
In Vitro and In Vivo Evidence towards Fibronectin’s Protective Effects against Prion Infection
 , ,
, ,  ,
, 

Abstract
:1. Introduction
2. Results
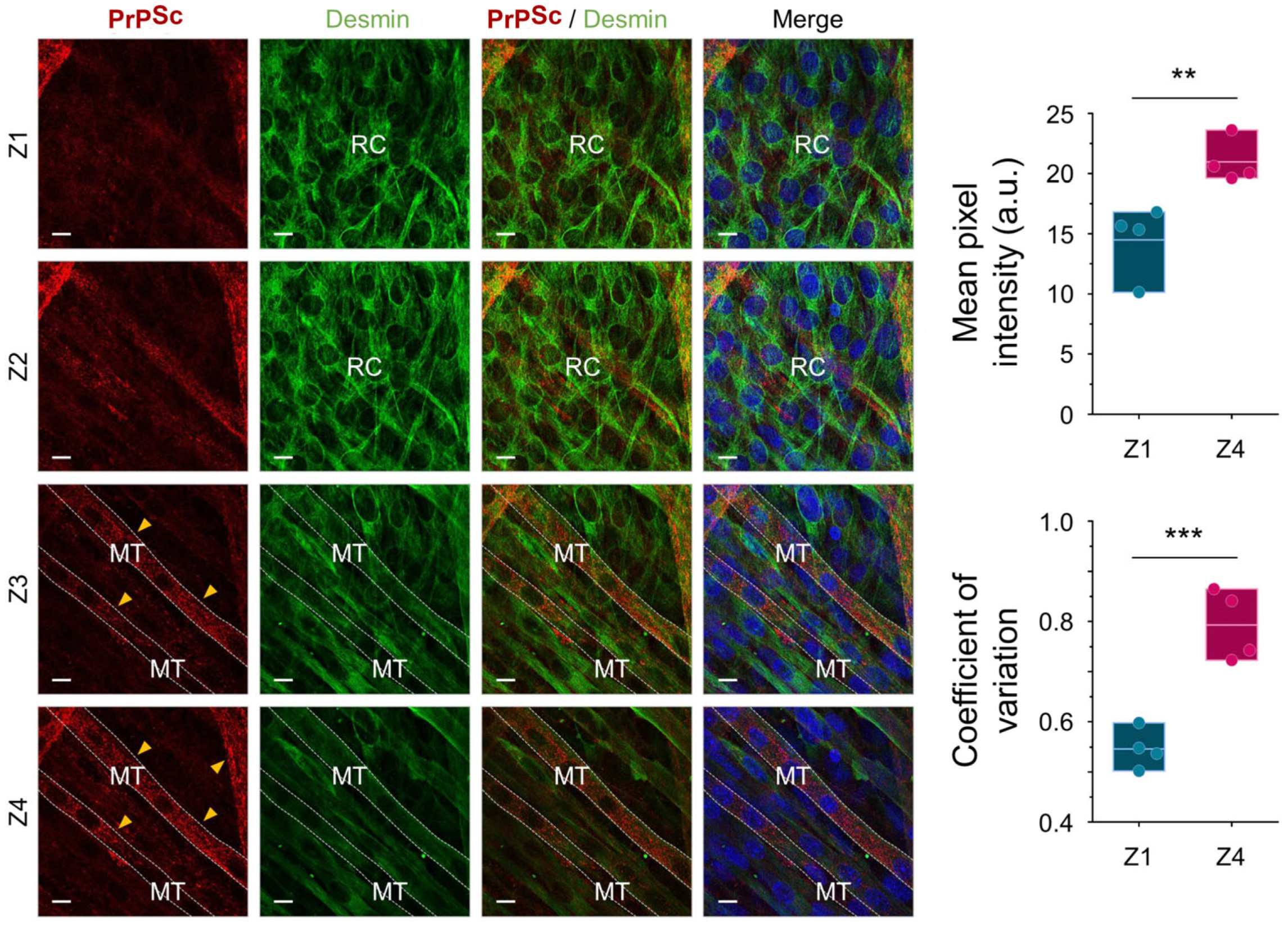
2.1. PrPSc Replicates and Accumulates Efficiently in Myotubes but Not in Reserve Cells
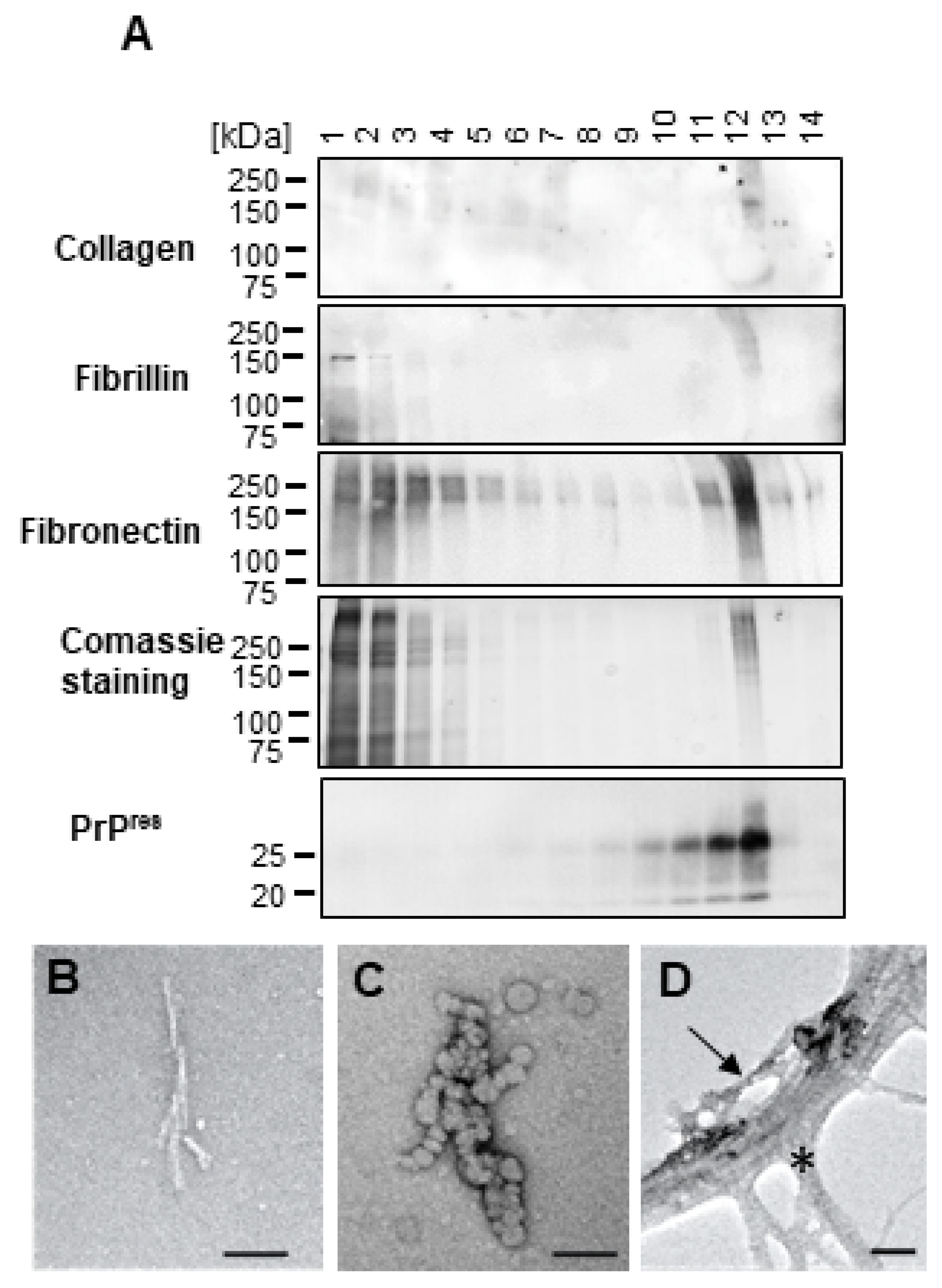
2.2. High Molecular Weight Proteins Co-Purify with dC2C12 PrPSc and Reduce Its Infectivity
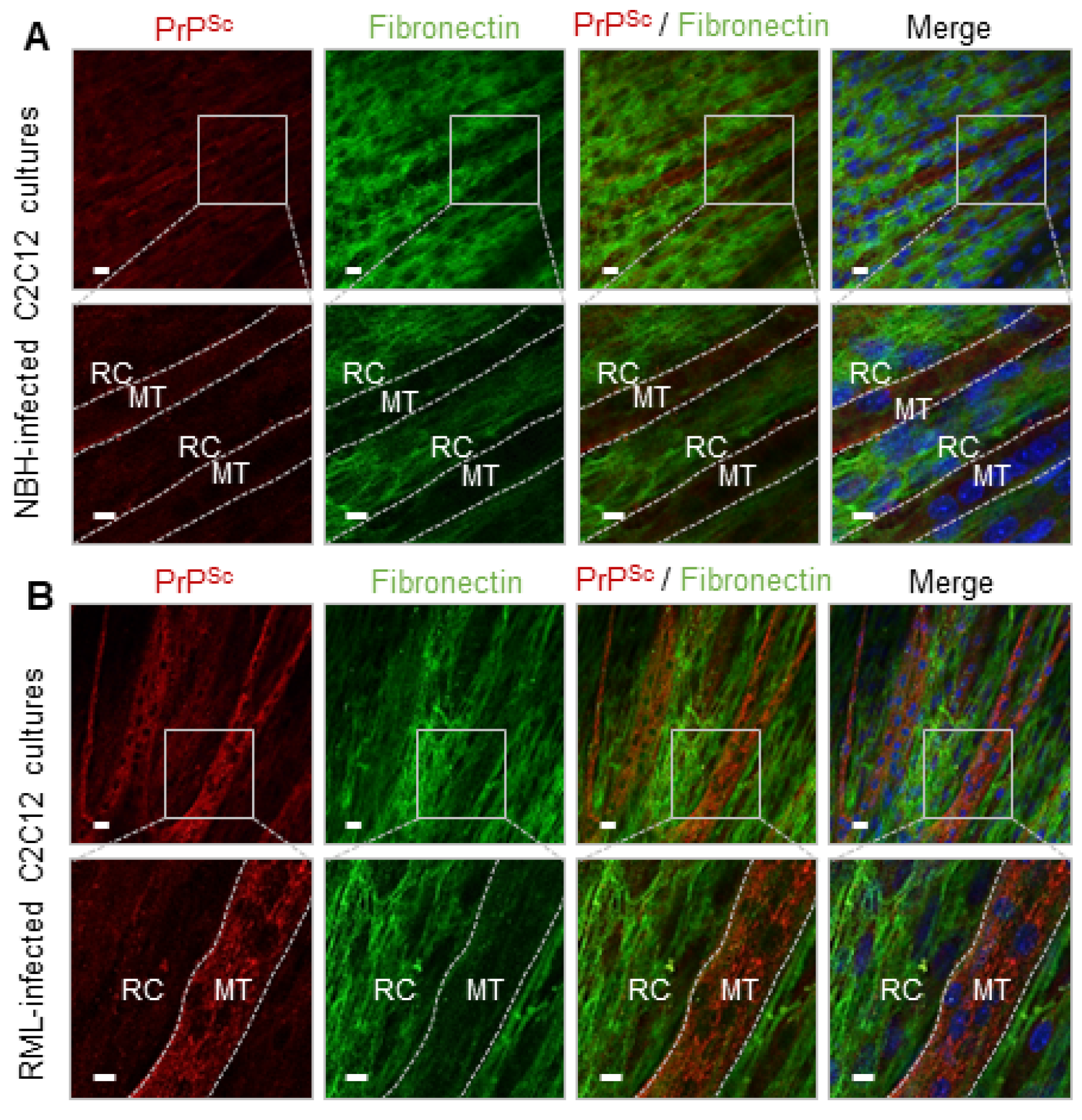
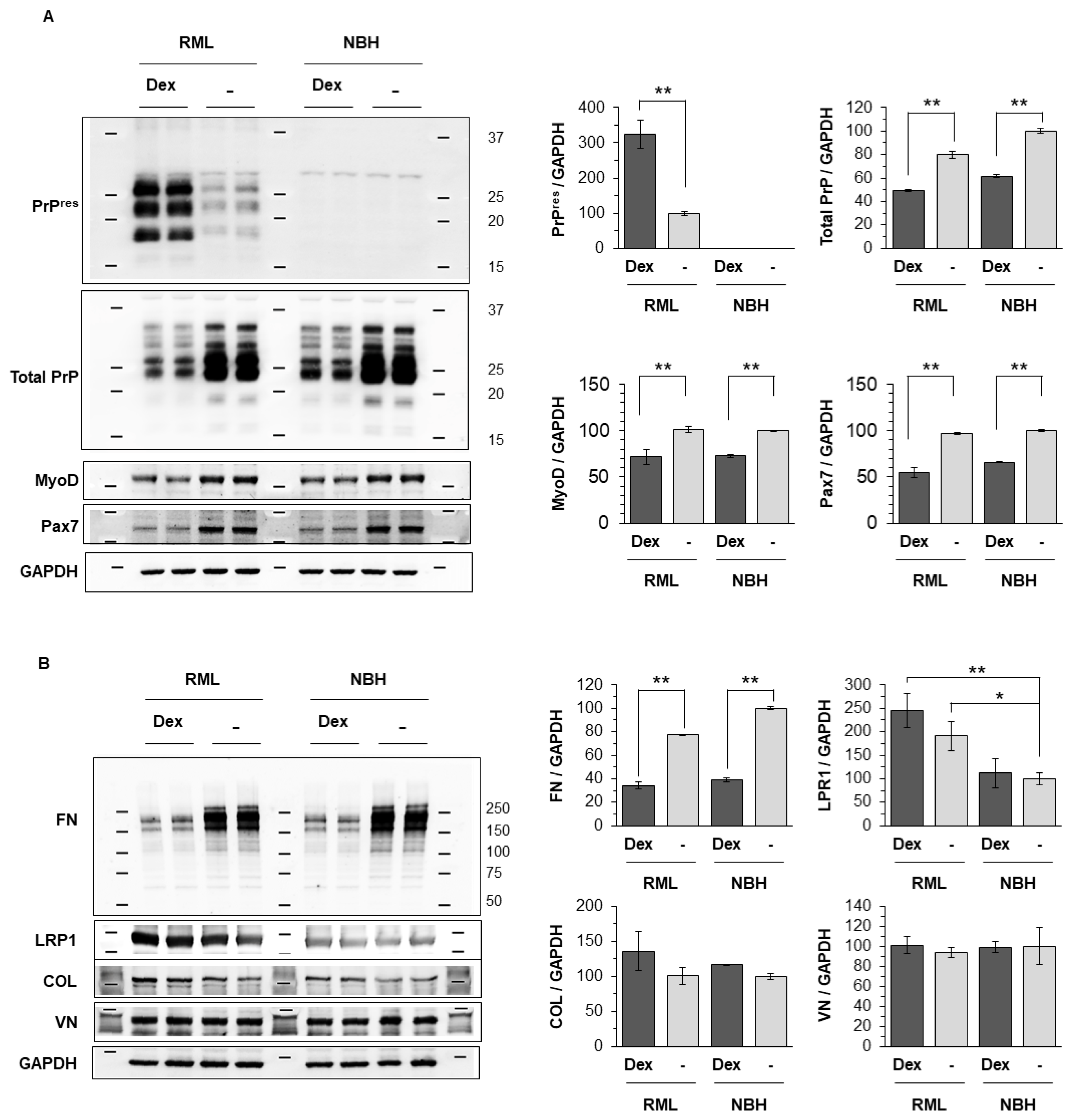
2.3. Extracellular Matrix Proteins Co-Precipitate and Co-Migrate with PrPSc from dC2C12
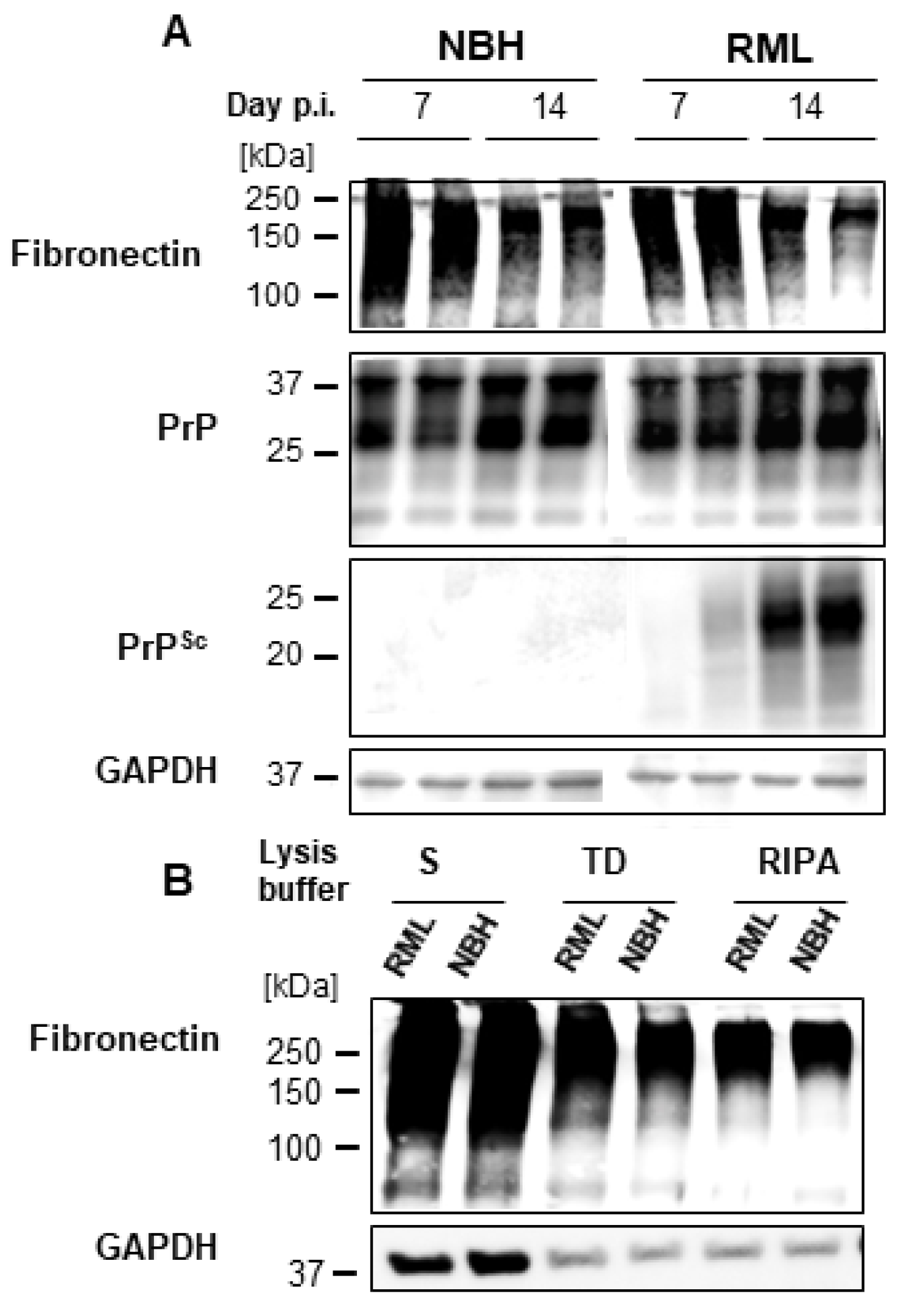
2.4. FN Negatively Correlates with PrPSc in dC2C12 Cultures
2.5. FN Does Not Interact with PrPC
2.6. Dexamethasone Treatment Renders Reserve Cells Permissive to Prion Infection
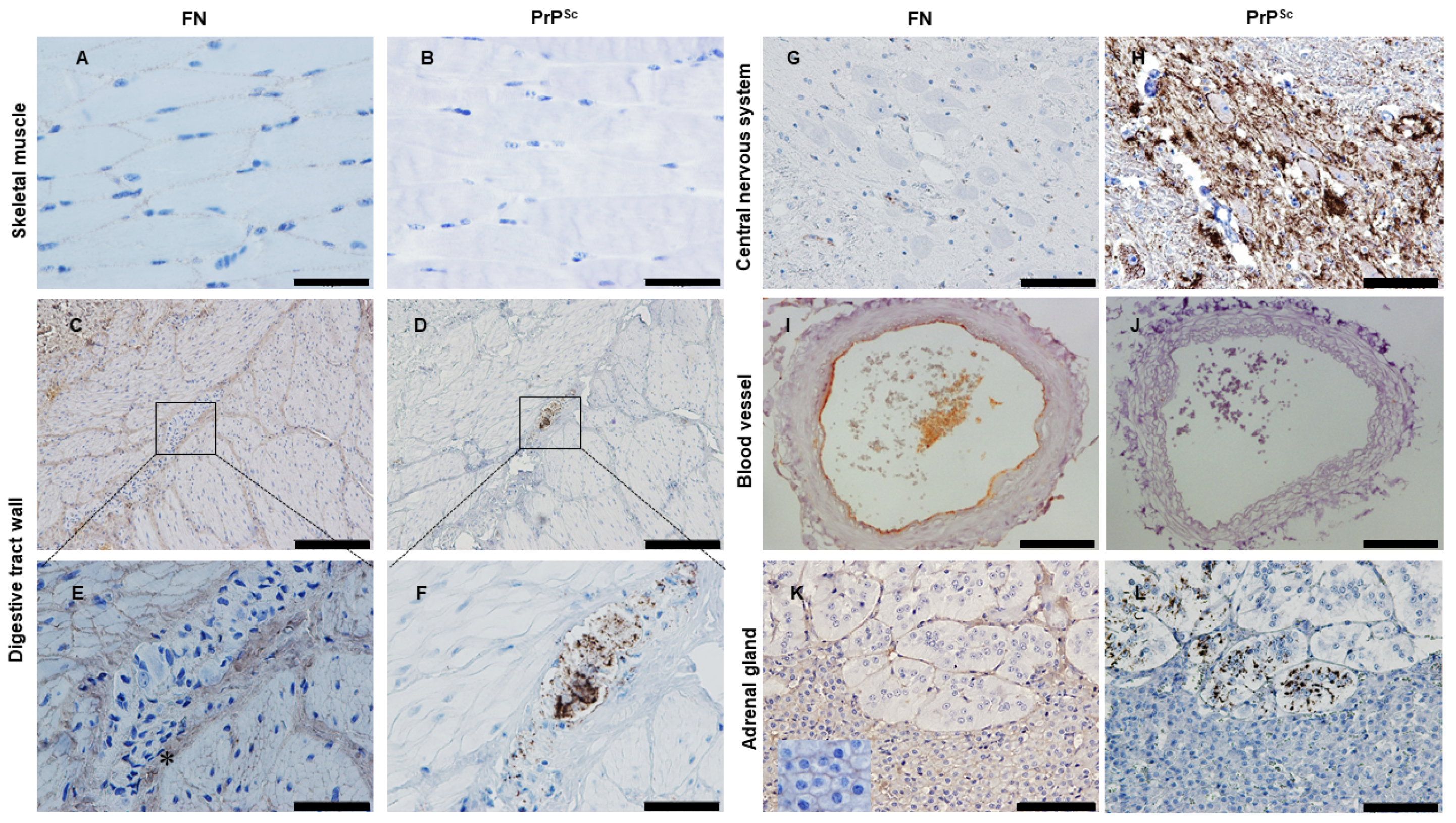
2.7. Looking for PrPSc and FN in Tissues from Naturally Infected Sheep
3. Discussion
3.1. Prion Tropism in Reserve Cells and Myotubes
3.2. Fibronectin and Prion Accumulation in Muscle
3.3. Fibronectin and Prion Accumulation in Nervous System and Other Peripheral Tissues
4. Materials and Methods
4.1. Infection of Cell Cultures and Sample Preparation
4.2. Immunocytochemistry
4.3. Measurements of Myotube Widths
4.4. PTA Based Purification Protocol
4.5. Velocity Sedimentation
4.6. Statistical Analysis
4.7. Negative Stain Electron Microscopy
4.8. Mass Spectrometry
4.8.1. Electrophoresis and In-Gel Protein Digestion
4.8.2. Mass Spectrometry and Database Search Parameters
4.9. Scrapie Cell Assay
4.10. Surface Plasmon Resonance Measurements
4.11. Immunohistochemistry for PrP and FN in Paraffin Tissues
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B.; Bolton, D.C.; Groth, D.F.; Bowman, K.A.; Cochran, S.P.; McKinley, M.P. Further purification and characterization of scrapie prions. Biochemistry 1982, 21, 6942–6950. [Google Scholar] [CrossRef] [PubMed]
- van Keulen, L.J.M.; Bossers, A.; van Zijderveld, F. TSE pathogenesis in cattle and sheep. Vet. Res. 2008, 39, 24. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Eiden, M.; Balkema-Buschmann, A.; Greenlee, J.; Schatzl, H.; Fast, C.; Richt, J.; Hildebrandt, J.-P.; Groschup, M.H. Detection of PrPSc in peripheral tissues of clinically affected cattle after oral challenge with bovine spongiform encephalopathy. J. Gen. Virol. 2012, 93, 2740–2748. [Google Scholar] [CrossRef] [PubMed]
- van Keulen, L.J.M.; Vromans, M.E.W.; van Zijderveld, F.G. Early and late pathogenesis of natural scrapie infection in sheep. APMIS 2002, 110, 23–32. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Williams, E.S.; Miller, M.W.; Spraker, T.R.; O’Rourke, K.I.; Hoover, E.A. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J. Gen. Virol. 1999, 80 Pt 10, 2757–2764. [Google Scholar] [CrossRef]
