
Translation Fidelity and Respiration Deficits in CLPP-Deficient Tissues: Mechanistic Insights from Mitochondrial Complexome Profiling


 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Migration Pattern and Abundance of AAA+ Proteins Indicate That CLPX and VWA8 Both Depend Strongly on CLPP
2.2. Co-Migration of mtSSU Monomeric Factors with ERAL1 as Expected, but Only mtLSU Intermediate Assemblies Co-Migrate with CLPX and VWA8
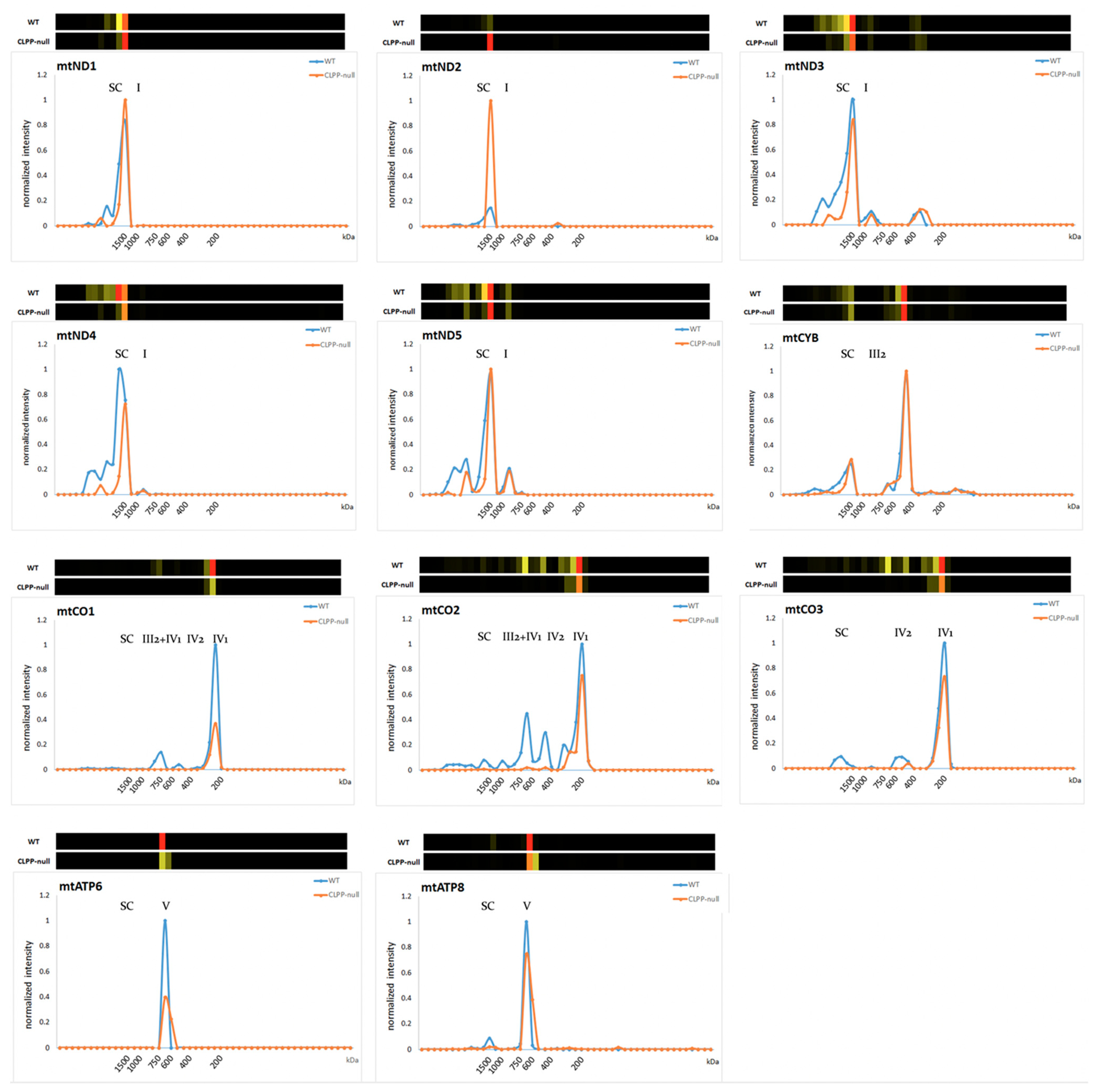
2.3. CLPP-Null OXPHOS Complexes Affected More by Assembly Than Translation Problems
2.4. Quantification of Heavy Metals in CLPP-Null Tissue Demonstrates Significant Increases in Iron, Molybdenum, Cobalt, and Manganese
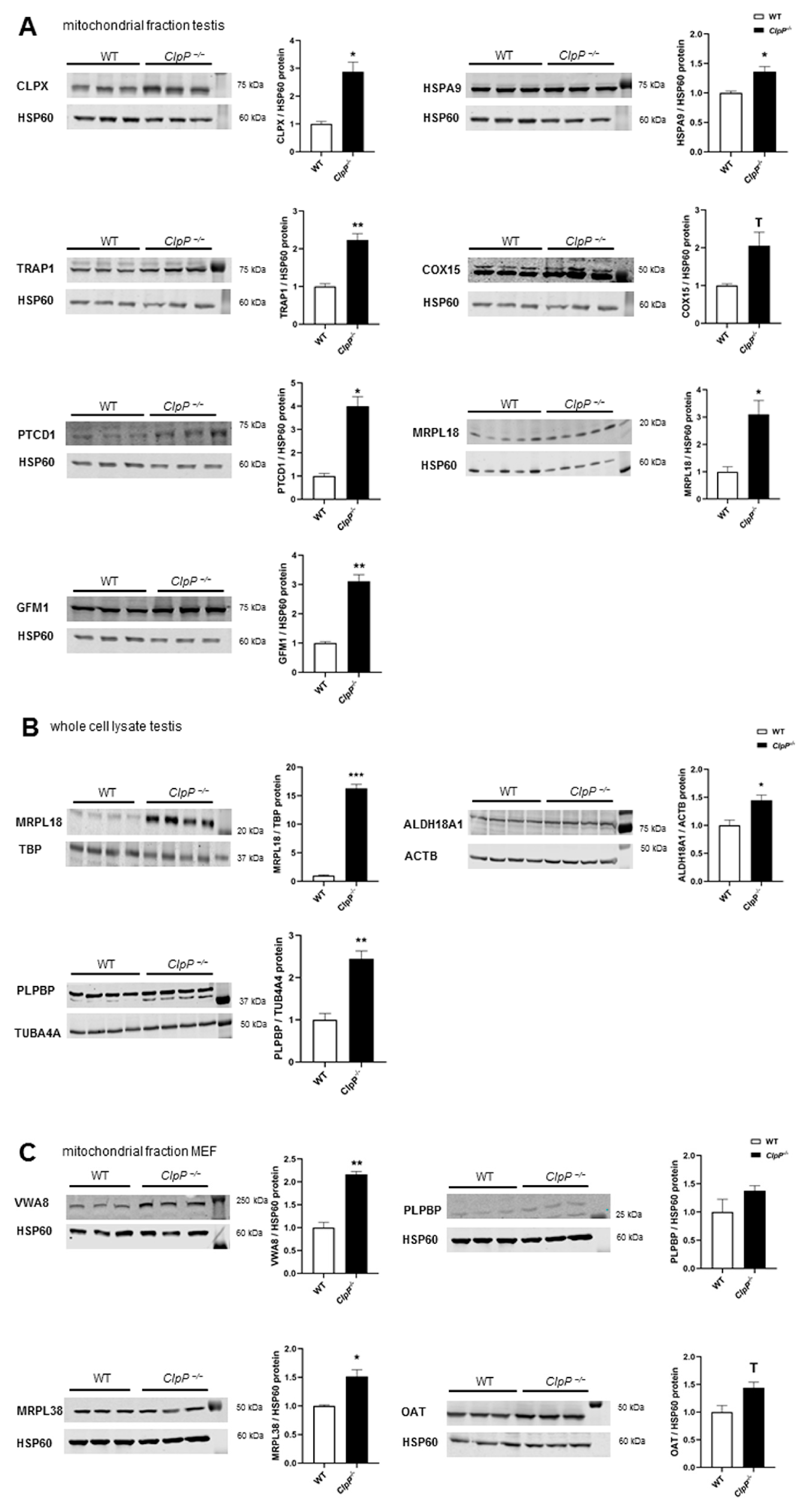
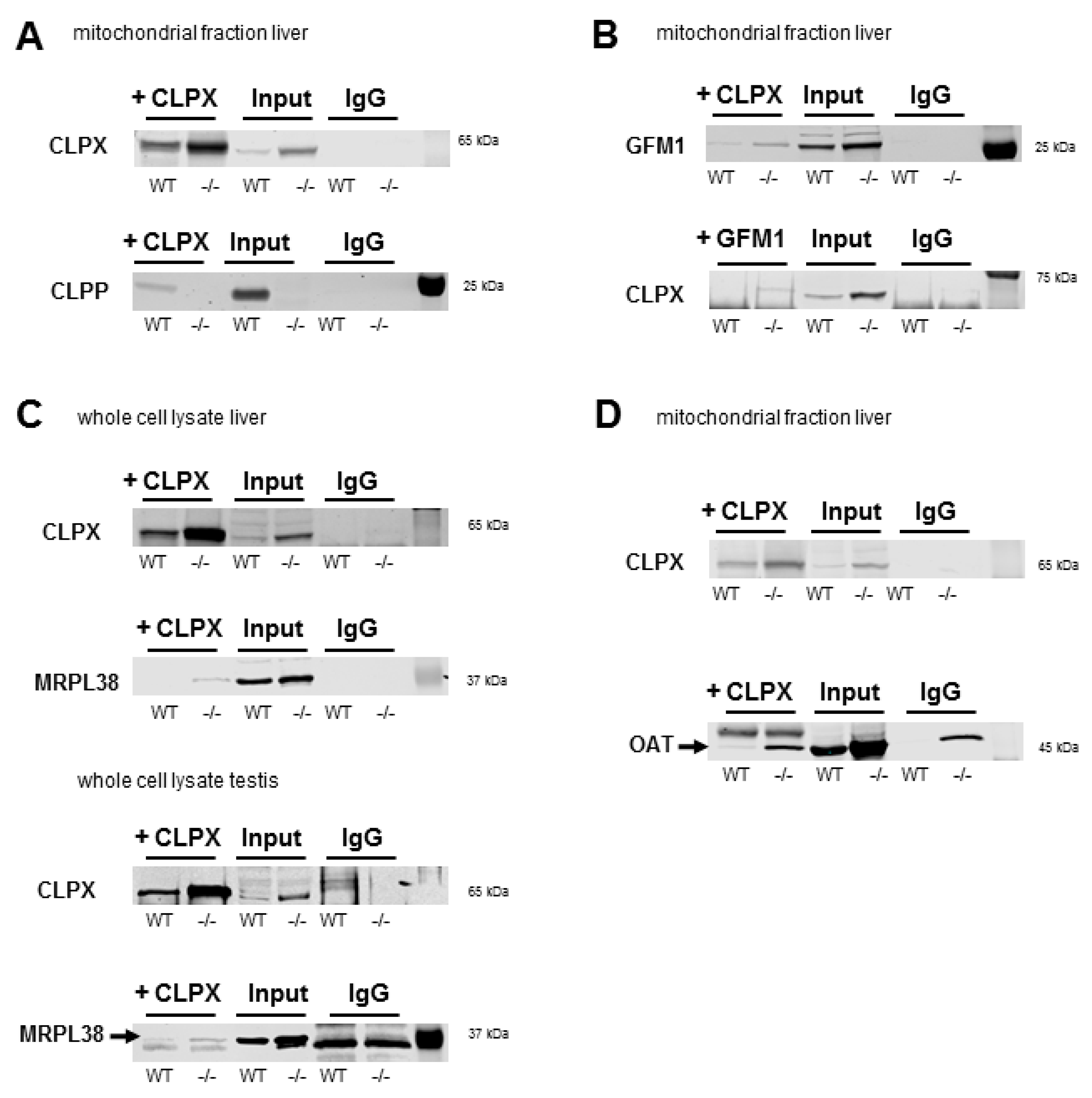
2.5. Validated Accumulation of VWA8, CLPX, PLPBP, GFM1, MRPL18, MRPL38, HSPA9, TRAP1, COX15, PTCD1, ALDH18A1, and OAT in Quantitative Immunoblots
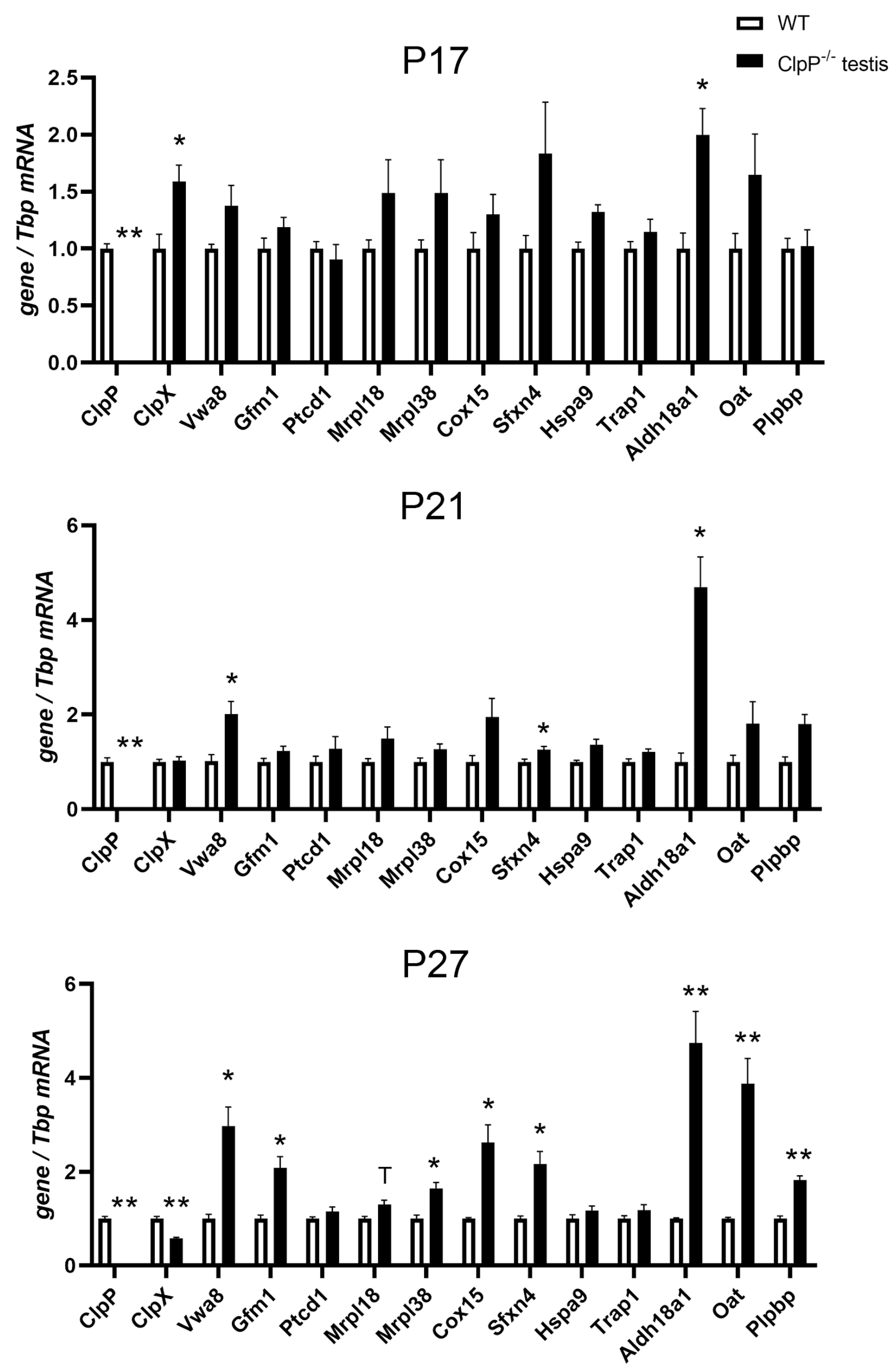
2.6. RT-qPCR Analysis of Transcriptional Regulations
2.7. Co-Immunoprecipitation of CLPX Confirms Interaction with MRPL18, GFM1, and OAT
3. Discussion
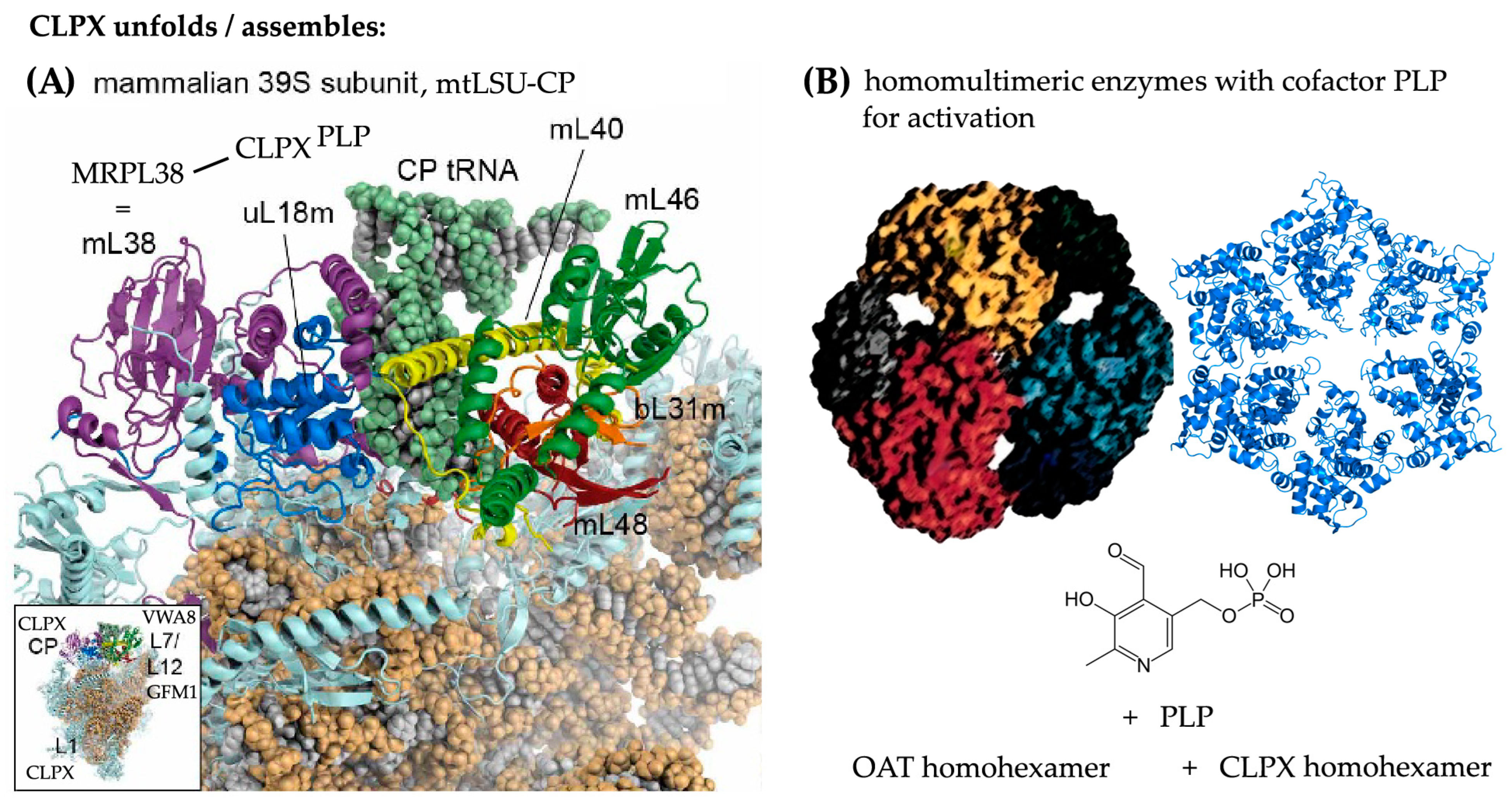
3.1. The Putative Role of CLPP-Dependent VWA8 for the mtLSU
3.2. The Putative Role of CLPX for the mtLSU Intermediate Assembly
3.3. Potential Functions of CLPX and VWA8 for Specific Translation/RNA Granule Factors, Matrix Enzymes, and PLP, as Well as Chaperones
3.4. Potential Functions of CLPX and VWA8 for Specific Respiratory Chain Complex Assembly
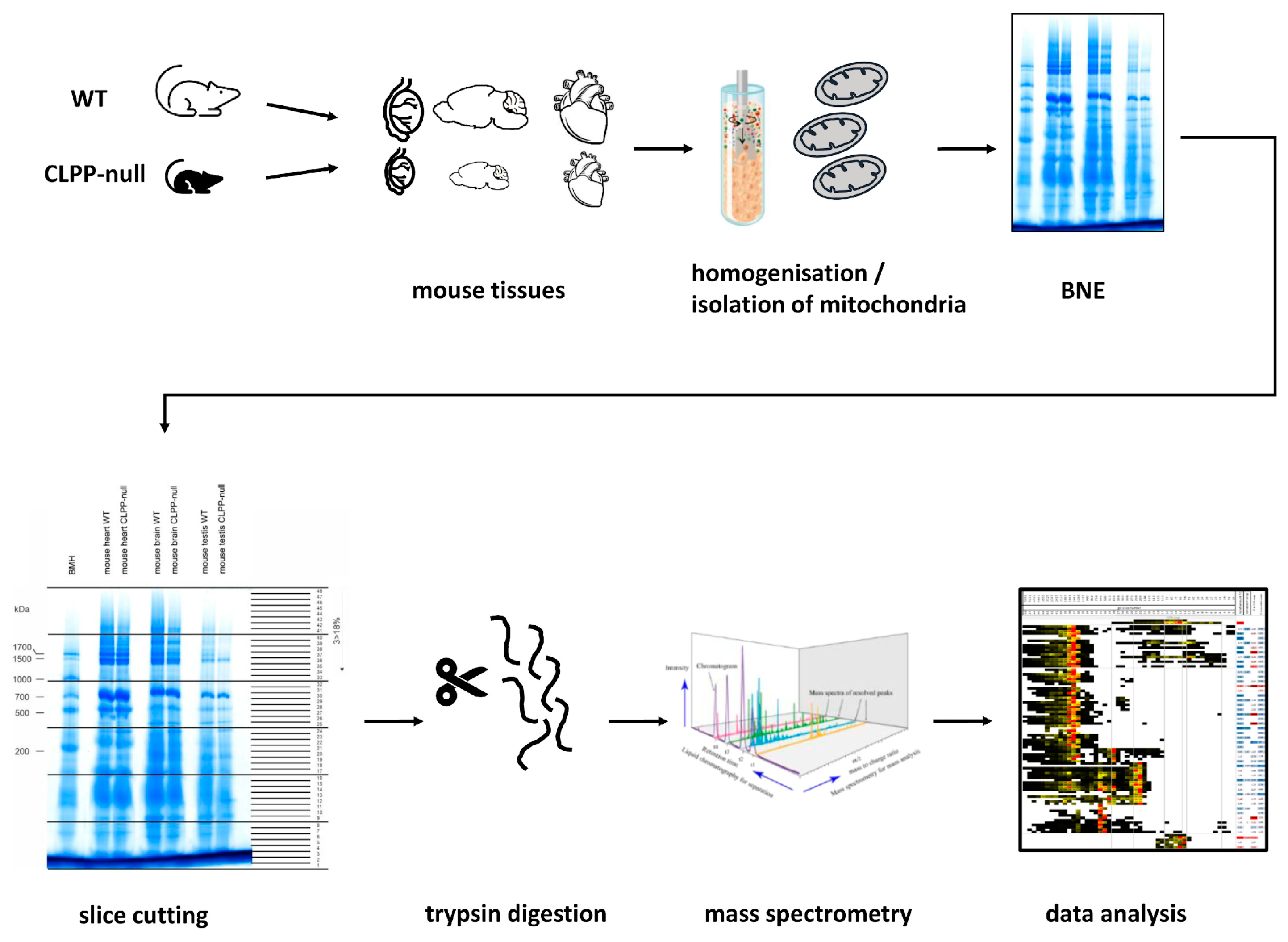
4. Materials and Methods
4.1. Animal Breeding, Aging, and Dissection
4.2. Experimental Design and Statistical Rationale
4.3. Isolation of Mitochondria in Sucrose Gradients
4.4. Sample Preparation for Complexome Profiling
4.5. Setting HPLC/MSMS Methods
4.6. Data Analysis with MaxQuant
4.7. Complexome Profiling for OXPHOS Subunits and Other Mitochondrial Assemblies
4.8. Protein–Protein Interaction Bioinformatics and Pathway Enrichment Analysis via STRING
4.9. Heavy Metal Analysis
4.10. Quantitative Immunoblots
4.11. RT-qPCR
4.12. Co-Immunoprecipitation
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 12S rRNA | ribosomal RNA component of SSU in bacteria |
| 16S rRNA | ribosomal RNA component of LSU in bacteria |
| 16S rRNA | ribosomal RNA component of mtSSU in mammals |
| 23S rRNA | ribosomal RNA component of mtLSU body |
| 28S | mtSSU of mammalian mitoribosomes |
| 39S | mtLSU of mammalian mitoribosomes |
| 55S | completely assembled mitoribosome |
| 5S rRNA | ribosomal RNA component of the mtLSU central protuberance |
| AAA+ | protein superfamily of ring-shaped P-loop NTPases |
| ABC | Ammonium Bicarbonate |
| ACAD9 | Acyl-Coenzyme A Dehydrogenase Family, Member 9, MCIA component |
| ACN | Acetonitrile |
| ACTB | beta-Actin protein |
| AFG3L2 | ATPase Family Member 3-Like 2, Matrix AAA Peptidase Subunit 2 |
| AGC | Automatic gain control |
| ALA | delta-Amino Levulinic Acid |
| ALAS | 5-AminoLevulinic Acid Synthase |
| ALAS2 | Delta-Amino-Levulinic-Acid-Synthase-2, erythroid-specific, mitochondrial |
| ALDH18A1 | P5CS, Aldehyde Dehydrogenase 18 Family Member A1 |
| ATAD3 | ATPase Family AAA Domain-Containing Protein 3 |
| ATPase | Adenosine 5’-TriPhosphatase |
| BCA | Bicinchoninic Acid |
| BCS1L | Ubiquinol-Cytochrome C Reductase Complex Chaperone |
| BNE | Blue Native gel Electrophoresis |
| CaCl2 | Calcium chloride |
| Cbb | Prokaryotic Rubisco activation protein |
| CbbQ | Prokaryotic Nitric oxide reductase NorQ protein; AAA ATPase |
| cDNA | complementary Deoxyribonucleic Acid |
| ChlD | Prokaryotic Magnesium protoporphyrin IX chelatase, d subunit |
| CI | Respiratory Complex I |
| CII | Respiratory Complex II |
| CIII | Respiratory Complex III |
| CIV | Respiratory Complex IV |
| CCDC12 | Coiled-Coil Domain-Containing Protein 127 |
| ClpA | Prokaryotic ATP-dependent specificity component of ClpAP protease |
| CLPB | Caseinolytic peptidase B protein homolog; AAA+ ATPase |
| CLPP | ATP-dependent Clp protease Proteolytic subunit, mitochondrial |
| CLPX | specificity component of Clp protease complex, AAA+ ATPase |
