
The Immune Signature of CSF in Multiple Sclerosis with and without Oligoclonal Bands: A Machine Learning Approach to Proximity Extension Assay Analysis

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
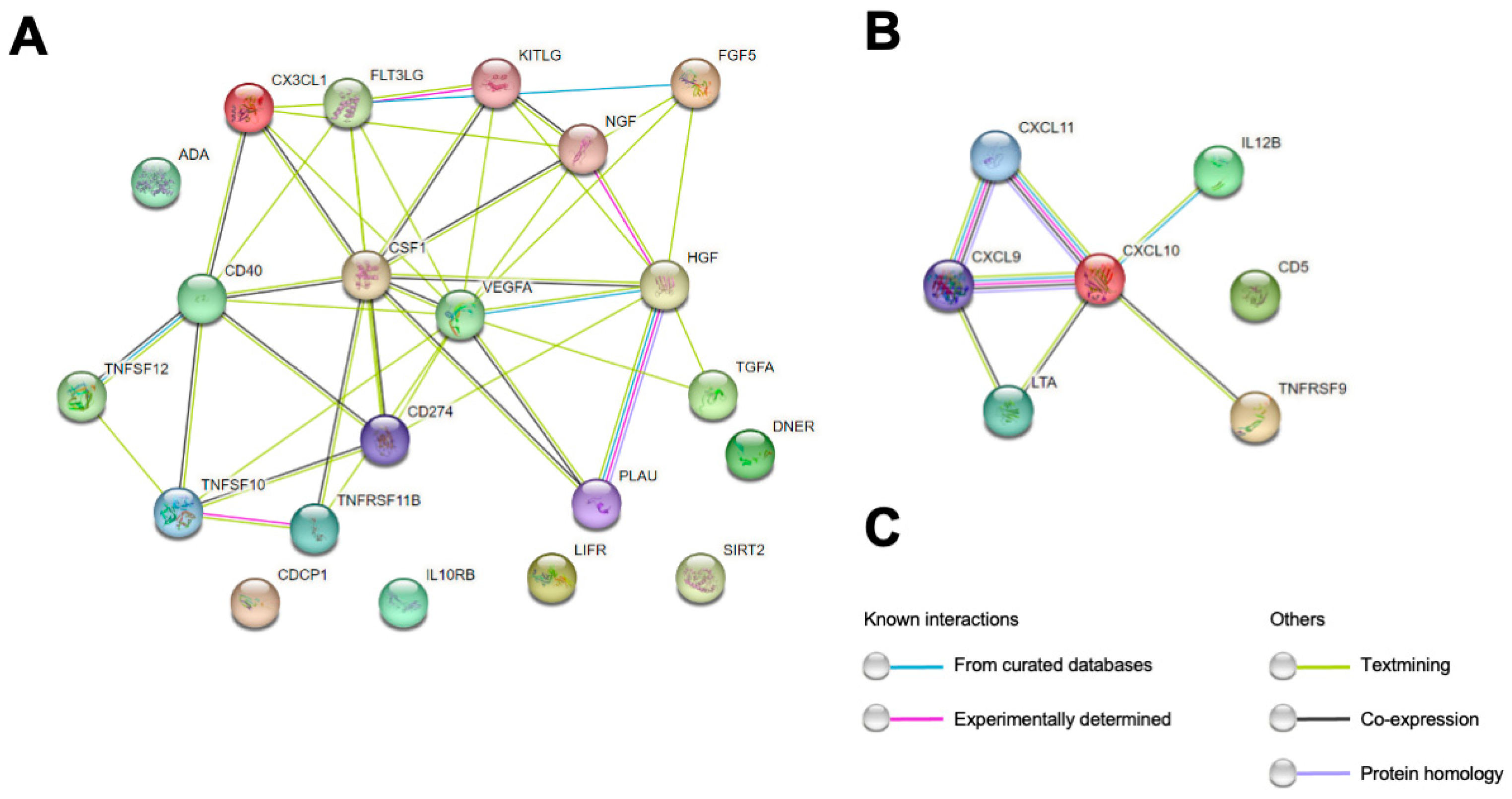
2. Results
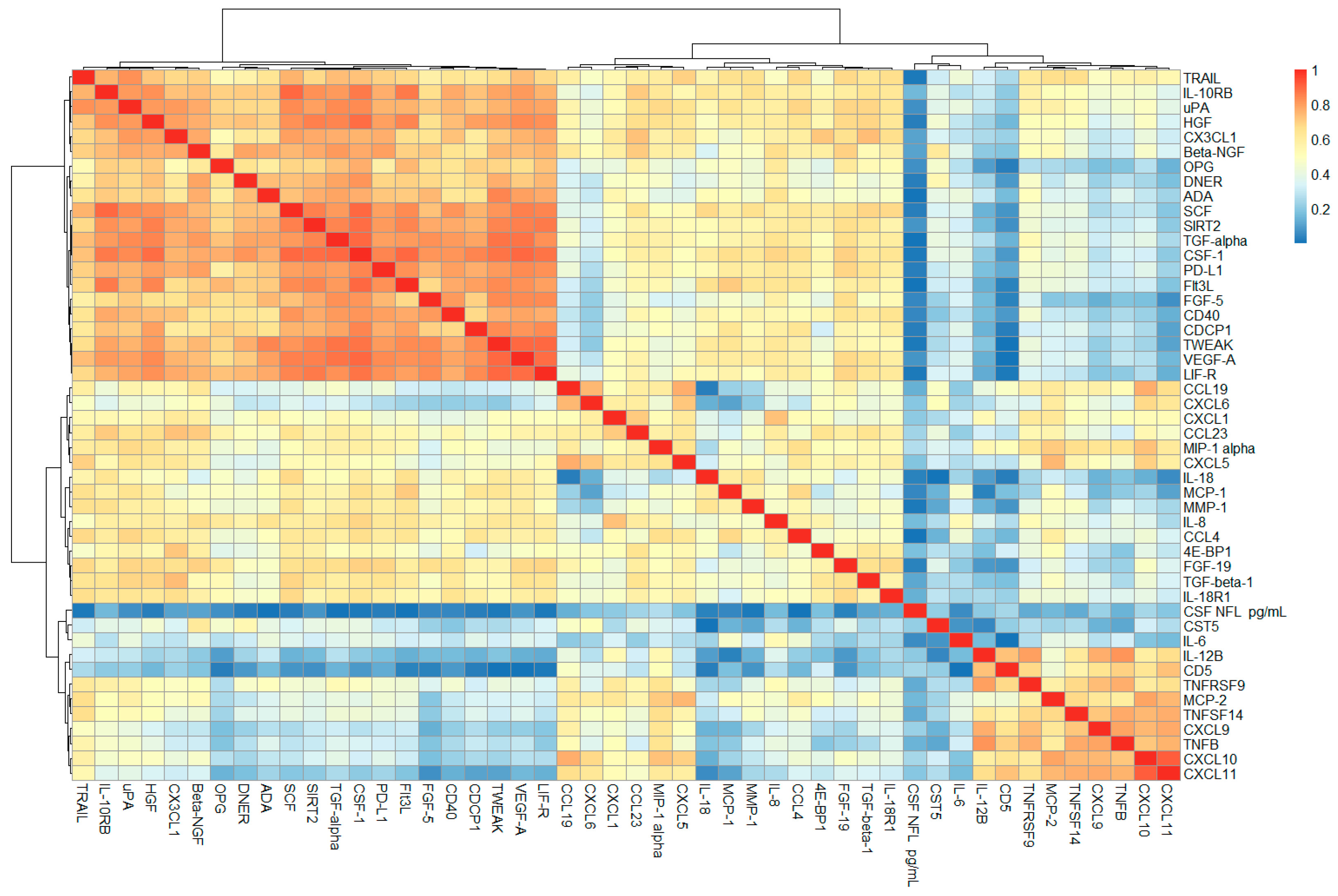
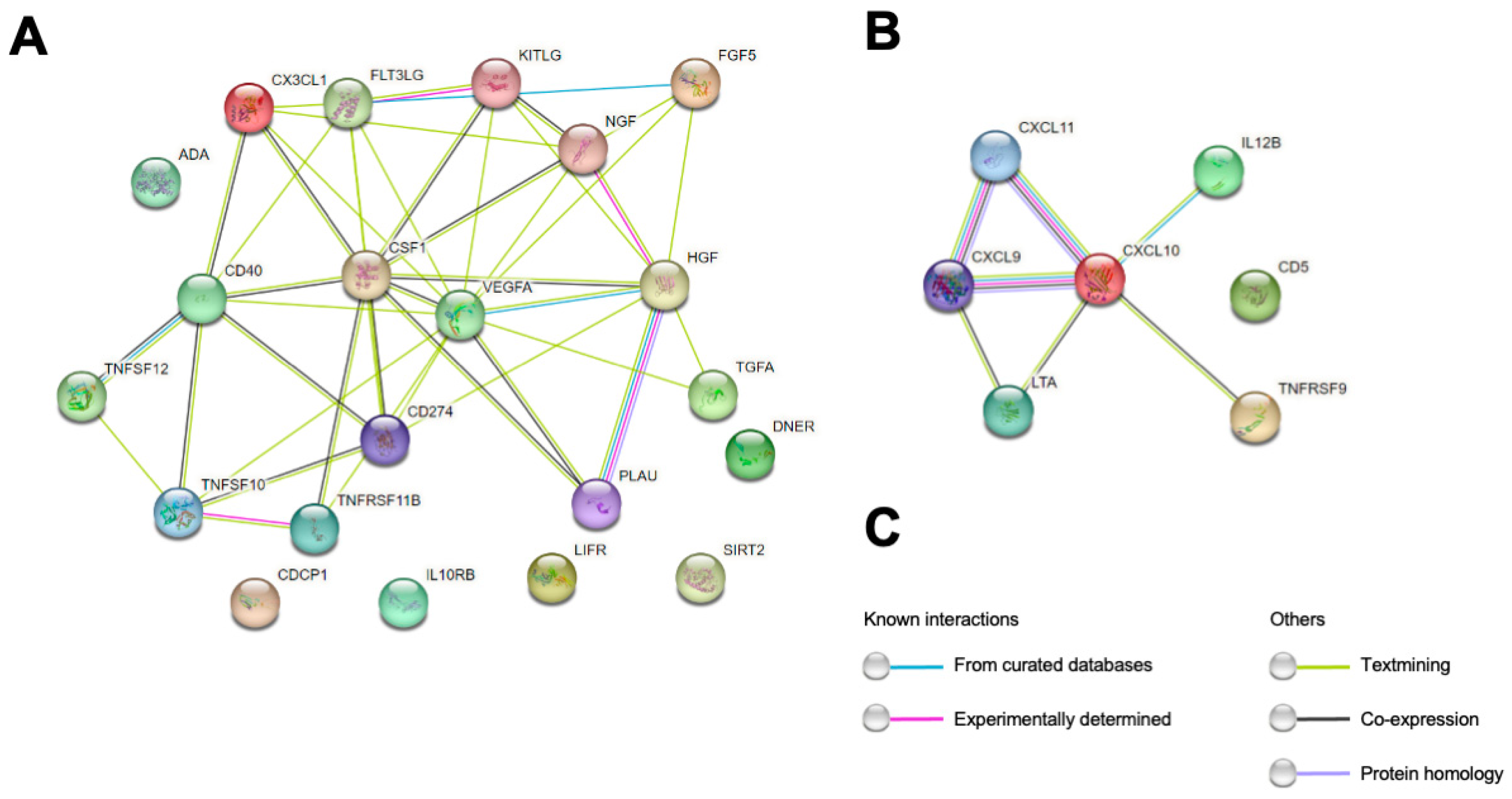
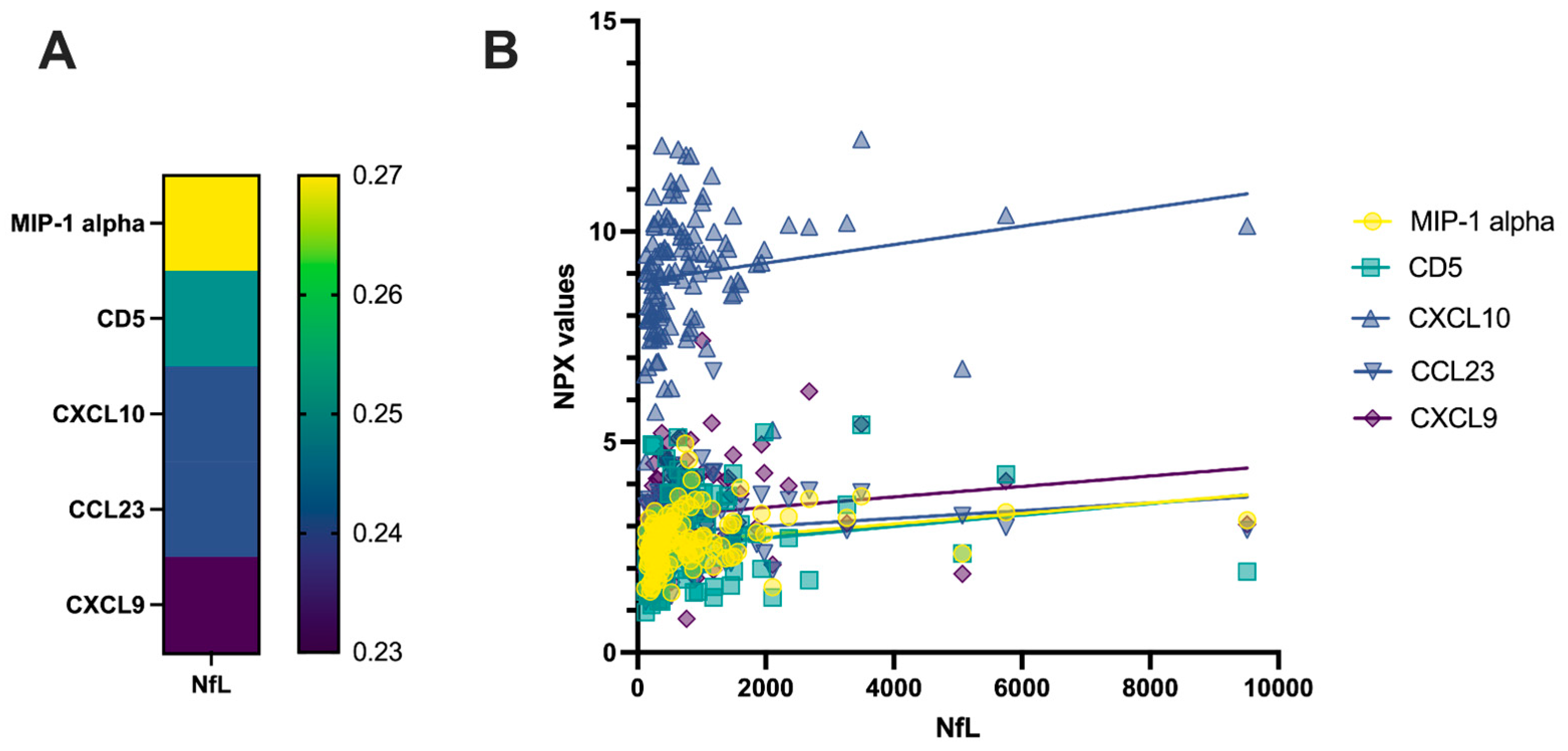
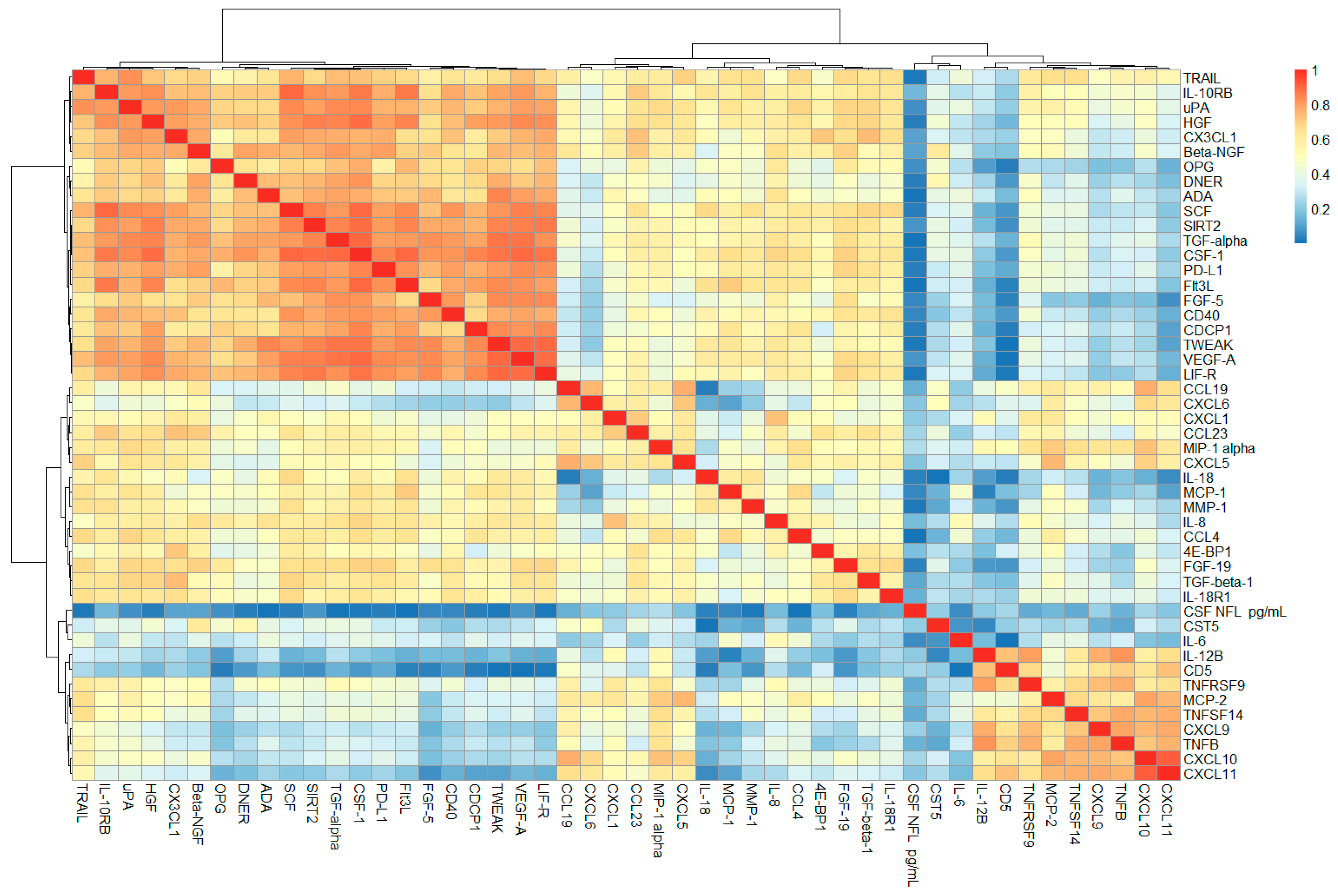
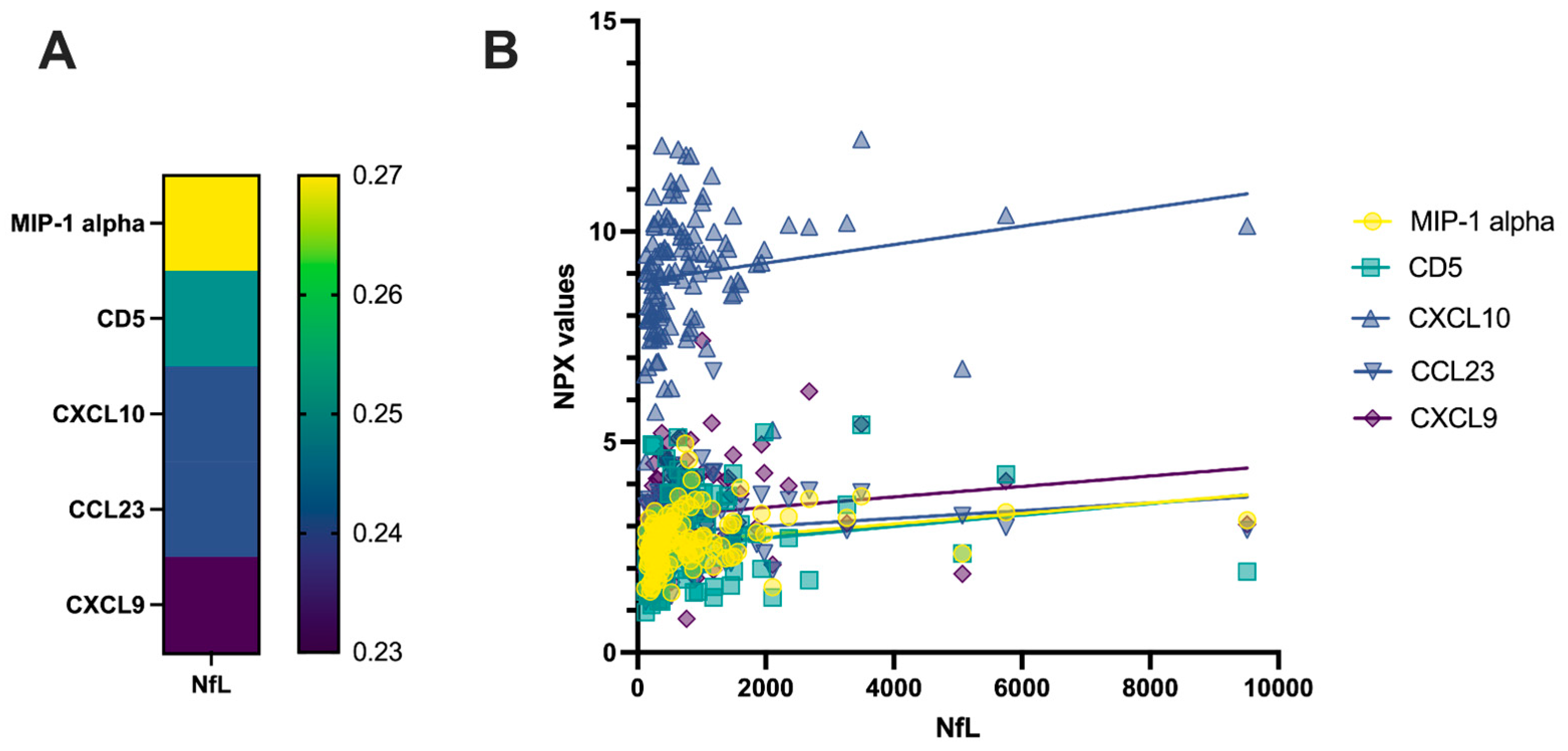
2.1. Correlation Analysis of CSF PEA-Tested Proteins and NfL
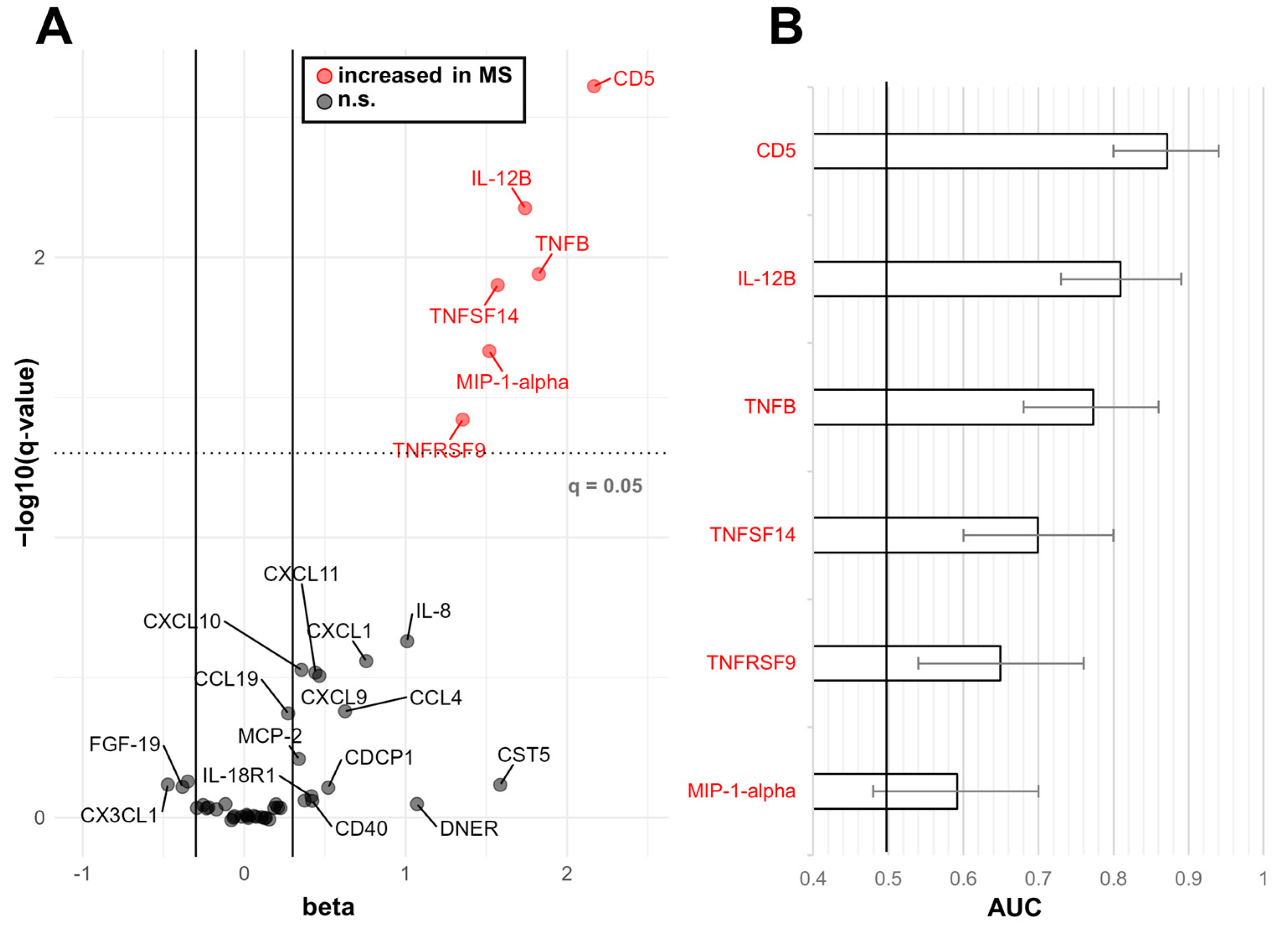
2.2. Biomarkers Efficacy in Discriminating between RRMS and OND
2.3. Multivariate Analysis of PEA-Tested Proteins in MS OCB+ vs. OND
2.4. Multivariate Analysis of PEA-Tested Proteins in MS OCB− vs. OND
3. Discussion
4. Materials and Methods
4.1. CSF Sampling
4.2. Selection of CSF Samples
4.3. Standard CSF Analysis
4.4. CSF NfL Measurement
4.5. CSF PEA Testing
4.6. Characteristics of the Patients
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jakimovski, D.; Bittner, S.; Zivadinov, R.; Morrow, S.A.; Benedict, R.H.; Zipp, F.; Weinstock-Guttman, B. Multiple sclerosis. Lancet, 2023; online first. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Solomon, A.J.; Arrambide, G.; Brownlee, W.J.; Flanagan, E.P.; Amato, M.P.; Amezcua, L.; Banwell, B.L.; Barkhof, F.; Corboy, J.R.; Correale, J.; et al. Differential diagnosis of suspected multiple sclerosis: An updated consensus approach. Lancet Neurol. 2023, 22, 750–768. [Google Scholar] [CrossRef] [PubMed]
- Tintore, M.; Rovira, À.; Río, J.; Otero-Romero, S.; Arrambide, G.; Tur, C.; Comabella, M.; Nos, C.; Arévalo, M.J.; Negrotto, L.; et al. Defining high, medium and low impact prognostic factors for developing multiple sclerosis. Brain 2015, 138, 1863–1874. [Google Scholar] [CrossRef]