- Andréoletti, O.; Berthon, P.; Marc, D.; Sarradin, P.; Grosclaude, J.; van Keulen, L.; Schelcher, F.; Elsen, J.-M.; Lantier, F. Early accumulation of PrP(Sc) in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J. Gen. Virol. 2000, 81, 3115–3126. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; González, L. Classical sheep transmissible spongiform encephalopathies: Pathogenesis, pathological phenotypes and clinical disease. Neuropathol. Appl. Neurobiol. 2007, 33, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Angers, R.C.; Browning, S.R.; Seward, T.S.; Sigurdson, C.J.; Miller, M.W.; Hoover, E.A.; Telling, G.C. Prions in Skeletal Muscles of Deer with Chronic Wasting Disease. Science 2006, 311, 1117. [Google Scholar] [CrossRef]
- Andréoletti, O.; Simon, S.; Lacroux, C.; Morel, N.; Tabouret, G.; Chabert, A.; Lugan, S.; Corbière, F.; Ferré, P.; Foucras, G.; et al. PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat. Med. 2004, 10, 591–593. [Google Scholar] [CrossRef]
- Garza, M.C.C.; Monzón, M.; Marín, B.; Badiola, J.J.; Monleón, E. Distribution of peripheral PrPSc in sheep with naturally acquired scrapie. PLoS ONE 2014, 9, e97768. [Google Scholar] [CrossRef]
- WHO. Tables on Tissue Infectivity Distribution in Transmissible Spongiform Encephalopathies; WHO: Geneva, Switzerland, 2010. [Google Scholar]
- Thomzig, A.; Schulz-Schaeffer, W.; Kratzel, C.; Mai, J.; Beekes, M. Preclinical deposition of pathological prion protein PrPSc in muscles of hamsters orally exposed to scrapie. J. Clin. Investig. 2004, 113, 1465–1472. [Google Scholar] [CrossRef]
- Krasemann, S.; Neumann, M.; Geissen, M.; Bodemer, W.; Kaup, F.-J.; Schulz-Schaeffer, W.; Morel, N.; Aguzzi, A.; Glatzel, M. Preclinical Deposition of Pathological Prion Protein in Muscle of Experimentally Infected Primates. Westermark P, editor. PLoS ONE 2010, 5, e13906. [Google Scholar] [CrossRef]
- Peden, A.H.; Ritchie, D.L.; Head, M.W.; Ironside, J.W. Detection and localization of PrPSc in the skeletal muscle of patients with variant, iatrogenic, and sporadic forms of Creutzfeldt-Jakob disease. Am. J. Pathol. 2006, 168, 927–935. [Google Scholar] [CrossRef]
- Honda, H.; Mori, S.; Watanabe, A.; Sasagasako, N.; Sadashima, S.; Đồng, T.; Satoh, K.; Nishida, N.; Iwaki, T. Abnormal prion protein deposits with high seeding activities in the skeletal muscle, femoral nerve, and scalp of an autopsied case of sporadic Creutzfeldt-Jakob disease. Neuropathology 2021, 41, 152–158. [Google Scholar] [CrossRef]
- Herzog, C.; Rivière, J.; Lescoutra-Etchegaray, N.; Charbonnier, A.; Leblanc, V.; Salès, N.; Deslys, J.-P.; Lasmézas, C.I. PrPTSE distribution in a primate model of variant, sporadic, and iatrogenic Creutzfeldt-Jakob disease. J. Virol. 2005, 79, 14339–14345. [Google Scholar] [CrossRef] [PubMed]