| CLPXP | proteolytic machine with components CLPP and CLPX |
| CO2 | Carbon dioxide |
| CobT | Prokaryotic Cobaltochelatase |
| CoIP | Co-immunoprecipitation |
| Cox1 | Cytochrome-c-Oxidase subunit 1, catalytic |
| COX15 | Cytochrome-c-Oxidase assembly factor; acts in heme-A biosynthesis |
| CP | Central Protuberance of mtLSU |
| CV | Respiratory Complex V |
| DNA | Deoxyribonucleic Acid |
| DnaJ | Prokaryotic co-chaperone Hsp40; acts with DnaK/GrpE in disassembly |
| DnaK | Prokaryotic chaperone Hsp70 |
| DAP3 | MRPS29, Death Associated Protein 3 |
| DNLZ | DNL-type Zinc-finger Protein; mtHsp70-escort protein |
| DTT | Dithiothreitol |
| ECSIT | Evolutionary Conserved Signal Intermediate In Toll pathway, mitochondrial |
| EDTA | Ethylenediaminetetraacetic Acid |
| EF-G | Translation Elongation Factor G, ortholog of GFM1 |
| EIF3 | Eukaryotic translation Initiation Factor 3 protein complex |
| EIF3C | Eukaryotic translation Initiation Factor 3, subunit C |
| ERAL1 | Era-Like 12S mitochondrial rRNA chaperone 1 |
| FASTKD4 | TBRG4, FAST Kinase Domain-Containing Protein 4 |
| FC | Fold Change |
| FDR | False Discovery Rate |
| FECH | Ferrochelatase, inserting iron into porphyrin rings to produce heme |
| Fe-S cluster | Iron–Sulfur cluster |
| FLAG | Octapeptide tag for proteins, with sequence DYKDDDDK |
| G4 | Guanosine-rich quadruplex structure of DNA/RNA |
| GABA | Gamma-Aminobutyric Acid, main inhibitory neurotransmitter |
| GAC | GTPase-AssociatedC |
| GAPDH | Glyceraldehyde 3-Phosphate Dehydrogenase, glycolysis enzyme |
| GFM1 | translation elongation Factor G 1, mitochondrial |
| GFM2 | translation elongation Factor G 2, mitochondrial |
| GRPEL1 | GrpE-Like 1 protein co-chaperone, mitochondrial |
| GRPEL2 | GrpE-Like 2 protein co-chaperone, mitochondrial |
| GRSF1 | G-Rich RNA Sequence binding Factor 1 |
| GroEL | Prokaryotic chaperonin Hsp60, peptide-dependent ATPase |
| GSA | Glutamate γ-Semialdehyde |
| GSAM | Glutamate-1-Semialdehyde-2,1-AminoMutase |
| GTP | Guanosine 5’-Triphosphate |
| GTPase | Guanosine 5’-Triphosphatase |
| HARS2 | Histidine-tRNA synthase, mitochondrial |
| HemL | Prokaryotic ortholog of GSAM, heme/porphyrin biosynthesis enzyme |
| HPLC | High-performance liquid chromatography |
| Hsp100 | Eukaryotic heat shock protein family, ~100 kDa size in yeast |
| HSP60 | Chaperonin family of heat shock proteins, ~60 kDa size in bacteria |
| Hsp70 | Ubiquitous family of heat shock proteins, homologous to DnaK |
| HSP90 | Bacterial/eukaryotic family of heat shock proteins |
| HSPA9 | Mortalin/GRP75, heat shock protein family A (Hsp70) member 9, mitochondrial |
| HssR | Prokaryotic Heme response Regulator |
| IBAQ | Intensity-Based Absolute Quantification |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| ID | Identity code |
| IF2 | Prokaryotic translation Initiation Factor 2, protects formylMet-tRNA |
| InfB | Prokaryotic translation Initiation Factor if-2 |
| IRP1 | IREB1/Aconitase-1, Iron Regulatory Protein 1 |
| kDa | kilodalton |
| KGD4 | MRPS36, Alpha-Ketoglutarate Dehydrogenase Component 4 |
| LARS2 | Leucine-tRNA synthase, mitochondrial |
| LC/MS | Liquid Chromatography/Mass Spectrometry |
| LepA | Prokaryotic elongation factor, back-translocating, stress fidelity, GTPase |
| LONP1 | Lon Peptidase 1, Mitochondrial |
| LRPPRC | Leucine Rich Pentatricopeptide Repeat Containing |
| MCIA | Mitochondrial Complex I Assembly complex |
| MDa | Megadalton |
| MEF | Murine Embryonic Fibroblasts |
| MICOS | Mitochondrial Contact site and cristae Organizing System |
| MIDAS | Metal Ion-Dependent Adhesion Site |
| Mg2+ | Magnesium ion |
| mM | millimolar |
| MoxR | Prokaryotic magnesium chelatase, AAA+ ATPase |
| MRPL18 | Mitochondrial Ribosomal Protein L18 |
| MRPL38 | Mitochondrial Ribosomal Protein L38 |
| MRPP1 | =TRMT10C, Mitochondrial Ribonuclease P Protein 1 |
| MS | Mass Spectrometry |
| MSMS | tandem Mass Spectrometry |
| MT-ATP8 | Mitochondrially encoded ATP synthase membrane subunit 8 |
| MTCO1 | COX1, Mitochondrially encoded Cytochrome c Oxidase I |
| MTCO2 | COX2, Mitochondrially encoded Cytochrome c Oxidase II |
| MTCO3 | COX3, Mitochondrially encoded Cytochrome c Oxidase III |