- Link, H.; Huang, Y.M. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: An update on methodology and clinical usefulness. J. Neuroimmunol. 2006, 180, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Malekzadeh, A.; Leurs, C.; Bridel, C.; Killestein, J. Body fluid biomarkers for multiple sclerosis—The long road to clinical application. Nat. Rev. Neurol. 2015, 11, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Ramagopalan, S.; Davis, A.; Giovannoni, G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: A meta-analysis of prevalence, prognosis and effect of latitude. J. Neurol. Neurosurg. Psychiatry 2013, 84, 909–914. [Google Scholar] [CrossRef]
- Katsarogiannis, E.; Landtblom, A.-M.; Kristoffersson, A.; Wikström, J.; Semnic, R.; Berntsson, S.G. Absence of Oligoclonal Bands in Multiple Sclerosis: A Call for Differential Diagnosis. J. Clin. Med. 2023, 12, 4656. [Google Scholar] [CrossRef]
- Andreasson, U.; Portelius, E.; Pannee, J.; Zetterberg, H.; Blennow, K. Multiplexing and multivariate analysis in neurodegeneration. Methods 2012, 56, 464–470. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Kimble, L.; Bayoumy, S.; Bolsewig, K.; Burtscher, F.; Coppens, S.; Das, S.; Gogishvili, D.; Gomes, B.F.; Gómez de San José, N.; et al. Methods to discover and validate biofluid-based biomarkers in neurodegenerative dementias. Mol. Cell. Proteom. 2023, 22, 100629. [Google Scholar] [CrossRef]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Rennel Dickens, E.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef]