- Suardi, S.; Vimercati, C.; Casalone, C.; Gelmetti, D.; Corona, C.; Iulini, B.; Mazza, M.; Lombardi, G.; Moda, F.; Ruggerone, M.; et al. Infectivity in Skeletal Muscle of Cattle with Atypical Bovine Spongiform Encephalopathy. PLoS ONE 2012, 7, e31449. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, E.R.; Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Prion Infection of Skeletal Muscle Cells and Papillae in the Tongue. J. Virol. 2004, 78, 6792–6798. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Banser, P.; Velasquez, C.D.; Mays, C.E.; Sim, V.L.; Westaway, D.; Aiken, J.M.; McKenzie, D. Infectious Prions Accumulate to High Levels in Non Proliferative C2C12 Myotubes. PLoS Pathog. 2013, 9, e1003755. [Google Scholar] [CrossRef] [PubMed]
- Stuelsatz, P.; Pouzoulet, F.; Lamarre, Y.; Dargelos, E.; Poussard, S.; Leibovitch, S.; Cottin, P.; Veschambre, P. Down-regulation of MyoD by calpain 3 promotes generation of reserve cells in C2C12 myoblasts. J. Biol. Chem. 2010, 285, 12670–12683. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-G.; Kim, C.; Aiken, J.; Yoo, H.S.; McKenzie, D. Dual MicroRNA to Cellular Prion Protein Inhibits Propagation of Pathogenic Prion Protein in Cultured Cells. Mol. Neurobiol. 2018, 55, 2384–2396. [Google Scholar] [CrossRef] [PubMed]
- Dlakic, W.M.; Grigg, E.; Bessen, R.A. Prion Infection of Muscle Cells In Vitro. J. Virol. 2007, 81, 4615–4624. [Google Scholar] [CrossRef]
- Yoshida, N.; Yoshida, S.; Koishi, K.; Masuda, K.; Nabeshima, Y. Cell heterogeneity upon myogenic differentiation: Down-regulation of MyoD and Myf-5 generates “reserve cells”. J. Cell Sci. 1998, 111 Pt 6, 769–779. [Google Scholar] [CrossRef]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Mahal, S.P.; Demczyk, C.A.; Smith, E.W.; Klohn, P.-C.; Weissmann, C. Assaying Prions in Cell Culture. Methods Mol. Biol. 2008, 459, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Tixador, P.; Herzog, L.; Reine, F.; Jaumain, E.; Chapuis, J.; Le Dur, A.; Laude, H.; Béringue, V. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 2010, 6, e1000859. [Google Scholar] [CrossRef]
- Erickson, H.P.; Carrell, N.; Mcdonagh, J. Fibronectin Molecule Visualized in Electron Microscopy: A Long, Thin, Flexible Strand. J. Cell Biol. 1981, 91, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Gilson, H.; Thissen, J.P. Mechanisms of glucocorticoid-induced myopathy. J. Endocrinol. 2008, 197, 1–10. [Google Scholar] [CrossRef]
- Massaccesi, L.; Goi, G.; Tringali, C.; Barassi, A.; Venerando, B.; Papini, N. Dexamethasone-Induced Skeletal Muscle Atrophy Increases O-GlcNAcylation in C2C12 Cells; Dexamethasone-Induced Skeletal Muscle Atrophy Increases O-GlcNAcylation in C2C12 Cells. J. Cell Biochem. 2016, 117, 1833–1842. [Google Scholar] [CrossRef]