| MTHFD2 | Methylenetetrahydrofolate Dehydrogenase (NADP+ dependent) 2 |
| mtLSU | mitochondrial Large Subunit of ribosome |
| MTND3 | Mitochondrially encoded NADH Dehydrogenase 3 |
| MTND4 | Mitochondrially encoded NADH Dehydrogenase 4 |
| MTND5 | Mitochondrially encoded NADH Dehydrogenase 5 |
| MTND6 | Mitochondrially encoded NADH Dehydrogenase 6 |
| mtSSU | mitochondrial Small Subunit of ribosome |
| m/z | ratio of mass versus charge number of ions |
| NaCl | Sodium Chloride salt |
| NADH | reduced form of Nicotinamide Adenine Dinucleotide |
| NDUFAB1 | mt-ACP2, NADH:Ubiquinone oxidoreductase subunit AB1 |
| NDUFAF1 | NADH:Ubiquinone oxidoreductase complex Assembly Factor 1 |
| NDUFAF3 | NADH:Ubiquinone oxidoreductase complex Assembly Factor 3 |
| NDUFS1 | NADH:Ubiquinone oxidoreductase core subunit S1 |
| NDUFV1 | NADH:Ubiquinone oxidoreductase core subunit V1 |
| NDUFV2 | NADH:Ubiquinone oxidoreductase core subunit V2 |
| NP40 | Nonyl Phenoxypolyethoxylethanol, non-ionic, non-denaturing detergent |
| NPET | Nascent Polypeptide Exit Tunnel in large subunit of ribosome |
| NTPase | Nucleoside-Triphosphatase |
| OAT | Ornithine delta-Aminotransferase, mitochondrial |
| p | Probability of error |
| P21 | Postnatal day 21 |
| P5C | Pyrroline-5-Carboxylate |
| P5CS | ALDH18A1, Pyrroline-5-Carboxylate Synthetase |
| PARL | Presenilin-Associated-Rhomboid-Like, mitochondrial intramembrane protease |
| PBS | Phosphate-buffered saline solution |
| PBS/T | Phosphate-buffered saline solution with Tween 20 |
| PCR | Polymerase chain reaction |
| PDK1 | Pyruvate Dehydrogenase Kinase 1 |
| PDK3 | Pyruvate Dehydrogenase Kinase 3 |
| PDPR | Pyruvate Dehydrogenase Phosphatase Regulatory subunit |
| pH | “potential of Hydrogen”, scale to quantify acidity of aqueous solutions |
| PHB1 | Prohibitin-1, forms large ring complexes in mitochondrial inner membrane |
| PLP | P5P, Pyridoxal 5’-Phosphate, vitamin B6 active form, enzyme cofactor |
| PLPBP | PROSC, Pyridoxal Phosphate Binding Protein |
| POLDIP2 | DNA Polymerase Delta Interacting Protein 2 |
| PP/PE | Polypropylene/Polyethylene |
| PPIX | Protoporphyrin IX |
| PPOX | Protoporphyrin IX oxidase |
| PRIDE | Proteomics Identification Database—EMBL/EBI |
| PRLTS3 | Perrault Syndrome type 3 |
| ProA | prokaryotic ortholog of ALDH18A1/P5CS |
| PRORP | MRPP3, Protein Only RNase-P Catalytic Subunit |
| PTCD1 | Pentatricopeptide repeat Domain 1 |
| PTCD2 | Pentatricopeptide repeat Domain 2 |
| PTCD3 | MRPS39/COXPD51, Pentatricopeptide repeat Domain 3 |
| RavA | Prokaryotic AAA+ moxr family ATPase, putative molecular chaperone |
| Rea1 | Midasin-1, yeast chaperone for maturation of pre-60S ribosome |
| RF3 | Prokaryotic translation Release Factor 3 |
| RIPA | Radio-Immuno-Precipitation Assay buffer |
| RMND1 | Required for Meiotic Nuclear Division 1 homolog |
| RNA | Ribo-Nucleic Acid |
| RpsA | Prokaryotic RNA chaperone to unfold structured mRNA on ribosome |
| rRNA | ribosomal RNA |
| RT | Room Temperature |
| RT-qPCR | Reverse Transcriptase real-time quantitative PCR |
| SC | Supercomplex |
| SDS | Sodium Dodecyl Sulfate, detergent |
| SEM | Standard Error of the Mean |
| SFXN4 | Sideroflexin 4 |
| SHMT2 | Serine Hydroxy Methyltransferase 2 |
| SLC25A28 | Solute Carrier family 25 member 28, mitochondrial iron transporter |
| SLIRP | SRA Stem-Loop Interacting RNA-binding Protein, mitochondrial |
| SLP2 | STOML2, Stomatin-Like Protein 2, at inner membrane rafts |
| SPATA5 | Spermatogenesis-associated factor protein, mitochondrial ribosome maturation |
| SPG7 | Matrix-AAA peptidase subunit, paraplegin |
| STRING | Search Tool for the Retrieval of Interacting genes/Proteins |
| T | statistical trend |
| Tbp | TATA-Binding Protein |
| TBRG4 | FASTKD4, Transforming growth factor Beta Regulator 4 |
| TIMMDC1 | Translocase of Inner Mitochondrial Membrane Domain Containing 1 |
| TMEM126B | Transmembrane protein 126B, mitochondrial complex I assembly factor |
| TMEM186 | Transmembrane protein 186, mitochondrial complex I assembly factor |
| TRAP1 | HSP90L, TNF Receptor Associated Protein 1, mitochondrial chaperone |
