]
- Okuda-Shimazaki, J.; Yoshida, H.; Sode, K. FAD Dependent Glucose Dehydrogenases—Discovery and Engineering of Representative Glucose Sensing Enzymes-. Bioelectrochemistry 2020, 132, 107414. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Gorton, L.; Antiochia, R. Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells. Sensors 2018, 18, 1319. [Google Scholar] [CrossRef] [PubMed]
- Schachinger, F.; Chang, H.; Scheiblbrandner, S.; Ludwig, R. Amperometric Biosensors Based on Direct Electron Transfer Enzymes. Molecules 2021, 26, 4525. [Google Scholar] [CrossRef]
- Kimura, H.; Miura, D.; Tsugawa, W.; Ikebukuro, K.; Sode, K.; Asano, R. Rapid and Homogeneous Electrochemical Detection by Fabricating a High Affinity Bispecific Antibody-Enzyme Complex Using Two Catcher/Tag Systems. Biosens. Bioelectron. 2021, 175, 112885. [Google Scholar] [CrossRef] [PubMed]
- Miura, D.; Kimura, H.; Tsugawa, W.; Ikebukuro, K.; Sode, K.; Asano, R. Rapid, Convenient, and Highly Sensitive Detection of Human Hemoglobin in Serum Using a High-Affinity Bivalent Antibody–Enzyme Complex. Talanta 2021, 234, 122638. [Google Scholar] [CrossRef] [PubMed]
- Jiaul Haque, A.; Kwon, J.; Kim, J.; Kim, G.; Lee, N.; Ho Yoon, Y.; Yang, H. Sensitive and Low-background Electrochemical Immunosensor Employing Glucose Dehydrogenase and 1,10-Phenanthroline-5,6-dione. Electroanalysis 2021, 33, 1877–1885. [Google Scholar] [CrossRef]
- Vogt, S.; Schneider, M.; Schäfer-Eberwein, H.; Nöll, G. Determination of the pH Dependent Redox Potential of Glucose Oxidase by Spectroelectrochemistry. Anal. Chem. 2014, 86, 7530–7535. [Google Scholar] [CrossRef]
- Wilson, G.S. Native Glucose Oxidase Does Not Undergo Direct Electron Transfer. Biosens. Bioelectron. 2016, 82, vii–viii. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Al-Lolage, F.A. There Is No Evidence to Support Literature Claims of Direct Electron Transfer (DET) for Native Glucose Oxidase (GOx) at Carbon Nanotubes or Graphene. J. Electroanal. Chem. 2018, 819, 26–37. [Google Scholar] [CrossRef]
- López Marzo, A.M.; Mayorga-Martinez, C.C.; Pumera, M. 3D-Printed Graphene Direct Electron Transfer Enzyme Biosensors. Biosens. Bioelectron. 2020, 151, 111980. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Fujii, T.; Honda, M.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of FAD-Dependent Glucose Dehydrogenase Using Porous Gold Electrodes and Enzymatically Implanted Platinum Nanoclusters. Bioelectrochemistry 2020, 133, 107457. [Google Scholar] [CrossRef] [PubMed]
- Hatada, M.; Loew, N.; Inose-Takahashi, Y.; Okuda-Shimazaki, J.; Tsugawa, W.; Mulchandani, A.; Sode, K. Development of a Glucose Sensor Employing Quick and Easy Modification Method with Mediator for Altering Electron Acceptor Preference. Bioelectrochemistry 2018, 121, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Hiraka, K.; Kojima, K.; Tsugawa, W.; Asano, R.; Ikebukuro, K.; Sode, K. Rational Engineering of Aerococcus viridans L-Lactate Oxidase for the Mediator Modification to Achieve Quasi-Direct Electron Transfer Type Lactate Sensor. Biosens. Bioelectron. 2020, 151, 111974. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Lee, J.; Loew, N.; Takahashi-Inose, Y.; Okuda-Shimazaki, J.; Kojima, K.; Mori, K.; Tsugawa, W.; Sode, K. Engineered Glucose Oxidase Capable of Quasi-Direct Electron Transfer after a Quick-and-Easy Modification with a Mediator. Int. J. Mol. Sci. 2020, 21, 1137. [Google Scholar] [CrossRef] [PubMed]