- Son, Y.H.; Lee, S.J.; Lee, K.B.; Lee, J.H.; Jeong, E.M.; Chung, S.G.; Park, S.-C.; Kim, I.-G. Dexamethasone downregulates caveolin-1 causing muscle atrophy via inhibited insulin signaling. J. Endocrinol. 2015, 225, 27–37. [Google Scholar] [CrossRef]
- Guller, S.; Markiewicz, L.; Wozniak, R.; Burnham, J.M.; Wang, E.Y.; Kaplan, P.; Lockwood, C.J. Developmental regulation of glucocorticoid-mediated effects on extracellular matrix protein expression in the human placenta. Endocrinology 1994, 134, 2064–2071. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nakagawa, K.; Sarkar, A.; Maruyama, J.; Iwasa, H.; Bao, Y.; Ishigami-Yuasa, M.; Ito, S.; Kagechika, H.; Hata, S.; et al. Screening with a Novel Cell-Based Assay for TAZ Activators Identifies a Compound That Enhances Myogenesis in C2C12 Cells and Facilitates Muscle Repair in a Muscle Injury Model. Mol. Cell Biol. 2014, 34, 1607. [Google Scholar] [CrossRef]
- Peltonen, S.; Alanne, M.; Peltonen, J. Barriers of the peripheral nerve. Tissue Barriers 2013, 1, e24956. [Google Scholar] [CrossRef]
- Bellail, A.C.; Hunter, S.B.; Brat, D.J.; Tan, C.; Van Meir, E.G. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 2004, 36, 1046–1069. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, J.D.; Joiner, S.; Hill, A.F.; Campbell, T.A.; Desbruslais, M.; Luthert, P.J.; Collinge, J. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 2001, 358, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ersdal, C.; Ulvund, M.J.; Espenes, A.; Benestad, S.L.; Sarradin, P.; Landsverk, T. Mapping PrPSc Propagation in Experimental and Natural Scrapie in Sheep with Different PrP Genotypes. Vet Pathol. 2005, 42, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Martin, S.; Thomson, J.R.; Dingwall, W.S.; Begara-McGorum, I.; González, L. Onset and distribution of tissue prp accumulation in scrapie-affected Suffolk sheep as demonstrated by sequential necropsies and tonsillar biopsies. J. Comp. Pathol. 2001, 125, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Spraker, T.R.; Miller, M.W.; Oesch, B.; Hoover, E.A. PrPCWD in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J. Gen. Virol. 2001, 82, 2327–2334. [Google Scholar] [CrossRef]
- Okada, H.; Iwamaru, Y.; Fukuda, S.; Yokoyama, T.; Mohri, S. Detection of Disease-associated Prion Protein in the Optic Nerve and the Adrenal Gland of Cattle with Bovine Spongiform Encephalopathy by Using Highly Sensitive Immunolabeling Procedures. J. Histochem. Cytochem. 2012, 60, 290–300. [Google Scholar] [CrossRef]
- Herbst, A.; Aiken, J.M.; McKenzie, D. Replication of prions in differentiated muscle cells. Prion 2014, 8, 166–168. [Google Scholar] [CrossRef]
- Kitzmann, M.; Carnac, G.; Vandromme, M.; Primig, M.; Lamb, N.J.C.; Fernandez, A. The muscle regulatory factors MyoD and myf-5 undergo distinct cell cycle-specific expression in muscle cells. J. Cell Biol. 1998, 142, 1447–1459. [Google Scholar] [CrossRef]
