
The Interplay between Helicobacter pylori and Gut Microbiota in Non-Gastrointestinal Disorders: A Special Focus on Atherosclerosis

 ,
, 
 , ,
, ,  ,
,  ,
, 
Abstract
:1. Methodology
2. Cardiovascular Disease: Risk Factors
2.1. Cardiovascular Disease: The Role of Microbiota
2.2. The Case of Atherosclerosis
3. H. pylori: Not Only Gastritis
4. H. pylori and Atherosclerosis
4.1. Cytotoxin-Associated Gene Antigen and Atherosclerosis
4.2. H. pylori, Inflammation and Hypercholesterolemia
4.3. H. pylori: Interaction with Platelets Aggregation
4.4. H. pylori and Other Mechanisms Affecting Atherosclerosis
5. Future Perspective
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 19 September 2023).
- Zhao, Z. Changing Mortality Patterns and Causes of Death. In Transition and Challenge: China’s Population at the Beginning of the 21st Century; Zhao, Z., Guo, F., Eds.; Oxford University Press: Oxford, UK, 2007; pp. 160–176. [Google Scholar]
- D’Agostino, R.B.; Vasan, R.S., Sr.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, A.; Michos, E.D.; Samad, Z.; Ballantyne, C.M.; Virani, S.S. The Use of Sex-Specific Factors in the Assessment of Women’s Cardiovascular Risk. Circulation 2020, 141, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Forouhi, N.G.; Sattar, N. CVD risk factors and ethnicity--A homogeneous relationship? Atheroscler. Suppl. 2006, 7, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Nakano, Y.; Adachi, S.; Murohara, T. Effects of Tobacco Smoking on Cardiovascular Disease. Circ. J. Off. J. Jpn. Circ. Soc. 2019, 83, 1980–1985. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Anic, G.M.; Rostron, B.L.; Tanwar, M.; Chang, C.M. Cigarette Smoking Reduction and Health Risks: A Systematic Review and Meta-analysis. Nicotine Tob. Res. Off. J. Soc. Res. Nicotine Tob. 2021, 23, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Steffen, L.M.; Selvin, E.; Rebholz, C.M. Diet quality, change in diet quality and risk of incident CVD and diabetes. Public Health Nutr. 2020, 23, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Chagas, P.; Chiva-Blanch, G. Diet and Cardiovascular Disease: Effects of Foods and Nutrients in Classical and Emerging Cardiovascular Risk Factors. Curr. Med. Chem. 2019, 26, 3639–3651. [Google Scholar] [CrossRef]
- Casas, R.; Castro-Barquero, S.; Estruch, R.; Sacanella, E. Nutrition and Cardiovascular Health. Int. J. Mol. Sci. 2018, 19, 3988. [Google Scholar] [CrossRef]
- Shivappa, N.; Godos, J.; Hébert, J.R.; Wirth, M.D.; Piuri, G.; Speciani, A.F.; Grosso, G. Dietary Inflammatory Index and Cardiovascular Risk and Mortality-A Meta-Analysis. Nutrients 2018, 10, 200. [Google Scholar] [CrossRef]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baβler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef]
- Christ, A.; Lauterbach, M.; Latz, E. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019, 51, 794–811. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickley, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e5. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: A review. Antonie Van Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef]
- Senthong, V.; Kiatchoosakun, S.; Wongvipaporn, C.; Phetcharaburanin, J.; Tatsanavivat, P.; Sritara, P.; Phromminitikul, A. Gut microbiota-generated metabolite, trimethylamine-N-oxide, and subclinical myocardial damage: A multicenter study from Thailand. Sci. Rep. 2021, 11, 14963. [Google Scholar] [CrossRef]