- Jabbari, E.; Woodside, J.; Guo, T.; Magdalinou, N.K.; Chelban, V.; Athauda, D.; Lees, A.J.; Foltynie, T.; Houlden, H.; Church, A.; et al. Proximity extension assay testing reveals novel diagnostic biomarkers of atypical parkinsonian syndromes. J. Neurol. Neurosurg. Psychiatry 2019, 90, 768–773. [Google Scholar] [CrossRef]
- Gaetani, L.; Bellomo, G.; Parnetti, L.; Blennow, K.; Zetterberg, H.; Di Filippo, M. Neuroinflammation and Alzheimer’s Disease: A Machine Learning Approach to CSF Proteomics. Cells 2021, 10, 1930. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Khademi, M.; Fugger, L.; Lindhe, Ö.; Novakova, L.; Axelsson, M.; Malmeström, C.; Constantinescu, C.; Lycke, J.; Piehl, F.; et al. Inflammation-related plasma and CSF biomarkers for multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 12952–12960. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Voisinne, G.; Gonzalez de Peredo, A.; Roncagalli, R. CD5, an Undercover Regulator of TCR Signaling. Front. Immunol. 2018, 9, 2900. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium (IMSGC). Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef]
- Myers, L.M.; Vella, A.T. Interfacing T-cell effector and regulatory function through CD137 (4-1BB) co-stimulation. Trends Immunol. 2005, 26, 440–446. [Google Scholar] [CrossRef]
- Sharief, M.K. Heightened intrathecal release of soluble CD137 in patients with multiple sclerosis. Eur. J. Neurol. 2002, 9, 49–54. [Google Scholar] [CrossRef]
- Balashov, K.E.; Rottman, J.B.; Weiner, H.L.; Hancock, W.W. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6873–6878. [Google Scholar] [CrossRef]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef]