- McGrath, M.J.; Mitchell, C.A.; Coghill, I.D.; Robinson, P.A.; Brown, S. Skeletal muscle LIM protein 1 (SLIM1/FHL1) induces alpha 5 beta 1-integrin-dependent myocyte elongation. Am. J. Physiol. Cell Physiol. 2003, 285, C1513–C1526. [Google Scholar] [CrossRef]
- Diel, P.; Baadners, D.; Schlüpmann, K.; Velders, M.; Schwarz, J.P. C2C12 myoblastoma cell differentiation and proliferation is stimulated by androgens and associated with a modulation of myostatin and Pax7 expression. J. Mol. Endocrinol. 2008, 40, 231–241. [Google Scholar] [CrossRef]
- Der Vartanian, A.; Audfray, A.; Al Jaam, B.; Janot, M.; Legardinier, S.; Maftah, A.; Germot, A. Protein O-fucosyltransferase 1 expression impacts myogenic C2C12 cell commitment via the Notch signaling pathway. Mol. Cell Biol. 2015, 35, 391–405. [Google Scholar] [CrossRef]
- Ohtake, Y.; Tojo, H.; Seiki, M. Multifunctional roles of MT1-MMP in myofiber formation and morphostatic maintenance of skeletal muscle. J. Cell Sci. 2006, 119, 3822–3832. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S.; Relaix, F.; Nagata, Y.; Ruiz, A.P.; Collins, C.A.; Partridge, T.A.; Beauchamp, J.R. Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci. 2006, 119, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Schollmeyer, J.E. Role of Ca2+ and Ca2+-activated protease in myoblast fusion. Exp. Cell Res. 1986, 162, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Meola, G.; Scarpini, E.; Velicogna, M.; Mottura, A.; Baron, P.L.; Beretta, S.; Scarlato, G. Analysis of fibronectin expression during human muscle differentiation. Basic Appl. Histochem. 1986, 30, 153–163. [Google Scholar] [PubMed]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.; Veiga, S.S.; A Juliano, M.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. Brain Res. 2000, 76, 85–92. [Google Scholar] [CrossRef]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Martins, V.R.; Jay, D.G.; Brentani, R.R. Laminin-induced PC-12 cell differentiation is inhibited following laser inactivation of cellular prion protein. FEBS Lett. 2000, 482, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Hajj, G.N.M.; Lopes, M.H.; Mercadante, A.F.; Veiga, S.S.; da Silveira, R.B.; Santos, T.G.; Ribeiro, K.C.B.; Juliano, M.A.; Jacchieri, S.G.; Zanata, S.M.; et al. Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J. Cell Sci. 2007, 120, 1915–1926. [Google Scholar] [CrossRef]
- White, A.R.; Enever, P.; Tayebi, M.; Mushens, R.; Linehan, J.; Brandner, S.; Anstee, D.; Collinge, J.; Hawke, S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003, 422, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Gutiérrez, J.F.; Aguilar Pierlé, S.; Schneider, D.A.; Baszler, T.V.; Stanton, J.B. Transcriptomic Determinants of Scrapie Prion Propagation in Cultured Ovine Microglia. PLoS ONE 2016, 11, e0147727. [Google Scholar] [CrossRef] [PubMed]