- Caldarelli, M.; Franza, L.; Rio, P.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Gut–Kidney–Heart: A Novel Trilogy. Biomedicines 2023, 11, 3063. [Google Scholar] [CrossRef]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef]
- Poesen, R.; Claes, K.; Evenepoel, P.; de Loor, H.; Augustijns, P.; Kuypers, D.; Meijers, B. Microbiota-Derived Phenylacetylglutamine Associates with Overall Mortality and Cardiovascular Disease in Patients with CKD. J. Am. Soc. Nephrol. JASN 2016, 27, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chen, W.-D.; Wang, Y.-D. TGR5, Not Only a Metabolic Regulator. Front. Physiol. 2016, 7, 646. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11, 584303. [Google Scholar] [CrossRef] [PubMed]
- Claudel, T.; Staels, B.; Kuipers, F. The Farnesoid X Receptor. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation – Protective or Causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Kotlo, K.; Anbazhagan, A.N.; Priyamvada, S.; Jayawardena, D.; Kumar, A.; Chen, Y.; Xia, Y.; Finn, P.W.; Perkin, D.L.; Dudeja, P.K.; et al. The olfactory G protein-coupled receptor (Olfr-78/OR51E2) modulates the intestinal response to colitis. Am. J. Physiol. Cell Physiol. 2020, 318, C502–C513. [Google Scholar] [CrossRef]
- Li, B.; Xia, Y.; Hu, B. Infection and atherosclerosis: TLR-dependent pathways. Cell. Mol. Life Sci. CMLS 2020, 77, 2751–2769. [Google Scholar] [CrossRef]
- Fan, J.; Watanabe, T. Atherosclerosis: Known and unknown. Pathol. Int. 2022, 72, 151–160. [Google Scholar] [CrossRef]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef]
- Libby, P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 2001, 104, 365–372. [Google Scholar] [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, e15–e29. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, A.L.; Bäckhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N. 23 years of the discovery of Helicobacter pylori: Is the debate over? Ann. Clin. Microbiol. Antimicrob. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.; Krogfelt, K.A. Helicobacter pylori: An invading microorganism? A review. FEMS Immunol. Med. Microbiol. 2003, 36, 117–126. [Google Scholar] [CrossRef]
- de Brito, B.B.; da Silva, F.A.F.; Soares, A.S.; Pereira, V.A.; Santos, M.L.C.; Sampaio, M.M.; Moreira Neves, P.H.; Freire de Melo, F. Pathogenesis and clinical management of Helicobacter pylori gastric infection. World J. Gastroenterol. 2019, 25, 5578–5589. [Google Scholar] [CrossRef]
- Worku, M.L.; Karim, Q.N.; Spencer, J.; Sidebotham, R.L. Chemotactic response of Helicobacter pylori to human plasma and bile. J. Med. Microbiol. 2004, 53, 807–811. [Google Scholar] [CrossRef]
- Ilver, D.; Arnqvist, A.; Ogren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Boren, T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef]
- Leylabadlo, H.E.; Yekani, M.; Ghotaslou, R. Helicobacter pylori hopQ alleles (type I and II) in gastric cancer. Biomed. Rep. 2016, 4, 601–604. [Google Scholar] [CrossRef]
- Fischbach, W.; Malfertheiner, P. Helicobacter Pylori Infection. Dtsch. Arztebl. Int. 2018, 115, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Gasbarrini, A.; Franceschi, F.; Does, H. Pylori infection play a role in idiopathic thrombocytopenic purpura and in other autoimmune diseases? Am. J. Gastroenterol. 2005, 100, 1271–1273. [Google Scholar] [CrossRef] [PubMed]