- Malmeström, C.; Gillett, A.; Jernås, M.; Khademi, M.; Axelsson, M.; Kockum, I.; Mattsson, N.; Zetterberg, H.; Blennow, K.; Alfredsson, L.; et al. Serum levels of LIGHT in MS. Mult. Scler. 2013, 19, 871–876. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef]
- Zuccalà, M.; Barizzone, N.; Boggio, E.; Gigliotti, L.; Sorosina, M.; Basagni, C.; Bordoni, R.; Clarelli, F.; Anand, S.; Mangano, E.; et al. Genomic and functional evaluation of TNFSF14 in multiple sclerosis susceptibility. J. Genet. Genom. 2021, 48, 497–507. [Google Scholar] [CrossRef]
- Gommerman, J.L.; Browning, J.L. Lymphotoxin/LIGHT, lymphoid microenvironments and autoimmune disease. Nat. Rev. Immunol. 2003, 3, 642–655. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.W.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Serafini, B.; Aloisi, F.; Reynolds, R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010, 68, 477–493. [Google Scholar] [CrossRef] [PubMed]
- James Bates, R.E.; Browne, E.; Schalks, R.; Jacobs, H.; Tan, L.; Parekh, P.; Magliozzi, R.; Calabrese, M.; Mazarakis, N.D.; Reynolds, R. Lymphotoxin-alpha expression in the meninges causes lymphoid tissue formation and neurodegeneration. Brain 2022, 145, 4287–4307. [Google Scholar] [CrossRef] [PubMed]
- Blauth, K.; Zhang, X.; Chopra, M.; Rogan, S.; Markovic-Plese, S. The role of fractalkine (CX3CL1) in regulation of CD4+ cell migration to the central nervous system in patients with relapsing-remitting multiple sclerosis. Clin. Immunol. 2015, 157, 121–132. [Google Scholar] [CrossRef]
- Hill, L.J.; Di Pietro, V.; Hazeldine, J.; Davies, D.; Toman, E.; Logan, A.; Belli, A. Cystatin D (CST5): An ultra-early inflammatory biomarker of traumatic brain injury. Sci. Rep. 2017, 7, 5002. [Google Scholar] [CrossRef]