- Hatada, M.; Saito, S.; Yonehara, S.; Tsugawa, W.; Asano, R.; Ikebukuro, K.; Sode, K. Development of Glycated Peptide Enzyme Sensor Based Flow Injection Analysis System for Haemoglobin A1c Monitoring Using Quasi-Direct Electron Transfer Type Engineered Fructosyl Peptide Oxidase. Biosens. Bioelectron. 2021, 177, 112984. [Google Scholar] [CrossRef]
- Takamatsu, S.; Lee, I.; Lee, J.; Asano, R.; Tsugawa, W.; Ikebukuro, K.; Dick, J.E.; Sode, K. Transient Potentiometry Based d-Serine Sensor Using Engineered D-Amino Acid Oxidase Showing Quasi-Direct Electron Transfer Property. Biosens. Bioelectron. 2022, 200, 113927. [Google Scholar] [CrossRef]
- Degani, Y.; Heller, A. Direct Electrical Communication between Chemically Modified Enzymes and Metal Electrodes. I. Electron Transfer from Glucose Oxidase to Metal Electrodes via Electron Relays, Bound Covalently to the Enzyme. J. Phys. Chem. 1987, 91, 1285–1289. [Google Scholar] [CrossRef]
- Degani, Y.; Heller, A. Direct Electrical Communication between Chemically Modified Enzymes and Metal Electrodes. 2. Methods for Bonding Electron-Transfer Relays to Glucose Oxidase and d-Amino-Acid Oxidase. J. Am. Chem. Soc. 1988, 110, 2615–2620. [Google Scholar] [CrossRef]
- Katz, E.; Bückmann, A.F.; Willner, I. Self-Powered Enzyme-Based Biosensors. J. Am. Chem. Soc. 2001, 123, 10752–10753. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, W.; Ohara, T.J.; Schmidt, H.L.; Heller, A. Electron Transfer between Glucose Oxidase and Electrodes via Redox Mediators Bound with Flexible Chains to the Enzyme Surface. J. Am. Chem. Soc. 1991, 113, 1394–1397. [Google Scholar] [CrossRef]
- Schuhmann, W. Electron-Transfer Pathways in Amperometric Biosensors. Ferrocene-Modified Enzymes Entrapped in Conducting-Polymer Layers. Biosens. Bioelectron. 1995, 10, 181–193. [Google Scholar] [CrossRef]
- Han, G.-C.; Su, X.; Hou, J.; Ferranco, A.; Feng, X.-Z.; Zeng, R.; Chen, Z.; Kraatz, H.-B. Disposable Electrochemical Sensors for Hemoglobin Detection Based on Ferrocenoyl Cysteine Conjugates Modified Electrode. Sens. Actuators B Chem. 2019, 282, 130–136. [Google Scholar] [CrossRef]
- Okuda, J.; Sode, K. PQQ Glucose Dehydrogenase with Novel Electron Transfer Ability. Biochem. Biophys. Res. Commun. 2004, 314, 793–797. [Google Scholar] [CrossRef]
- Oubrie, A.; Rozeboom, H.J.; Kalk, K.H.; Huizinga, E.G.; Dijkstra, B.W. Crystal Structure of Quinohemoprotein Alcohol Dehydrogenase from Comamonas testosteroni. J. Biol. Chem. 2002, 277, 3727–3732. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Okuda-Shimazaki, J.; Mori, K.; Kojima, K.; Tsugawa, W.; Ikebukuro, K.; Lin, C.E.; La Belle, J.; Yoshida, H.; Sode, K. Designer Fungus FAD Glucose Dehydrogenase Capable of Direct Electron Transfer. Biosens. Bioelectron. 2019, 123, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Yanase, T.; Okuda-Shimazaki, J.; Mori, K.; Kojima, K.; Tsugawa, W.; Sode, K. Creation of a Novel DET Type FAD Glucose Dehydrogenase Harboring Escherichia coli Derived Cytochrome b562 as an Electron Transfer Domain. Biochem. Biophys. Res. Commun. 2020, 530, 82–86. [Google Scholar] [CrossRef]
- Hiraka, K.; Tsugawa, W.; Asano, R.; Yokus, M.A.; Ikebukuro, K.; Daniele, M.A.; Sode, K. Rational Design of Direct Electron Transfer Type l-Lactate Dehydrogenase for the Development of Multiplexed Biosensor. Biosens. Bioelectron. 2021, 176, 112933. [Google Scholar] [CrossRef]
- Viehauser, M.C.; Breslmayr, E.; Scheiblbrandner, S.; Schachinger, F.; Ma, S.; Ludwig, R. A Cytochrome b-Glucose Dehydrogenase Chimeric Enzyme Capable of Direct Electron Transfer. Biosens. Bioelectron. 2022, 196, 113704. [Google Scholar] [CrossRef]