- Waldum, H.; Fossmark, R. Gastritis, Gastric Polyps and Gastric Cancer. Int. J. Mol. Sci. 2021, 22, 6548. [Google Scholar] [CrossRef] [PubMed]
- Cianci, R.; Franza, L.; Schinzari, G.; Rossi, E.; Ianiro, G.; Tortora, G.; Gasbarrini, A.; Gambassi, G.; Cammarota, G. The Interplay between Immunity and Microbiota at Intestinal Immunological Niche: The Case of Cancer. Int. J. Mol. Sci. 2019, 20, 501. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, Y.; Ouyang, Q.; Li, R.; Li, J.; Chen, W.; Hu, W.; He, L.; Bao, Q.; Li, P.; et al. Helicobacter pylori and unignorable extragastric diseases: Mechanism and implications. Front. Microbiol. 2022, 13, 972777. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, F.; Covino, M.; Baudron, C.R. Review: Helicobacter pylori and extragastric diseases. Helicobacter. 2019, 24 (Suppl. 1), e12636. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, F.; Satta, M.A.; Mentella, M.C.; Penland, R.; Candelli, M.; Grillo, R.L.; Leo, D.; Fini, L.; Nista, E.C.; Cazzato, I.A.; et al. Helicobacter pylori infection in patients with Hashimoto’s thyroiditis. Helicobacter. 2004, 9, 369. [Google Scholar] [PubMed]
- Lando, V.; Calciano, L.; Minelli, C.; Bombieri, C.; Ferrari, M.; Malerba, G.; Margagliotti, A.; Murgia, N.; Nicolis, M.; Olivieri, M.; et al. IL18 Gene Polymorphism Is Associated with Total IgE in Adult Subjects with Asthma. J. Clin. Med. 2023, 12, 3963. [Google Scholar] [CrossRef]
- Zuo, Z.T.; Ma, Y.; Sun, Y.; Bai, C.Q.; Ling, C.H.; Yuan, F.L. The Protective Effects of Helicobacter pylori Infection on Allergic Asthma. Int. Arch. Allergy Immunol. 2020, 182, 53–64. [Google Scholar] [CrossRef]
- Miftahussurur, M.; Nusi, I.A.; Graham, D.Y.; Yamaoka, Y. Helicobacter, Hygiene, Atopy, and Asthma. Front. Microbiol. 2017, 8, 1034. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kang, H.R.; Lee, J.K.; Heo, E.Y.; Choi, S.H.; Kim, D.K. The effect of Helicobacter pylori infection on the decline of lung function in a health screening population. Ann. Palliat. Med. 2020, 9, 3115–3122. [Google Scholar] [CrossRef] [PubMed]
- Oster, P.; Vaillant, L.; Riva, E.; McMillan, B.; Begka, C.; Truntzer, C.; Richard, C.; Leblond, M.M.; Messaoudene, M.; Machremi, E.; et al. Helicobacter pylori infection has a detrimental impact on the efficacy of cancer immunotherapies. Gut 2022, 71, 457–466. [Google Scholar] [CrossRef]
- Wang, J.; Dong, F.; Su, H.; Zhu, L.; Shao, S.; Wu, J.; Liu, H.H. pylori is related to NAFLD but only in female: A Cross-sectional Study. Int. J. Med. Sci. 2021, 18, 2303–2311. [Google Scholar] [CrossRef] [PubMed]
- Okushin, K.; Tsutsumi, T.; Ikeuchi, K.; Kado, A.; Enooku, K.; Fujinaga, H.; Moriya, K.; Yotsuyanagi, K.; Koike, K. Helicobacter pylori infection and liver diseases: Epidemiology and insights into pathogenesis. World J. Gastroenterol. 2018, 24, 3617–3625. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Liou, J.M.; Lee, Y.C.; Hong, T.C.; El-Omar, E.M.; Wu, M.S. The interplay between Helicobacter pylori and gastrointestinal microbiota. Gut Microbes 2021, 13, 1909459. [Google Scholar] [CrossRef] [PubMed]
- Lolekha, P.; Sriphanom, T.; Vilaichone, R.K. Helicobacter pylori eradication improves motor fluctuations in advanced Parkinson’s disease patients: A prospective cohort study (HP-PD trial). PLoS ONE 2021, 16, e0251042. [Google Scholar] [CrossRef] [PubMed]