- Marbiah, M.M.; Harvey, A.; West, B.T.; Louzolo, A.; Banerjee, P.; Alden, J.; Grigoriadis, A.; Hummerich, H.; Kan, H.; Cai, Y.; et al. Identification of a gene regulatory network associated with prion replication. EMBO J. 2014, 33, 1527–1547. [Google Scholar] [CrossRef] [PubMed]
- Imberdis, T.; Harris, D.A. Prion permissive pathways: Extracellular matrix genes control susceptibility to prion infection. EMBO J. 2014, 33, 1506–1508. [Google Scholar] [CrossRef] [PubMed]
- Bosque, P.J.; Ryou, C.; Telling, G.; Peretz, D.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Prions in skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 3812–3817. [Google Scholar] [CrossRef]
- Hynes, R.O. Fibronectin and the Cytoskeleton; Springer: New York, NY, USA, 1990; pp. 249–280. [Google Scholar] [CrossRef]
- Gulati, A.K.; Reddi, A.H.; Zalewski, A.A. Changes in the basement membrane zone components during skeletal muscle fiber degeneration and regeneration. J. Cell Biol. 1983, 97, 957–962. [Google Scholar] [CrossRef]
- Roman, W.; Martins, J.P.; Gomes, E.R. Local Arrangement of Fibronectin by Myofibroblasts Governs Peripheral Nuclear Positioning in Muscle Cells. Dev. Cell 2018, 46, 102–111.e6. [Google Scholar] [CrossRef] [PubMed]
- Glatzel, M.; Abela, E.; Maissen, M.; Aguzzi, A. Extraneural Pathologic Prion Protein in Sporadic Creutzfeldt–Jakob Disease. N. Engl. J. Med. 2003, 349, 1812–1820. [Google Scholar] [CrossRef]
- Cadot, B.; Gache, V.; Gomes, E.R. Moving and positioning the nucleus in skeletal muscle—One step at a time. Nucleus 2015, 6, 373–381. [Google Scholar] [CrossRef]
- Farhadian, F.; Contard, F.; Corbier, A.; Barrieux, A.; Rappaport, L.; Samuel, J.L. Fibronectin expression during physiological and pathological cardiac growth. J. Mol. Cell Cardiol. 1995, 27, 981–990. [Google Scholar] [CrossRef]
- Willems, I.; Arends, J.-W.; Daemen, M.J.A.P. Tenascin and fibronectin expression in healing human myocardial scars. J. Pathol. 1996, 179, 321–325. [Google Scholar] [CrossRef]
- Heng, B.C.; Haider, H.K.; Sim, E.K.-W.; Cao, T.; Ng, S.C. Strategies for directing the differentiation of stem cells into the cardiomyogenic lineage in vitro. Cardiovasc. Res. 2004, 62, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.E.; Brown, J.; Kreeger, T.; Williams, E.S. Prion protein in cardiac muscle of elk (Cervus elaphus nelsoni) and white-tailed deer (Odocoileus virginianus) infected with chronic wasting disease. J. Gen. Virol. 2006, 87, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Wiley, C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–585. [Google Scholar] [CrossRef]