- Collins, A.R.; Grubb, A. Cystatin D, a natural salivary cysteine protease inhibitor, inhibits coronavirus replication at its physiologic concentration. Oral Microbiol. Immunol. 1998, 13, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, R.; Böttiger, G.; Stadelmann, C.; Karnati, S.; Berghoff, M. FGF/FGFR Pathways in Multiple Sclerosis and in Its Disease Models. Cells 2021, 10, 884. [Google Scholar] [CrossRef]
- Passaponti, S.; Ermini, L.; Acconci, G.; Severi, F.M.; Romagnoli, R.; Cutrupi, S.; Clerico, M.; Guerrera, G.; Ietta, F. Rank-Rankl-Opg Axis in Multiple Sclerosis: The Contribution of Placenta. Cells 2022, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Glasnović, A.; O’Mara, N.; Kovačić, N.; Grčević, D.; Gajović, S. RANK/RANKL/OPG Signaling in the Brain: A Systematic Review of the Literature. Front. Neurol. 2020, 11, 590480. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Gasperini, C.; Tortorella, C.; Schiavi, G.; Frisullo, G.; Ragonese, P.; Fantozzi, R.; Prosperini, L.; Annovazzi, P.; Cordioli, C.; et al. “Better explanations” in multiple sclerosis diagnostic workup: A 3-year longitudinal study. Neurology 2019, 92, e2527–e2537. [Google Scholar] [CrossRef]
- Martin, S.-J.; McGlasson, S.; Hunt, D.; Overell, J. Cerebrospinal fluid neurofilament light chain in multiple sclerosis and its subtypes: A meta-analysis of case-control studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1059–1067. [Google Scholar] [CrossRef]
- Sørensen, T.L.; Tani, M.; Jensen, J.; Pierce, V.; Lucchinetti, C.; Folcik, V.A.; Qin, S.; Rottman, J.; Sellebjerg, F.; Strieter, R.M.; et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J. Clin. Investig. 1999, 103, 807–815. [Google Scholar] [CrossRef]