- Yu, K.; Liu, C.; Kim, B.-G.; Lee, D.-Y. Synthetic Fusion Protein Design and Applications. Biotechnol. Adv. 2015, 33, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.J.; Geeson, M.B.; Journeaux, T.; Bernardes, G.J.L. Chemical and Enzymatic Methods for Post-Translational Protein-Protein Conjugation. J. Am. Chem. Soc. 2022, 144, 14404–14419. [Google Scholar] [CrossRef] [PubMed]
- Thöny-Meyer, L.; Kunzler, P. Translocation to the Periplasm and Signal Sequence Cleavage of Preapocytochrome c Depend on sec and lep, but Not on the ccm Gene Products. Eur. J. Biochem. 1997, 799, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Thöny-Meyer, L. Biogenesis of Respiratory Cytochromes in Bacteria. Microbiol. Mol. Biol. Rev. 1997, 61, 337–376. [Google Scholar] [CrossRef] [PubMed]
- Mavridou, D.A.I.; Ferguson, S.J.; Stevens, J.M. Cytochrome c Assembly. IUBMB Life 2013, 65, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Arslan, E.; Schulz, H.; Zufferey, R.; Künzler, P.; Thöny-Meyer, L. Overproduction of the Bradyrhizobium japonicum c-Type Cytochrome Subunits of the cbb3 Oxidase in Escherichia coli. Biochem. Biophys. Res. Commun. 1998, 251, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Pollock, W.B.R.; Rosell, F.I.; Twitchett, M.B.; Dumont, M.E.; Mauk, A.G. Bacterial Expression of a Mitochondrial Cytochrome c. Trimethylation of Lys72 in Yeast Iso-1-Cytochrome c and the Alkaline Conformational Transition. Biochemistry 1998, 37, 6124–6131. [Google Scholar] [CrossRef]
- Asher, W.B.; Bren, K.L. Cytochrome c Heme Lyase Can Mature a Fusion Peptide Composed of the Amino-Terminal Residues of Horse Cytochrome c. Chem. Commun. 2012, 48, 8344. [Google Scholar] [CrossRef]
- Sanders, C.; Lill, H. Expression of Prokaryotic and Eukaryotic Cytochromes c in Escherichia coli. Biochim. Biophys. Acta—Bioenerg. 2000, 1459, 131–138. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide Tag Forming a Rapid Covalent Bond to a Protein, through Engineering a Bacterial Adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef]
- Reddington, S.C.; Howarth, M. Secrets of a Covalent Interaction for Biomaterials and Biotechnology: SpyTag and SpyCatcher. Curr. Opin. Chem. Biol. 2015, 29, 94–99. [Google Scholar] [CrossRef]
- Kimura, H.; Asano, R.; Tsukamoto, N.; Tsugawa, W.; Sode, K. Convenient and Universal Fabrication Method for Antibody-Enzyme Complexes as Sensing Elements Using the SpyCatcher/SpyTag System. Anal. Chem. 2018, 90, 14500–14506. [Google Scholar] [CrossRef]
- Miyazaki, R.; Yamazaki, T.; Yoshimatsu, K.; Kojima, K.; Asano, R.; Sode, K.; Tsugawa, W. Elucidation of the Intra- and Inter-Molecular Electron Transfer Pathways of Glucoside 3-Dehydrogenase. Bioelectrochemistry 2018, 122, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Sakai, G.; Kojima, K.; Mori, K.; Oonishi, Y.; Sode, K. Stabilization of Fungi-Derived Recombinant FAD-Dependent Glucose Dehydrogenase by Introducing a Disulfide Bond. Biotechnol. Lett. 2015, 37, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Sakai, G.; Mori, K.; Kojima, K.; Kamitori, S.; Sode, K. Structural Analysis of Fungus-Derived FAD Glucose Dehydrogenase. Sci. Rep. 2015, 5, 13498. [Google Scholar] [CrossRef]
- Saam, J.; Rosini, E.; Molla, G.; Schulten, K.; Pollegioni, L.; Ghisla, S. O2 Reactivity of Flavoproteins: Dynamic Access of Dioxygen to the Active Site and Role of a H+ Relay System in d-Amino Acid Oxidase. J. Biol. Chem. 2010, 285, 24439–24446. [Google Scholar] [CrossRef]