- Bandtlow, C.E.; Zimmermann, D.R. Proteoglycans in the Developing Brain: New Conceptual Insights for Old Proteins. Physiol Rev. 2000, 80, 1267–1290. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M. Extracellular matrix molecules and synaptic plasticity. Nat. Rev. Neurosci. 2003, 4, 456–468. [Google Scholar] [CrossRef]
- Yamaguchi, Y. Lecticans: Organizers of the brain extracellular matrix. Cell. Mol. Life Sci. 2000, 57, 276–289. [Google Scholar] [CrossRef]
- Gardiner, N.J. Integrins and the extracellular matrix: Key mediators of development and regeneration of the sensory nervous system. Dev. Neurobiol. 2011, 71, 1054–1072. [Google Scholar] [CrossRef]
- de Luca, A.; di Summa, P.; Lacour, S.; Raffoul, W. Extracellular matrix components in peripheral nerve repair: How to affect neural cellular response and nerve regeneration. Neural. Regen Res. 2014, 9, 1943–1948. [Google Scholar] [CrossRef]
- Vaz, R.; Martins, G.G.; Thorsteinsdóttir, S.; Rodrigues, G. Fibronectin promotes migration, alignment and fusion in an in vitro myoblast cell model. Cell Tissue Res. 2012, 348, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.L.; Hopkins, L.J.; Halliday, S.; Houston, F.; Hunter, N.; McConnell, I. Architecture of secondary lymphoid tissue in sheep experimentally challenged with scrapie. Immunology 2004, 111, 230–236. [Google Scholar] [CrossRef] [PubMed]
- McGovern, G.; Jeffrey, M. Scrapie-specific pathology of sheep lymphoid tissues. PLoS ONE 2007, 2, e1304. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Dagleish, M.P.; Bellworthy, S.J.; Sisó, S.; Stack, M.J.; Chaplin, M.J.; Davis, L.A.; Hawkins, S.A.C.; Hughes, J.; Jeffrey, M. Postmortem diagnosis of preclinical and clinical scrapie in sheep by the detection of disease-associated PrP in their rectal mucosa. Vet. Rec. 2006, 158, 325–331. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Aguzzi, A. Prions and lymphoid organs: Solved and remaining mysteries. Prion 2013, 7, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Notari, S.; Moleres, F.J.; Hunter, S.B.; Belay, E.D.; Schonberger, L.B.; Cali, I.; Parchi, P.; Shieh, W.-J.; Brown, P.; Zaki, S.; et al. Multiorgan Detection and Characterization of Protease-Resistant Prion Protein in a Case of Variant CJD Examined in the United States. Goletti D, editor. PLoS ONE 2010, 5, e8765. [Google Scholar] [CrossRef]
- Sisó, S.; González, L.; Jeffrey, M. Neuroinvasion in prion diseases: The roles of ascending neural infection and blood dissemination. Interdiscip. Perspect. Infect. Dis. 2010, 2010, 747892. [Google Scholar] [CrossRef] [PubMed]
- Shikiya, R.A.; Langenfeld, K.A.; Eckland, T.E.; Trinh, J.; Holec, S.A.M.; Mathiason, C.K.; Kincaid, A.E.; Bartz, J.C. PrPSc formation and clearance as determinants of prion tropism. PLoS Pathog. 2017, 13, e1006298. [Google Scholar] [CrossRef]