| TRMT10C | MRPP1, tRNA Methyltransferase 10C, mitochondrial RNase-P Subunit |
| tRNA | transfer RNA |
| tRNAPhe | tRNA for the amino acid Phenylalanine |
| tRNAVal | tRNA for the amino acid Valine |
| TUBA4A | Tubulin Alpha 4a protein |
| TWNK | Twinkle mtDNA helicase, Ataxin-8 |
| UPR | Unfolded Protein Response pathway |
| UPRmt | Unfolded Protein Response in mitochondria |
| v/v | ratio volume per volume |
| VCP | Valosin Containing Protein, transitional ER AAA+ ATPase |
| ViaA | Prokaryotic VWA-domain-protein, CoxE-like family, AAA+ interacting |
| VWA | von Willebrand factor type A domain |
| VWA8 | Von Willebrand factor A domain containing 8 |
| WT | wildtype |
| w/v | ratio weight per volume |
| YME1L1 | ATP-dependent zinc metalloprotease, mitochondrial intermembrane space |
| ZFE | Central Animal Facility, University Hospital Frankfurt am Main |
References
- Schirmer, E.C.; Glover, J.R.; Singer, M.A.; Lindquist, S. HSP100/Clp proteins: A common mechanism explains diverse functions. Trends Biochem. Sci. 1996, 21, 289–296. [Google Scholar] [CrossRef]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanical Protein Unfolding and Degradation. Annu. Rev. Physiol. 2018, 80, 413–429. [Google Scholar] [CrossRef]
- De Gaetano, A.; Gibellini, L.; Bianchini, E.; Borella, R.; De Biasi, S.; Nasi, M.; Boraldi, F.; Cossarizza, A.; Pinti, M. Impaired Mitochondrial Morphology and Functionality in Lonp1(wt/-) Mice. J. Clin. Med. 2020, 9, 1783. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef]
- Shin, M.; Watson, E.R.; Song, A.S.; Mindrebo, J.T.; Novick, S.J.; Griffin, P.R.; Wiseman, R.L.; Lander, G.C. Structures of the human LONP1 protease reveal regulatory steps involved in protease activation. Nat. Commun. 2021, 12, 3239. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-C.; Jia, B.; Yang, J.-K.; Le Van, D.; Shao, Y.G.; Han, S.W.; Jeon, Y.-J.; Chung, C.H.; Cheong, G.-W. Oligomeric structure of the ATP-dependent protease La (Lon) of Escherichia coli. Mol. Cells 2006, 21, 129–134. [Google Scholar]
- Fischer, F.; Weil, A.; Hamann, A.; Osiewacz, H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat. Commun. 2013, 4, 1397. [Google Scholar] [CrossRef]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.; Famulla, K.; Dahmen, M.; Göbl, C.; Malik, I.; Richter, K.; Korotkov, V.S.; Sass, P.; Rübsamen-Schaeff, H.; Madl, T.; et al. AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat. Commun. 2015, 6, 6320. [Google Scholar] [CrossRef]
- Wawrzynow, A.; Wojtkowiak, D.; Marszalek, J.; Banecki, B.; Jonsen, M.; Graves, B.; Georgopoulos, C.; Zylicz, M. The ClpX heat-shock protein of Escherichia coli, the ATP-dependent substrate specificity component of the ClpP-ClpX protease, is a novel molecular chaperone. EMBO J. 1995, 14, 1867–1877. [Google Scholar] [CrossRef]
- Laut, C.L.; Leasure, C.S.; Pi, H.; Carlin, S.M.; Chu, M.L.; Hillebrand, G.H.; Lin, H.K.; Yi, X.I.; Stauff, D.L.; Skaar, E.P. DnaJ and ClpX Are Required for HitRS and HssRS Two-Component System Signaling in Bacillus anthracis. Infect. Immun. 2022, 90, e0056021. [Google Scholar] [CrossRef]
- Mangla, N.; Singh, R.; Agarwal, N. HtpG Is a Metal-Dependent Chaperone Which Assists the DnaK/DnaJ/GrpE Chaperone System of Mycobacterium tuberculosis via Direct Association with DnaJ2. Microbiol. Spectr. 2023, 11, e0031223. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.J.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Regulation of host hemoglobin binding by the Staphylococcus aureus Clp proteolytic system. J. Bacteriol. 2013, 195, 5041–5050. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.J.; Friedman, D.B.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Proteomic analyses of iron-responsive, Clp-dependent changes in Staphylococcus aureus. Pathog. Dis. 2015, 73, ftv004. [Google Scholar] [CrossRef] [PubMed]
- Kobe, C.; Kracht, L.W.; Timmermann, L.; Bachmann, J.; Schmidt, M.C. Perrault Syndrome with progressive nervous system involvement. Clin. Nucl. Med. 2008, 33, 922–924. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Gispert, S.; Koornneef, L.; Sleddens-Linkels, E.; Kohli, A.; Torres-Odio, S.; Koepf, G.; Amr, S.; Reichlmeir, M.; Harter, P.N.; et al. CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2. Cells 2022, 12, 52. [Google Scholar] [CrossRef]