- Piekut, T.; Hurła, M.; Banaszek, N.; Szejn, P.; Dorszewska, J.; Kozubski, W.; Prendecki, M. Infectious agents and Alzheimer’s disease. J. Integr. Neurosci. 2022, 21, 73. [Google Scholar] [CrossRef]
- Xie, J.; Cools, L.; Van Imschoot, G.; Van Wonterghem, E.; Pauwels, M.J.; Vlaeminck, I.; De Witte, C.; El Andaloussi, S.; Wierda, K.; De Groef, L.; et al. Helicobacter pylori-derived outer membrane vesicles contribute to Alzheimer’s disease pathogenesis via C3-C3aR signalling. J. Extracell. Vesicles 2023, 12, e12306. [Google Scholar] [CrossRef]
- Willison, H.J.; Jacobs, B.C.; van Doorn, P.A. Guillain-Barré syndrome. Lancet 2016, 388, 717–727. [Google Scholar] [CrossRef]
- Dardiotis, E.; Sokratous, M.; Tsouris, Z.; Siokas, V.; Mentis, A.A.; Aloizou, A.M.; Michalopoulou, A.; Bogdanos, D.P.; Xirmerisiou, G.; Deretzi, G.; et al. Association between Helicobacter pylori infection and Guillain-Barré Syndrome: A meta-analysis. Eur. J. Clin. Investig. 2020, 50, e13218. [Google Scholar] [CrossRef]
- Pedrini, M.J.; Seewann, A.; Bennett, K.A.; Wood, A.J.; James, I.; Burton, J.; Marshall, B.J.; Carrol, W.M.; Kermode, A.G. Helicobacter pylori infection as a protective factor against multiple sclerosis risk in females. J. Neurol. Neurosurg. Psychiatry 2015, 86, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Jaruvongvanich, V.; Sanguankeo, A.; Jaruvongvanich, S.; Upala, S. Association between Helicobacter pylori infection and multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2016, 7, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Arjmandi, D.; Abdollahi, A.; Ardekani, A.; Razavian, I.; Razavian, E.; Sartip, B.; Mahjour, S.; Parsa, H.; Azizi Kyvanani, N.; Marhommirzabak, E.; et al. Helicobacter pylori infection and risk of multiple sclerosis: An updated meta-analysis. Helicobacter 2022, 27, e12927. [Google Scholar] [CrossRef] [PubMed]
- Gasbarrini, A.; De Luca, A.; Fiore, G.; Franceschi, F.; Ojetti, V.V.; Torre, E.S.; Di Campli, C.; Candelli, M.; Pola, R.; Serricchio, M.; et al. Primary Headache and Helicobacter Pylori. Int. J. Angiol. 1998, 7, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Bawand, R.; Ghiasian, M.; Samadyan, M.; Qaderi, S. Association of Helicobacter pylori with migraine headaches and the effects of this infection and its eradication on the migraine characteristics in adults: A comprehensive systematic review and meta-analysis. Helicobacter 2023, 28, e13010. [Google Scholar] [CrossRef]
- Mendall, M.A.; Patel, P.; Ballam, L.; Strachan, D.; Northfield, T.C. C reactive protein and its relation to cardiovascular risk factors: A population based cross sectional study. BMJ (Clin. Res. Ed) 1996, 312, 1061–1065. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Tan, G.; Wu, F.; Zhi, F.C.H. pylori attenuates TNBS-induced colitis via increasing mucosal Th2 cells in mice. Oncotarget 2017, 8, 73810–73816. [Google Scholar] [CrossRef]
- Codolo, G.; Coletta, S.; D’Elios, M.M.; de Bernard, M. HP-NAP of Helicobacter pylori: The Power of the Immunomodulation. Front. Immunol. 2022, 13, 944139. [Google Scholar] [CrossRef]
- Foegeding, N.J.; Caston, R.R.; McClain, M.S.; Ohi, M.D.; Cover, T.L. An Overview of Helicobacter pylori VacA Toxin Biology. Toxins 2016, 8, 173. [Google Scholar] [CrossRef]
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B 2017, 93, 196–219. [Google Scholar] [CrossRef]