- Kramer, D.A.; Eldeeb, M.A.; Wuest, M.; Mercer, J.; Fahlman, R.P. Proteomic characterization of EL4 lymphoma-derived tumors upon chemotherapy treatment reveals potential roles for lysosomes and caspase-6 during tumor cell death in vivo. Proteomics 2017, 17, 1700060. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction 14 of Sedimentation Velocity Gradient from C2C12 in Sarkosyl (Figure 3) | ||
| Accession | Description | Percentage of Contamination |
| P11087 | Collagen alpha-1(I) chain (GN = COL1A1) | 75.6 |
| Q01149 | Collagen alpha-2(I) chain (GN = COL1A2) | 11.9 |
| P04104 | Keratin, type II cytoskeletal 1 (GN = KRT1) | 8.9 |
| P02535 | Keratin, type I cytoskeletal 10 (GN = KRT10) | 3.5 |
| P11276 | Fibronectin (GN = FN1) * | 0.04 * |
| Fraction 12 of Sedimentation Velocity gradient from C2C12 in Triton X-100 Na-Deoxicholate (Figure 3) | ||
| Accession | Description | Percentage of Contamination |
| P11087 | Collagen alpha-1(I) chain (GN = COL1A1) | 55.4 |
| P04104 | Keratin, type II cytoskeletal 1 (GN = KRT1) | 14.7 |
| Q01149 | Collagen alpha-2(I) chain (GN = COL1A2) | 8.1 |
| Q61554 | Fibrillin-1 (GN = FBN1) | 7.4 |
| P11276 | Fibronectin (GN = FN1) | 4.7 |
| P02535 | Keratin, type I cytoskeletal 10 (GN = KRT10) | 4.3 |
| Q05793 | Basement membrane-specific heparan sulfate proteoglycan core protein (GN = HSPG2) | 2.0 |
| P62806 | Histone H4 (GN = HIST1H4A) | 1.6 |
| Q02788 | Collagen alpha-2(VI) chain (GN = COL6A2) | 1.1 |
| P04247 | Myoglobin (GN = MB) | 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garza, M.C.; Kang, S.-G.; Kim, C.; Monleón, E.; van der Merwe, J.; Kramer, D.A.; Fahlman, R.; Sim, V.L.; Aiken, J.; McKenzie, D.; et al. In Vitro and In Vivo Evidence towards Fibronectin’s Protective Effects against Prion Infection. Int. J. Mol. Sci. 2023, 24, 17525. https://doi.org/10.3390/ijms242417525
Garza MC, Kang S-G, Kim C, Monleón E, van der Merwe J, Kramer DA, Fahlman R, Sim VL, Aiken J, McKenzie D, et al. In Vitro and In Vivo Evidence towards Fibronectin’s Protective Effects against Prion Infection. International Journal of Molecular Sciences. 2023; 24(24):17525. https://doi.org/10.3390/ijms242417525
Chicago/Turabian StyleGarza, M. Carmen, Sang-Gyun Kang, Chiye Kim, Eva Monleón, Jacques van der Merwe, David A. Kramer, Richard Fahlman, Valerie L. Sim, Judd Aiken, Debbie McKenzie, and et al. 2023. "In Vitro and In Vivo Evidence towards Fibronectin’s Protective Effects against Prion Infection" International Journal of Molecular Sciences 24, no. 24: 17525. https://doi.org/10.3390/ijms242417525
APA StyleGarza, M. C., Kang, S.-G., Kim, C., Monleón, E., van der Merwe, J., Kramer, D. A., Fahlman, R., Sim, V. L., Aiken, J., McKenzie, D., Cortez, L. M., & Wille, H. (2023). In Vitro and In Vivo Evidence towards Fibronectin’s Protective Effects against Prion Infection. International Journal of Molecular Sciences, 24(24), 17525. https://doi.org/10.3390/ijms242417525