- Faridi, R.; Rea, A.; Fenollar-Ferrer, C.; O’keefe, R.T.; Gu, S.; Munir, Z.; Khan, A.A.; Riazuddin, S.; Hoa, M.; Naz, S.; et al. New insights into Perrault syndrome, a clinically and genetically heterogeneous disorder. Hum. Genet. 2022, 141, 805–819. [Google Scholar] [CrossRef]
- Hochberg, I.; Demain, L.A.; Richer, J.; Thompson, K.; Urquhart, J.E.; Rea, A.; Pagarkar, W.; Rodríguez-Palmero, A.; Schlüter, A.; Verdura, E.; et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am. J. Hum. Genet. 2021, 108, 2195–2204. [Google Scholar] [CrossRef]
- Yien, Y.Y.; Ducamp, S.; van der Vorm, L.N.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in human CLPX elevates levels of delta-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc. Natl. Acad. Sci. USA 2017, 114, E8045–E8052. [Google Scholar] [CrossRef]
- Ducamp, S.; Luscieti, S.; Ferrer-Cortès, X.; Nicolas, G.; Manceau, H.; Peoc’h, K.; Yien, Y.Y.; Kannengiesser, C.; Gouya, L.; Puy, H.; et al. A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in a patient suffering from CLPX associated erythropoietic protoporphyria. Haematologica 2021, 106, 2030–2033. [Google Scholar] [CrossRef]
- van der Vorm, L.N.; Paw, B.H. Studying disorders of vertebrate iron and heme metabolism using zebrafish. Methods Cell Biol. 2017, 138, 193–220. [Google Scholar]
- Shanley, B.C.; Neethling, A.C.; Percy, V.A.; Carstens, M. Neurochemical aspects of porphyria. Studies on the possible neurotoxicity of delta-aminolaevulinic acid. S. Afr. Med. J. 1975, 49, 576–580. [Google Scholar]
- Jiménez-Jiménez, F.J.; Agúndez, J.A.G.; Martínez, C.; Navacerrada, F.; Plaza-Nieto, J.F.; Pilo-De-La-Fuente, B.; Alonso-Navarro, H.; García-Martín, E. Hereditary Coproporphyria Associated with the Q306X Mutation in the Coproporphyrin Oxidase Gene Presenting with Acute Ataxia. Tremor Hyperkinet. Mov. 2013, 3, tre-03-151-4162-1. [Google Scholar] [CrossRef]
- Kevelam, S.H.; Neeleman, R.A.; Waisfisz, Q.; Friesema, E.C.; Langendonk, J.G.; van der Knaap, M.S. Acute intermittent porphyria-related leukoencephalopathy. Neurology 2016, 87, 1258–1265. [Google Scholar] [CrossRef]
- Yasuda, M.; Gan, L.; Chen, B.; Yu, C.; Zhang, J.; Gama-Sosa, M.A.; Pollak, D.D.; Berger, S.; Phillips, J.D.; Edelmann, W.; et al. Homozygous hydroxymethylbilane synthase knock-in mice provide pathogenic insights into the severe neurological impairments present in human homozygous dominant acute intermittent porphyria. Hum. Mol. Genet. 2019, 28, 1755–1767. [Google Scholar] [CrossRef]
- Ferreira, G.C.; Oberstaller, J.; Fonseca, R.; Keller, T.E.; Adapa, S.R.; Gibbons, J.; Wang, C.; Liu, X.; Li, C.; Pham, M.; et al. Iron Hack—A symposium/hackathon focused on porphyrias, Friedreich’s ataxia, and other rare iron-related diseases. F1000Res 2019, 8, 1135. [Google Scholar] [CrossRef] [PubMed]
- Stutterd, C.A.; Kidd, A.; Florkowski, C.; Janus, E.; Fanjul, M.; Raizis, A.; Wu, T.Y.; Archer, J.; Leventer, R.J.; Amor, D.J.; et al. Expanding the clinical and radiological phenotypes of leukoencephalopathy due to biallelic HMBS mutations. Am. J. Med. Genet. A 2021, 185, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Sedel, F. [Inborn errors of metabolism in adult neurology]. Rev. Neurol. 2013, 169 (Suppl. S1), S63–S69. [Google Scholar] [CrossRef] [PubMed]
- Kardon, J.R.; Yien, Y.Y.; Huston, N.C.; Branco, D.S.; Hildick-Smith, G.J.; Rhee, K.Y.; Paw, B.H.; Baker, T.A. Mitochondrial ClpX Activates a Key Enzyme for Heme Biosynthesis and Erythropoiesis. Cell 2015, 161, 858–867. [Google Scholar] [CrossRef]
- Cellini, B.; Montioli, R.; Oppici, E.; Astegno, A.; Voltattorni, C.B. The chaperone role of the pyridoxal 5′-phosphate and its implications for rare diseases involving B6-dependent enzymes. Clin. Biochem. 2014, 47, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, K.; Kaneko, K.; Vargas, P.D. Heme as a magnificent molecule with multiple missions: Heme determines its own fate and governs cellular homeostasis. Tohoku J. Exp. Med. 2007, 213, 1–16. [Google Scholar] [CrossRef]