- Li, N.; Tang, B.; Jia, Y.-P.; Zhu, P.; Zhuang, Y.; Fang, Y.; Li, Q.; Wang, K.; Zhang, W.J.; Guo, G.; et al. Helicobacter pylori CagA Protein Negatively Regulates Autophagy and Promotes Inflammatory Response via c-Met-PI3K/Akt-mTOR Signaling Pathway. Front. Cell. Infect. Microbiol. 2017, 7, 417. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhang, L.; Wu, H.; Chen, F.; Liu, X.; Xu, H.; Cui, Y.; Zhu, Q.; Wang, M.; Hao, H.; et al. CagA+Helicobacter pylori, Not CagA–Helicobacter pylori, Infection Impairs Endothelial Function through Exosomes-Mediated ROS Formation. Front. Cardiovasc. Med. 2022, 9, 881372. [Google Scholar] [CrossRef] [PubMed]
- Rožanković, P.B.; Huzjan, A.L.; Cupić, H.; Benčić, I.J.; Bašić, S.; Demarin, V. Influence of CagA-positive Helicobacter pylori strains on atherosclerotic carotid disease. J. Neurol. 2011, 258, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, F.; Sepulveda, A.R.; Gasbarrini, A.; Pola, P.; Silveri, N.G.; Gasbarrini, G.; Graham, D.Y.; Genta, R.M. Cross-reactivity of anti-CagA antibodies with vascular wall antigens: Possible pathogenic link between Helicobacter pylori infection and atherosclerosis. Circulation 2002, 106, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Chmiela, M.; Gonciarz, W. Molecular mimicry in Helicobacter pylori infections. World J. Gastroenterol. 2017, 23, 3964–3977. [Google Scholar] [CrossRef]
- Amedei, A.; Munari, F.; Bella, C.D.; Niccolai, E.; Benagiano, M.; Bencini, L.; Cianchi, F.; Farsi, M.; Emmi, G.; Zanotti, G.; et al. Helicobacter pylori secreted peptidyl prolyl cis, trans-isomerase drives Th17 inflammation in gastric adenocarcinoma. Intern. Emerg. Med. 2014, 9, 303–309. [Google Scholar] [CrossRef]
- Pandolfi, F.; Franza, L.; Carusi, V.; Altamura, S.; Andriollo, G.; Nucera, E. Interleukin-6 in Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 5238. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Goldstein, D.R. Ageing and atherosclerosis: Vascular intrinsic and extrinsic factors and potential role of IL-6. Nat. Rev. Cardiol. 2021, 18, 58–68. [Google Scholar] [CrossRef]
- Yang, S.; Xia, Y.P.; Luo, X.Y.; Chen, S.L.; Li, B.W.; Ye, Z.M.; Chen, S.C.; Mao, L.; Jin, H.J.; Li, Y.n.; et al. Exosomal CagA derived from Helicobacter pylori-infected gastric epithelial cells induces macrophage foam cell formation and promotes atherosclerosis. J. Mol. Cell. Cardiol. 2019, 135, 40–51. [Google Scholar] [CrossRef]
- Tahmina, K.; Hikawa, N.; Takahashi-Kanemitsu, A.; Knight, C.T.; Sato, K.; Itoh, F.; Hatakeyama, N. Transgenically expressed Helicobacter pylori CagA in vascular endothelial cells accelerates arteriosclerosis in mice. Biochem. Biophys. Res. Commun. 2022, 618, 79–85. [Google Scholar] [CrossRef]
- Shimoda, A.; Ueda, K.; Nishiumi, S.; Murata-Kamiya, N.; Mukai, S.A.; Sawada, S.; Azuma, T.; Hatakeyama, M.; Akyoshi, K. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 2016, 6, 18346. [Google Scholar] [CrossRef] [PubMed]
- Li, B.W.; Liu, Y.; Zhang, L.; Guo, X.Q.; Wen, C.; Zhang, F.; Luo, X.Y.; Xia, Y.P. Cytotoxin-associated gene A (CagA) promotes aortic endothelial inflammation and accelerates atherosclerosis through the NLRP3/caspase-1/IL-1β axis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21942. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, J.; Xue, Z.; Chang, M.; Feng, X.; Cai, Y.; Bai, L.; Wang, W.; Liu, E.; Zhao, S.; et al. Deficiency of protein inhibitor of activated STAT3 exacerbates atherosclerosis by modulating VSMC phenotypic switching. Atherosclerosis 2023, 380, 117195. [Google Scholar] [CrossRef] [PubMed]