- Kanno, Y. The uPA/uPAR System Orchestrates the Inflammatory Response, Vascular Homeostasis, and Immune System in Fibrosis Progression. Int. J. Mol. Sci. 2023, 24, 1796. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, F.-Y.; Ma, T.-L.; Shih, H.-Y.; Lin, S.-J.; Huang, C.-W.; Wang, H.-Y.; Cheng, Y.-C. Dner inhibits neural progenitor proliferation and induces neuronal and glial differentiation in zebrafish. Dev. Biol. 2013, 375, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.; Lai, Y.; Baella, N.; Dalmau, J.; Lancaster, E. Antibodies to Delta/Notch-like Epidermal Growth Factor–Related Receptor in Patients With Anti-Tr, Paraneoplastic Cerebellar Degeneration, and Hodgkin Lymphoma. JAMA Neurol. 2014, 71, 1003. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Hong, T.; Chen, X.; Cui, L. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res. Rev. 2019, 55, 100961. [Google Scholar] [CrossRef]
- Hossain, M.Z.; Daskalaki, E.; Brüstle, A.; Desborough, J.; Lueck, C.J.; Suominen, H. The role of machine learning in developing non-magnetic resonance imaging based biomarkers for multiple sclerosis: A systematic review. BMC Med. Inform. Decis. Mak. 2022, 22, 242. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Petzold, A.; Bennett, J.L.; Berven, F.S.; Brundin, L.; Comabella, M.; Franciotta, D.; Frederiksen, J.L.; Fleming, J.O.; Furlan, R.; et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 2009, 73, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Deisenhammer, F.; Bartos, A.; Egg, R.; Gilhus, N.E.; Giovannoni, G.; Rauer, S.; Sellebjerg, F. Guidelines on routine cerebrospinal fluid analysis. Report from an EFNS task force. Eur. J. Neurol. 