- Hedtke, B.; Alawady, A.; Chen, S.; Börnke, F.; Grimm, B. HEMA RNAi silencing reveals a control mechanism of ALA biosynthesis on Mg chelatase and Fe chelatase. Plant Mol. Biol. 2007, 64, 733–742. [Google Scholar] [CrossRef]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Mabanglo, M.F.; Houry, W.A. Recent structural insights into the mechanism of ClpP protease regulation by AAA+ chaperones and small molecules. J. Biol. Chem. 2022, 298, 101781. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-I.; Levchenko, I.; Fraczkowska, K.; Woodruff, R.V.; Sauer, R.T.; Baker, T.A. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat. Struct. Biol. 2001, 8, 230–233. [Google Scholar] [CrossRef]
- Kang, S.G.; Dimitrova, M.N.; Ortega, J.; Ginsburg, A.; Maurizi, M.R. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J. Biol. Chem. 2005, 280, 35424–35432. [Google Scholar] [CrossRef]
- Gribun, A.; Kimber, M.S.; Ching, R.; Sprangers, R.; Fiebig, K.M.; Houry, W.A. The ClpP double ring tetradecameric protease exhibits plastic ring-ring interactions, and the N termini of its subunits form flexible loops that are essential for ClpXP and ClpAP complex formation. J. Biol. Chem. 2005, 280, 16185–16196. [Google Scholar] [CrossRef]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef]
- Prattes, M.; Lo, Y.-H.; Bergler, H.; Stanley, R.E. Shaping the Nascent Ribosome: AAA-ATPases in Eukaryotic Ribosome Biogenesis. Biomolecules 2019, 9, 715. [Google Scholar] [CrossRef]
- De Lonlay, P.; Valnot, I.; Barrientos, A.; Gorbatyuk, M.; Tzagoloff, A.; Taanman, J.-W.; Benayoun, E.; Chrétien, D.; Kadhom, N.; Lombès, A.; et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat. Genet. 2001, 29, 57–60. [Google Scholar] [CrossRef]
- Juhola, M.; Shah, Z.H.; Grivell, L.A.; Jacobs, H.T. The mitochondrial inner membrane AAA metalloprotease family in metazoans. FEBS Lett. 2000, 481, 91–95. [Google Scholar] [CrossRef]
- Escobar-Henriques, M.; Anton, V. Mitochondrial Surveillance by Cdc48/p97: MAD vs. Membrane Fusion. Int. J. Mol. Sci. 2020, 21, 6841. [Google Scholar] [CrossRef]
- Issop, L.; Fan, J.; Lee, S.; Rone, M.B.; Basu, K.; Mui, J.; Papadopoulos, V. Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: Role of ATAD3. Endocrinology 2015, 156, 334–345. [Google Scholar] [CrossRef]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.-T.; Wibom, R.; et al. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 2016, 35, 2566–2583. [Google Scholar] [CrossRef] [PubMed]
- Dennerlein, S.; Rozanska, A.; Wydro, M.; Chrzanowska-Lightowlers, Z.M.A.; Lightowlers, R.N. Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem. J. 2010, 430, 551–558. [Google Scholar] [CrossRef]
- Uchiumi, T.; Ohgaki, K.; Yagi, M.; Aoki, Y.; Sakai, A.; Matsumoto, S.; Kang, D. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res. 2010, 38, 5554–5568. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Kohli, A.; Bárcena, C.; López-Otín, C.; Heidler, J.; Wittig, I.; Auburger, G. Global Proteome of LonP1(+/-) Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. Int. J. Mol. Sci. 2019, 20, 4523. [Google Scholar] [CrossRef]
- Auburger, G.; Key, J.; Gispert, S. The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes. Cells 2022, 11, 2370. [Google Scholar] [CrossRef] [PubMed]
- Rumyantseva, A.; Popovic, M.; Trifunovic, A. CLPP deficiency ameliorates neurodegeneration caused by impaired mitochondrial protein synthesis. Brain 2022, 145, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Dröse, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef]
- Lee, M.; Matsunaga, N.; Akabane, S.; Yasuda, I.; Ueda, T.; Takeuchi-Tomita, N. Reconstitution of mammalian mitochondrial translation system capable of correct initiation and long polypeptide synthesis from leaderless mRNA. Nucleic Acids Res. 2021, 49, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Senft, K.; Heidler, J.; Herholz, M.; Kukat, A.; Höhne, M.N.; Hofsetz, E.; Becker, C.; Kaspar, S.; Giese, H.; et al. A salvage pathway maintains highly functional respiratory complex I. Nat. Commun. 2020, 11, 1643. [Google Scholar] [CrossRef] [PubMed]