- Tabata, N.; Sueta, D.; Arima, Y.; Okamoto, K.; Shono, T.; Hanatani, S.; Takashio, S.; Oniki, K.; Saruwatari, J.; Sakamoto, K.; et al. Cytotoxin-associated gene-A-seropositivity and Interleukin-1 polymorphisms influence adverse cardiovascular events. Int. J. Cardiol. Heart Vasc. 2020, 27, 100498. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Li, Y.; Dong, C.; Si, G.; Xu, Y.; Peng, M.; Li, Y. Helicobacter pylori infection and the progression of atherosclerosis: A systematic review and meta-analysis. Helicobacter 2022, 27, e12865. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhou, F.; Chen, C.; Luo, H.; Guo, J.; Wang, W.; Yang, J.; Li, L. Role of Outer Membrane Vesicles From Helicobacter pylori in Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 673993. [Google Scholar] [CrossRef]
- Ninomiya, R.; Kubo, S.; Baba, T.; Kajiwara, T.; Tokunaga, A.; Nabeka, H.; Doihara, T.; Shimokawa, T.; Matsuda, S.; Murakami, K.; et al. Inhibition of low-density lipoprotein uptake by Helicobacter pylori virulence factor CagA. Biochem. Biophys. Res. Commun. 2021, 556, 192–198. [Google Scholar] [CrossRef]
- Kim, D.H.; Son, B.K.; Min, K.W.; Han, S.K.; Na, J.U.; Choi, P.C.; Kim, H.L.; Kwon, M.J.; Oh, Y.H.; Jung, W.Y.; et al. Chronic Gastritis Is Associated with a Decreased High-Density Lipid Level: Histological Features of Gastritis Based on the Updated Sydney System. J. Clin. Med. 2020, 9, 1856. [Google Scholar] [CrossRef]
- Gutierrez, P.S. Foam Cells in Atherosclerosis. Arq. Bras. De Cardiol. 2022, 119, 542–543. [Google Scholar] [CrossRef]
- Hirooka, Y.; Nozaki, Y. Interleukin-18 in Inflammatory Kidney Disease. Front. Med. 2021, 8, 639103. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Yi, J.; Lu, J.; Nie, M.; Huang, M.; Rong, J.; Zhu, Z.; Chen, J.; Zhou, X.; Li, B.; et al. N-Acetylcysteine Reduces ROS-Mediated Oxidative DNA Damage and PI3K/Akt Pathway Activation Induced by Helicobacter pylori Infection. Oxidative Med. Cell. Longev. 2018, 2018, 1874985. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Spor, A.; Felin, J.; Fåk, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4592–4598. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.J. Helicobacter Pylori Infection Induces Intestinal Dysbiosis That Could Be Related to the Onset of Atherosclerosis. BioMed Res. Int. 2022, 2022, 9943158. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, A.C.; Grayson, B.E.; Boger, A.; Bakke, D.; Carmody, E.M.; Bales, C.E.; Paski, S.C.; Murphy, S.F.; Dethlefs, C.R.; Shannon, K.J.; et al. Gastrokine-1, an anti-amyloidogenic protein secreted by the stomach, regulates diet-induced obesity. Sci. Rep. 2021, 11, 9477. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Okamoto, A. Helicobacter pylori Infection and Chronic Immune Thrombocytopenia. J. Clin. Med. 2022, 11, 4822. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.F.; Kerrigan, S.W.; Corcoran, P.A.; Atherton, J.C.; Murray, F.E.; Fitzgerald, D.J.; Cox, D. M:. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology 2003, 124, 1846–1854. [Google Scholar] [CrossRef]
- Takeuchi, H.; Islam, J.M.; Kaneko, A.; Kimura, A.; Shida, T.; Oboshi, W.; Katayama, H.; Oishi, T.; Fujieda, M.; Morimoto, N. Helicobacter pylori protein that binds to and activates platelet specifically reacts with sera of H. pylori-associated chronic immune thrombocytopenia. Platelets 2021, 32, 1120–1123. [Google Scholar] [CrossRef]