2006, 13, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, L.; Höglund, K.; Parnetti, L.; Pujol-Calderon, F.; Becker, B.; Eusebi, P.; Sarchielli, P.; Calabresi, P.; Di Filippo, M.; Zetterberg, H.; et al. A new enzyme-linked immunosorbent assay for neurofilament light in cerebrospinal fluid: Analytical validation and clinical evaluation. Alzheimers Res. Ther. 2018, 10, 8. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Condition | ||||
|---|---|---|---|---|
| Total 118 | MS OCB+ 67 | MS OCB− 5 | OND 46 | |
| Actual condition | MS OCB+ 58 | 55 | 10 | 2 |
| MS OCB− 24 | 1 | 3 | 1 | |
| OND 36 | 2 | 11 | 33 | |
| A | Predicted Condition | B | Predicted Condition | ||||
|---|---|---|---|---|---|---|---|
| Total 40 | MS OCB+ 22 | OND 18 | Total 54 | MS OCB+ 36 | OND 18 | ||
| Actual condition | MS OCB+ 22 | 20 | 2 | Actual condition | MS OCB+ 36 | 29 | 7 |
| OND 18 | 2 | 16 | OND 18 | 7 | 11 | ||
| A | Predicted Condition | B | Predicted Condition | ||||
|---|---|---|---|---|---|---|---|
| Total 30 | MS OCB− 15 | OND 15 | Total 30 | MS OCB− 9 | OND 21 | ||
| Actual condition | MS OCB− 15 | 13 | 2 | Actual condition | MS OCB− 9 | 5 | 4 |
| OND 15 | 2 | 13 | OND 21 | 4 | 17 | ||
| OND | RRMS | p-Value * | MS OCB+ | MS OCB− | p-Value ** | |
|---|---|---|---|---|---|---|
| N | 36 | 82 | - | 58 | 24 | - |
| Sex–F | 18 (50%) | 58 (70.7%) | <0.05 | 41 (70.7%) | 17 (70.8%) | n.s. |
| Age (years) | 57.9 ± 16.6 | 37.9 ± 10.6 | <0.01 | 37 ± 10.3 | 40.2 ± 11.4 | n.s. |
| EDSS | - | 1.9 ± 1 | - | 1.8 ± 0.8 | 2.3 ± 1.5 | n.s. |
| Disease duration (months) | - | 3 (23.5) | - | 3 (23.6) | 2 (23.5) | n.s. |
| Recent relapse (<30 days) | - | 60 (73.2%) | - | 43 (74.1%) | 17 (70.8%) | n.s. |