- Mabanglo, M.F.; Bhandari, V.; Houry, W.A. Houry, Substrates and interactors of the ClpP protease in the mitochondria. Curr. Opin. Chem. Biol. 2022, 66, 102078. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Wong, K.S.; Zhou, J.L.; Mabanglo, M.F.; Batey, R.A.; Houry, W.A. The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem. Biol. 2018, 13, 1413–1425. [Google Scholar] [CrossRef]
- Key, J.; Torres-Odio, S.; Bach, N.C.; Gispert, S.; Koepf, G.; Reichlmeir, M.; West, A.P.; Prokisch, H.; Freisinger, P.; Newman, W.G.; et al. Inactivity of Peptidase ClpP Causes Primary Accumulation of Mitochondrial Disaggregase ClpX with Its Interacting Nucleoid Proteins, and of mtDNA. Cells 2021, 10, 3354. [Google Scholar] [CrossRef] [PubMed]
- Hofsetz, E.; Demir, F.; Szczepanowska, K.; Kukat, A.; Kizhakkedathu, J.N.; Trifunovic, A.; Huesgen, P.F. The Mouse Heart Mitochondria N Terminome Provides Insights into ClpXP-Mediated Proteolysis. Mol. Cell Proteomics 2020, 19, 1330–1345. [Google Scholar] [CrossRef] [PubMed]
- Strack, P.R.; Brodie, E.J.; Zhan, H.; Schuenemann, V.J.; Valente, L.J.; Saiyed, T.; Lowth, B.R.; Angley, L.M.; Perugini, M.A.; Zeth, K.; et al. Polymerase delta-interacting protein 38 (PDIP38) modulates the stability and activity of the mitochondrial AAA+ protease CLPXP. Commun. Biol. 2020, 3, 646. [Google Scholar] [CrossRef]
- Lasserre, J.-P.; Beyne, E.; Pyndiah, S.; Lapaillerie, D.; Claverol, S.; Bonneu, M. A complexomic study of Escherichia coli using two-dimensional blue native/SDS polyacrylamide gel electrophoresis. Electrophoresis 2006, 27, 3306–3321. [Google Scholar] [CrossRef]
- Maurizi, M.R.; Thompson, M.W.; Singh, S.K.; Kim, S.H. Endopeptidase Clp: ATP-dependent Clp protease from Escherichia coli. Methods Enzymol. 1994, 244, 314–331. [Google Scholar]
- Wai, T.; Saita, S.; Nolte, H.; Müller, S.; König, T.; Richter-Dennerlein, R.; Sprenger, H.; Madrenas, J.; Mühlmeister, M.; Brandt, U.; et al. The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep. 2016, 17, 1844–1856. [Google Scholar] [CrossRef]
- Liu, H.; Fan, H.; He, P.; Zhuang, H.; Liu, X.; Chen, M.; Zhong, W.; Zhang, Y.; Zhen, C.; Li, Y.; et al. Prohibitin 1 regulates mtDNA release and downstream inflammatory responses. EMBO J. 2022, 41, e111173. [Google Scholar] [CrossRef]
- Manon, S.; Priault, M.; Camougrand, N. Mitochondrial AAA-type protease Yme1p is involved in Bax effects on cytochrome c oxidase. Biochem. Biophys. Res. Commun. 2001, 289, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Wessels, H.J.C.T.; Vogel, R.O.; Lightowlers, R.N.; Spelbrink, J.N.; Rodenburg, R.J.; Heuvel, L.P.v.D.; van Gool, A.J.; Gloerich, J.; Smeitink, J.A.M.; Nijtmans, L.G. Analysis of 953 human proteins from a mitochondrial HEK293 fraction by complexome profiling. PLoS ONE 2013, 8, e68340. [Google Scholar] [CrossRef] [PubMed]
- Gardeitchik, T.; Mohamed, M.; Ruzzenente, B.; Karall, D.; Guerrero-Castillo, S.; Dalloyaux, D.; van den Brand, M.; van Kraaij, S.; van Asbeck, E.; Assouline, Z.; et al. Bi-allelic Mutations in the Mitochondrial Ribosomal Protein MRPS2 Cause Sensorineural Hearing Loss, Hypoglycemia, and Multiple OXPHOS Complex Deficiencies. Am. J. Hum. Genet. 2018, 102, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Chatzispyrou, I.A.; Alders, M.; Guerrero-Castillo, S.; Zapata Perez, R.; Haagmans, M.A.; Mouchiroud, L.; Koster, J.; Ofman, R.; Baas, F.; Waterham, H.R.; et al. A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum. Mol. Genet. 2017, 26, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Van Strien, J.; Guerrero-Castillo, S.A.; Chatzispyrou, I.; Houtkooper, R.H.; Brandt, U.; Huynen, M.A. COmplexome Profiling ALignment (COPAL) reveals remodeling of mitochondrial protein complexes in Barth syndrome. Bioinformatics 2019, 35, 3083–3091. [Google Scholar] [CrossRef]
- Aibara, S.; Singh, V.; Modelska, A.; Amunts, A. Structural basis of mitochondrial translation. Elife 2020, 9, e58362. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Ostermeyer-Fay, A.G.; Haley, J.D.; Garcia-Diaz, M. Kinetics and Mechanism of Mammalian Mitochondrial Ribosome Assembly. Cell Rep. 2018, 22, 1935–1944. [Google Scholar] [CrossRef]