- Wassermann, G.E.; Olivera-Severo, D.; Uberti, A.F.; Carlini, C.R. Helicobacter pylori urease activates blood platelets through a lipoxygenase-mediated pathway. J. Cell. Mol. Med. 2010, 14, 2025–2034. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Gong, R.; Yao, H.; Fan, M.; Zeng, J.; Xu, S.; Lin, R. Eradication of Helicobacter pylori alleviates lipid metabolism deterioration: A large-cohort propensity score-matched analysis. Lipids Health Dis. 2022, 21, 34. [Google Scholar] [CrossRef]
- Watanabe, J.; Hamasaki, M.; Kotani, K. The Effect of Helicobacter pylori Eradication on Lipid Levels: A Meta-Analysis. J. Clin. Med. 2021, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, L.; Gabrielli, M.; Cremonini, F.; Santoliquido, A.; Candelli, M.; Nista, E.C.; Pola, P.; Gasbarrini, G.; Gasbarini, A. Atrophic gastritis as a cause of hyperhomocysteinaemia. Aliment. Pharmacol. Ther. 2004, 19, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Bloemenkamp, D.G.; Mali, W.P.; Tanis, B.C.; Rosendaal, F.R.; van den Bosch, M.A.; Kemmeren, J.M.; Algra, A.; Visseren, F.L.J.; van der Graaf, Y. The relation between Helicobacter pylori and atherosclerosis cannot be explained by a high homocysteine concentration. Eur. J. Clin. Investig. 2002, 32, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Hu, J.; Tian, M.; Li, Y.; Ren, C.; Deng, Y.; Jiang, Y. Extracellular vesicles from helicobacter pylori-infected cells and helicobacter pylori outer membrane vesicles in atherosclerosis. Helicobacter 2022, 27, e12877. [Google Scholar] [CrossRef] [PubMed]
- Buzás, G.M. Metabolic consequences of Helicobacter pylori infection and eradication. World J. Gastroenterol. 2014, 20, 5226–5234. [Google Scholar] [CrossRef]
- Aydemir, S.; Eren, H.; Tekin, I.O.; Harmandar, F.A.; Demircan, N.; Cabuk, M. Helicobacter pylori eradication lowers serum asymmetric dimethylarginine levels. Mediat. Inflamm. 2010, 2010, 685903. [Google Scholar] [CrossRef]
- Iwai, N.; Okuda, T.; Oka, K.; Hara, T.; Inada, Y.; Tsuji, T.; Komaki, T.; Inoue, K.; Dohi, O.; Konishi, H.; et al. Helicobacter pylori eradication increases the serum high density lipoprotein cholesterol level in the infected patients with chronic gastritis: A single-center observational study. PLoS ONE 2019, 14, e0221349. [Google Scholar] [CrossRef]
- Kanbay, M.; Gür, G.; Yücel, M.; Yilmaz, U.; Boyacioğlu, S. Does eradication of Helicobacter pylori infection help normalize serum lipid and CRP levels? Dig. Dis. Sci. 2005, 50, 1228–1231. [Google Scholar] [CrossRef]
- Migneco, A.; Ojetti, V.; Specchia, L.; Franceschi, F.; Candelli, M.; Mettimano, M.; Montebelli, R.; Savi, G.; Gasbarrini, G. Eradication of Helicobacter pylori infection improves blood pressure values in patients affected by hypertension. Helicobacter 2003, 8, 585–589. [Google Scholar] [CrossRef]
- Alba, C.; Blanco, A.; Alarcón, T. Antibiotic resistance in Helicobacter pylori. Curr. Opin. Infect. Dis. 2017, 30, 489–497. [Google Scholar] [CrossRef]
- Nista, E.C.; Pellegrino, A.; Giuli, L.; Candelli, M.; Schepis, T.; De Lucia, S.S.; Ojetti, V.; Franceschi, F.; Gasbarrini, A. Clinical Implications of Helicobacter pylori Antibiotic Resistance in Italy: A Review of the Literature. Antibiotics 2022, 11, 1452. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; O’Morain, C.; McNamara, D. Helicobacter pylori resistance to current therapies. Curr. Opin. Gastroenterol. 2019, 35, 6–13. [Google Scholar] [CrossRef] [PubMed]