- Hiraka, K.; Kojima, K.; Lin, C.E.; Tsugawa, W.; Asano, R.; La Belle, J.T.; Sode, K. Minimizing the Effects of Oxygen Interference on l-Lactate Sensors by a Single Amino Acid Mutation in Aerococcus viridans l-Lactate Oxidase. Biosens. Bioelectron. 2018, 103, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Okuda-Shimazaki, J.; Loew, N.; Hirose, N.; Kojima, K.; Mori, K.; Tsugawa, W.; Sode, K. Construction and Characterization of Flavin Adenine Dinucleotide Glucose Dehydrogenase Complex Harboring a Truncated Electron Transfer Subunit. Electrochim. Acta 2018, 277, 276–286. [Google Scholar] [CrossRef]
- Ito, K.; Okuda-Shimazaki, J.; Kojima, K.; Mori, K.; Tsugawa, W.; Asano, R.; Ikebukuro, K.; Sode, K. Strategic Design and Improvement of the Internal Electron Transfer of Heme b Domain-Fused Glucose Dehydrogenase for Use in Direct Electron Transfer-Type Glucose Sensors. Biosens. Bioelectron. 2021, 176, 112911. [Google Scholar] [CrossRef]
- Li, L.; Fierer, J.O.; Rapoport, T.A.; Howarth, M. Structural Analysis and Optimization of the Covalent Association between SpyCatcher and a Peptide Tag. J. Mol. Biol. 2014, 426, 309–317. [Google Scholar] [CrossRef]
- Studier, F.W. Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Lai, R.Y.; Plaxco, K.W. Preparation of Electrode-Immobilized, Redox-Modified Oligonucleotides for Electrochemical DNA and Aptamer-Based Sensing. Nat. Protoc. 2007, 2, 2875–2880. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidoreductase | Type | Cofactor | Origin | Dehydrogenase Activity (U/mg) | Dehydrogenase Activity (U/µmol) | Quaternary Structure | Molecular Weight of Monomer (kDa) | Reference |
|---|---|---|---|---|---|---|---|---|
| Glucose dehydrogenase V149C/G190C (GDH) | Dehydrogenase thermostable mutant | FAD | Aspergillus flavus | 2.0 × 102 | 5.6 | Monomer | 60 | [44] |
| d-Amino acid oxidase G52V (DAAOx) | Oxygen-insensitive oxidase mutant | FAD | Rhodotorula glacilis | 8.4 | 0.20 | Homodimer | 41 | [18,46] |
| l-Lactate oxidase A96L/N212K (LOx) | Oxygen-insensitive oxidase mutant | FMN | Aerococcus viridans | 1.7 × 102 | 3.8 | Homotetramer | 44 | [15] |
| KM (mM) | Vmax (U/µmol) | ||
|---|---|---|---|
| GDH | ST-GDH | 56 | 3.2 |
| CYTc-SC/ST-GDH | 44 | 2.0 | |
| DAAOx | ST-DAAOx | 2.6 | 0.22 |
| CYTc-SC/ST-DAAOx | 1.9 | 0.10 | |
| LOx | ST-LOx | 3.1 | 2.0 |
| CYTc-SC/ST-LOx | 3.3 | 1.4 |
| KMapp (mM) | Imaxapp (nA) | |
|---|---|---|
| CYTc-SC/ST-GDH | 5.6 | 55 |
| CYTc-SC/ST-DAAOx | 0.3 | 7.5 |
| CYTc-SC/ST-LOx | 1.8 | 410 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanase, T.; Okuda-Shimazaki, J.; Asano, R.; Ikebukuro, K.; Sode, K.; Tsugawa, W. Development of a Versatile Method to Construct Direct Electron Transfer-Type Enzyme Complexes Employing SpyCatcher/SpyTag System. Int. J. Mol. Sci. 2023, 24, 1837. https://doi.org/10.3390/ijms24031837
Yanase T, Okuda-Shimazaki J, Asano R, Ikebukuro K, Sode K, Tsugawa W. Development of a Versatile Method to Construct Direct Electron Transfer-Type Enzyme Complexes Employing SpyCatcher/SpyTag System. International Journal of Molecular Sciences. 2023; 24(3):1837. https://doi.org/10.3390/ijms24031837
Chicago/Turabian StyleYanase, Takumi, Junko Okuda-Shimazaki, Ryutaro Asano, Kazunori Ikebukuro, Koji Sode, and Wakako Tsugawa. 2023. "Development of a Versatile Method to Construct Direct Electron Transfer-Type Enzyme Complexes Employing SpyCatcher/SpyTag System" International Journal of Molecular Sciences 24, no. 3: 1837. https://doi.org/10.3390/ijms24031837
APA StyleYanase, T., Okuda-Shimazaki, J., Asano, R., Ikebukuro, K., Sode, K., & Tsugawa, W. (2023). Development of a Versatile Method to Construct Direct Electron Transfer-Type Enzyme Complexes Employing SpyCatcher/SpyTag System. International Journal of Molecular Sciences, 24(3), 1837. https://doi.org/10.3390/ijms24031837