| CSF NfL (pg/mL) | 361 (504) | 361 (504) | 361 (504) | 633 (929) | 637.5 (558) | n.s. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaetani, L.; Bellomo, G.; Di Sabatino, E.; Sperandei, S.; Mancini, A.; Blennow, K.; Zetterberg, H.; Parnetti, L.; Di Filippo, M. The Immune Signature of CSF in Multiple Sclerosis with and without Oligoclonal Bands: A Machine Learning Approach to Proximity Extension Assay Analysis. Int. J. Mol. Sci. 2024, 25, 139. https://doi.org/10.3390/ijms25010139
Gaetani L, Bellomo G, Di Sabatino E, Sperandei S, Mancini A, Blennow K, Zetterberg H, Parnetti L, Di Filippo M. The Immune Signature of CSF in Multiple Sclerosis with and without Oligoclonal Bands: A Machine Learning Approach to Proximity Extension Assay Analysis. International Journal of Molecular Sciences. 2024; 25(1):139. https://doi.org/10.3390/ijms25010139
Chicago/Turabian StyleGaetani, Lorenzo, Giovanni Bellomo, Elena Di Sabatino, Silvia Sperandei, Andrea Mancini, Kaj Blennow, Henrik Zetterberg, Lucilla Parnetti, and Massimiliano Di Filippo. 2024. "The Immune Signature of CSF in Multiple Sclerosis with and without Oligoclonal Bands: A Machine Learning Approach to Proximity Extension Assay Analysis" International Journal of Molecular Sciences 25, no. 1: 139. https://doi.org/10.3390/ijms25010139
APA StyleGaetani, L., Bellomo, G., Di Sabatino, E., Sperandei, S., Mancini, A., Blennow, K., Zetterberg, H., Parnetti, L., & Di Filippo, M. (2024). The Immune Signature of CSF in Multiple Sclerosis with and without Oligoclonal Bands: A Machine Learning Approach to Proximity Extension Assay Analysis. International Journal of Molecular Sciences, 25(1), 139. https://doi.org/10.3390/ijms25010139