- D’Achille, G.; Morroni, G. Side effects of antibiotics and perturbations of mitochondria functions. Int. Rev. Cell Mol. Biol. 2023, 377, 121–139. [Google Scholar] [PubMed]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.J.; van der Velden, S.; Rios-Morales, M.; van Faassen, M.J.R.; Loreti, M.G.; de Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef]
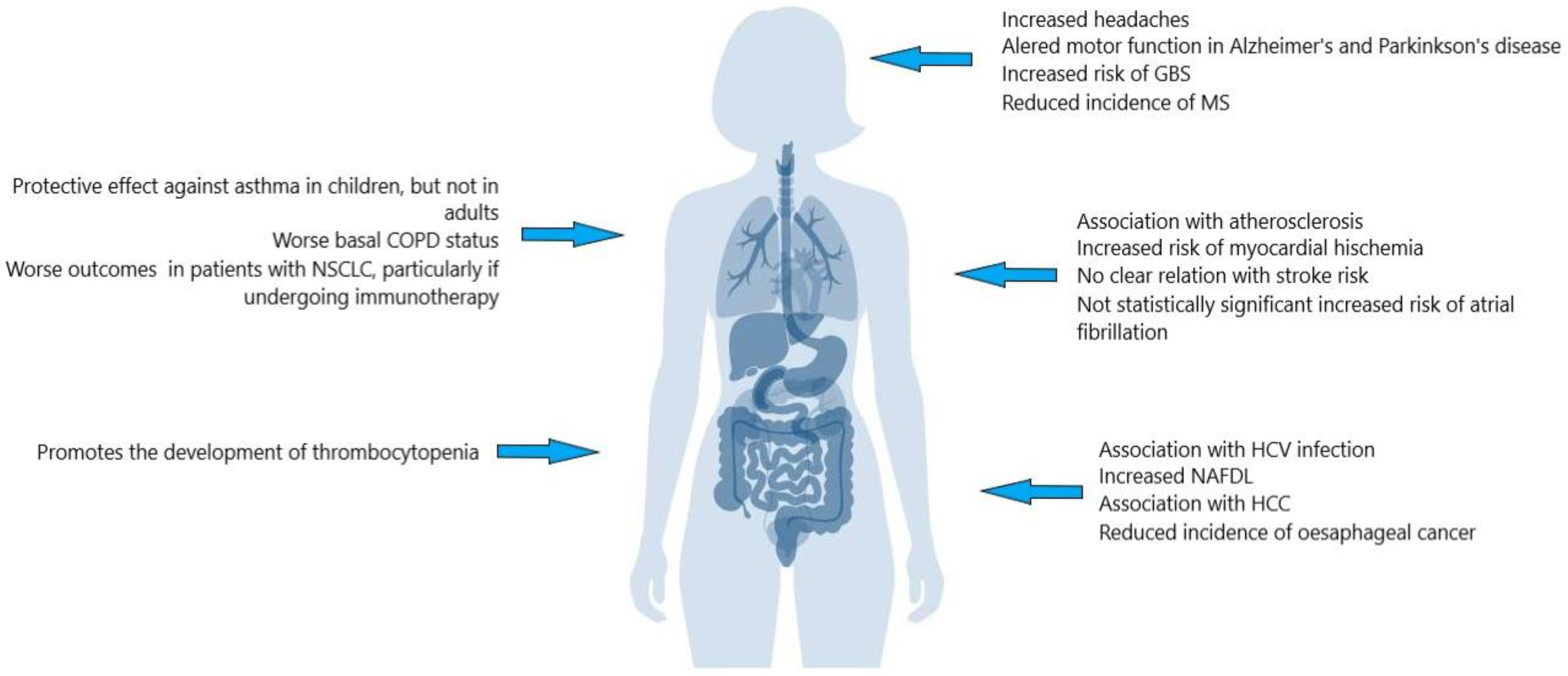

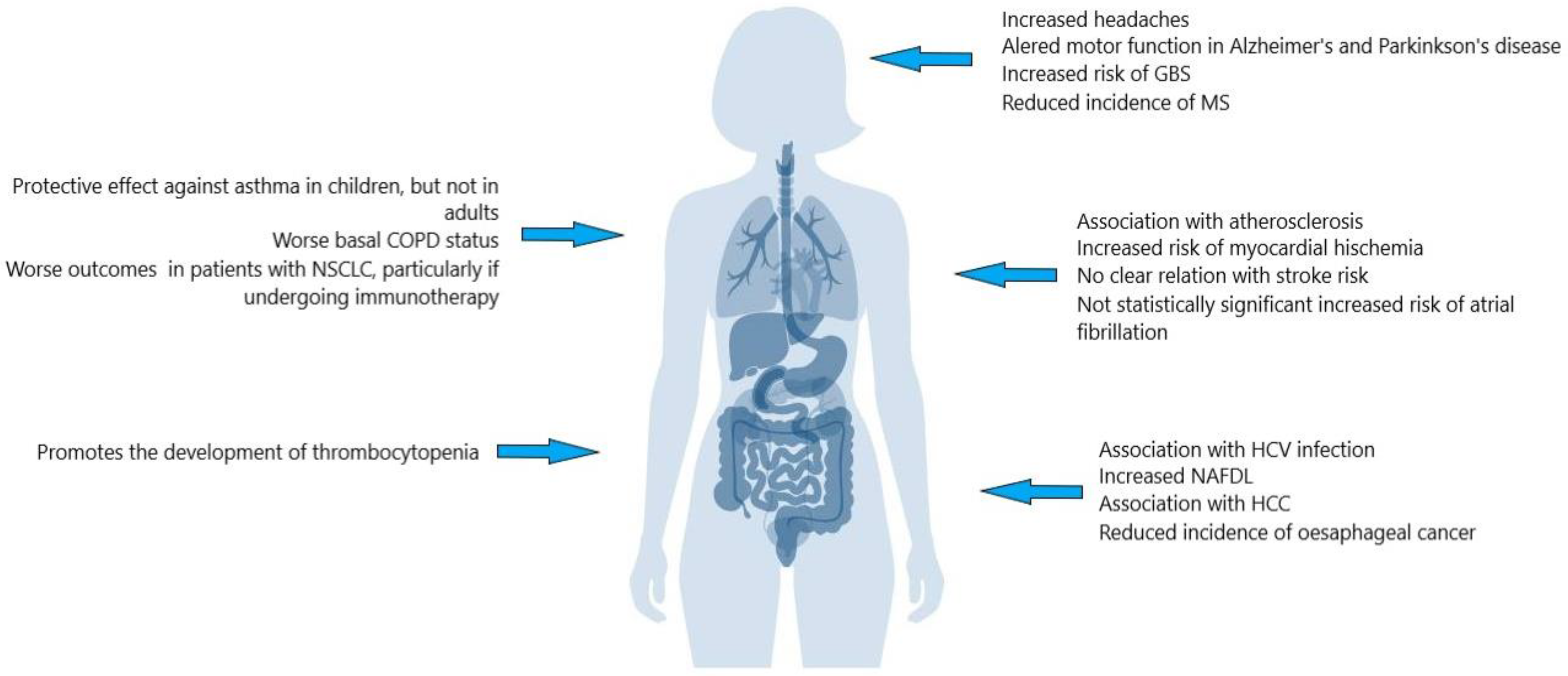{kind=link}
| Pathogenic Pathway | Effect | Model | Reference |
|---|---|---|---|
| LPS | Th1/Th2-activation Low grade inflammation through DCSIGN/lectin pathway | Murine | [69] |
| HP-NAP | IL-8, IL-12, IL-23 ↑ MIP-1α, and MIP-1 ↑ Mast cell activation Increased IL-6 and TNF-α | In vitro Murine | [70] |
| VacA | TNF-α, MIP-1α, IL-1, IL-6, IL-10, and IL-1 ↑ Inhibited differentiation of T lymphocytes | In vitro Murine Human | [71] |
| CagA | Autophagy in host cells ↓ IL-1α, IL-6, IL-8, and IL-18 ↑ c-Met activation PI3K/AKT/mTOR pathway activation Accumulation of SQSTM1 ROS ↑ Cross-reactivity PPARγ and LXRα ↑ Foam cells ↑ | In vitro Murine Human | [73,74,75,78,79,80,81,91] |
| NLRP3 | Pyroptosis ↑ ROS ↑ Activation NADPH oxidase NF-κB pathway activation PI3K/Akt pathway activation | In vitro Murine | [94] |
| GKN-1 | Microbiota modulation Leaky gut Firmicutes↑ TMA/TMAO↑ | Human | [96] |
| Platelet activation | Interaction between von Willebrand factor and platelet surface glycoproteins Ib/IX ↑ 12-lipoxigenase pathway | Rabbit Human | [100,101] |
| Lpp20 | Platelet immune complexes | Human | [100] |
| Extracellular vesicles Outer membrane vesicles | Transport of pathogenic factors | Murine Human | [88,106] |
| Lipid metabolism | HDL ↑ after eradication | In vitro Murine Human | [103] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candelli, M.; Franza, L.; Cianci, R.; Pignataro, G.; Merra, G.; Piccioni, A.; Ojetti, V.; Gasbarrini, A.; Franceschi, F. The Interplay between Helicobacter pylori and Gut Microbiota in Non-Gastrointestinal Disorders: A Special Focus on Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 17520. https://doi.org/10.3390/ijms242417520
Candelli M, Franza L, Cianci R, Pignataro G, Merra G, Piccioni A, Ojetti V, Gasbarrini A, Franceschi F. The Interplay between Helicobacter pylori and Gut Microbiota in Non-Gastrointestinal Disorders: A Special Focus on Atherosclerosis. International Journal of Molecular Sciences. 2023; 24(24):17520. https://doi.org/10.3390/ijms242417520
Chicago/Turabian StyleCandelli, Marcello, Laura Franza, Rossella Cianci, Giulia Pignataro, Giuseppe Merra, Andrea Piccioni, Veronica Ojetti, Antonio Gasbarrini, and Francesco Franceschi. 2023. "The Interplay between Helicobacter pylori and Gut Microbiota in Non-Gastrointestinal Disorders: A Special Focus on Atherosclerosis" International Journal of Molecular Sciences 24, no. 24: 17520. https://doi.org/10.3390/ijms242417520
APA StyleCandelli, M., Franza, L., Cianci, R., Pignataro, G., Merra, G., Piccioni, A., Ojetti, V., Gasbarrini, A., & Franceschi, F. (2023). The Interplay between Helicobacter pylori and Gut Microbiota in Non-Gastrointestinal Disorders: A Special Focus on Atherosclerosis. International Journal of Molecular Sciences, 24(24), 17520. https://doi.org/10.3390/ijms242417520