- Brown, A.; Amunts, A.; Bai, X.-C.; Sugimoto, Y.; Edwards, P.C.; Murshudov, G.; Scheres, S.H.W.; Ramakrishnan, V. Structure of the large ribosomal subunit from human mitochondria. Science 2014, 346, 718–722. [Google Scholar] [CrossRef]
- Greber, B.J.; Boehringer, D.; Leibundgut, M.; Bieri, P.; Leitner, A.; Schmitz, N.; Aebersold, R.; Ban, N. The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 2014, 515, 283–286. [Google Scholar] [CrossRef]
- Itoh, Y.; Naschberger, A.; Mortezaei, N.; Herrmann, J.M.; Amunts, A. Analysis of translating mitoribosome reveals functional characteristics of translation in mitochondria of fungi. Nat. Commun. 2020, 11, 5187. [Google Scholar] [CrossRef] [PubMed]
- Rorbach, J.; Gao, F.; Powell, C.A.; D’Souza, A.; Lightowlers, R.N.; Minczuk, M.; Chrzanowska-Lightowlers, Z.M. Human mitochondrial ribosomes can switch their structural RNA composition. Proc. Natl. Acad. Sci. USA 2016, 113, 12198–12201. [Google Scholar] [CrossRef]
- Barrio-Garcia, C.; Thoms, M.; Flemming, D.; Kater, L.; Berninghausen, O.; Baßler, J.; Beckmann, R.; Hurt, E. Architecture of the Rix1-Rea1 checkpoint machinery during pre-60S-ribosome remodeling. Nat. Struct. Mol. Biol. 2016, 23, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.P.; Sharma, M.R.; Qi, L.; Frank, J.; Agrawal, R.K. Interaction of the G’ domain of elongation factor G and the C-terminal domain of ribosomal protein L7/L12 during translocation as revealed by cryo-EM. Mol. Cell 2005, 20, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Heublein, M.; Burguillos, M.A.; Vögtle, F.N.; Teixeira, P.F.; Imhof, A.; Meisinger, C.; Ott, M. The novel component Kgd4 recruits the E3 subunit to the mitochondrial alpha-ketoglutarate dehydrogenase. Mol. Biol. Cell 2014, 25, 3342–3349. [Google Scholar] [CrossRef]
- Heublein, M.; Ndi, M.; Vazquez-Calvo, C.; Vögtle, F.-N.; Ott, M. Alternative Translation Initiation at a UUG Codon Gives Rise to Two Functional Variants of the Mitochondrial Protein Kgd4. J. Mol. Biol. 2019, 431, 1460–1467. [Google Scholar] [CrossRef]
- Obayashi, E.; Luna, R.E.; Nagata, T.; Martin-Marcos, P.; Hiraishi, H.; Singh, C.R.; Erzberger, J.P.; Zhang, F.; Arthanari, H.; Morris, J.; et al. Molecular Landscape of the Ribosome Pre-initiation Complex during mRNA Scanning: Structural Role for eIF3c and Its Control by eIF5. Cell Rep. 2017, 18, 2651–2663. [Google Scholar] [CrossRef]
- Ujino, S.; Nishitsuji, H.; Sugiyama, R.; Suzuki, H.; Hishiki, T.; Sugiyama, K.; Shimotohno, K.; Takaku, H. The interaction between human initiation factor eIF3 subunit c and heat-shock protein 90: A necessary factor for translation mediated by the hepatitis C virus internal ribosome entry site. Virus Res. 2012, 163, 390–395. [Google Scholar] [CrossRef]
- Villa, N.; Do, A.; Hershey, J.W.; Fraser, C.S. Human eukaryotic initiation factor 4G (eIF4G) protein binds to eIF3c, -d, and -e to promote mRNA recruitment to the ribosome. J. Biol. Chem. 2013, 288, 32932–32940. [Google Scholar] [CrossRef]
- Gildea, D.E.; Luetkemeier, E.S.; Bao, X.; Loftus, S.K.; Mackem, S.; Yang, Y.; Pavan, W.J.; Biesecker, L.G. The pleiotropic mouse phenotype extra-toes spotting is caused by translation initiation factor Eif3c mutations and is associated with disrupted sonic hedgehog signaling. FASEB J. 2011, 25, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Del’olio, S.; Barrientos, A. The Diseased Mitoribosome. FEBS Lett. 2021, 595, 1025–1061. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lepage, P.; Miller, K.; Bunkenborg, J.; Reich, M.; Hjerrild, M.; Delmonte, T.; Villeneuve, A.; Sladek, R.; Xu, F.; et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA 2003, 100, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Morin, C.; Mitchell, G.; Ackerley, C.; Robinson, B.H. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: Mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem. J. 2004, 382 Pt 1, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Addis, J.B.L.; Cameron, J.M.; Robinson, B.H. LRPPRC mutation suppresses cytochrome oxidase activity by altering mitochondrial RNA transcript stability in a mouse model. Biochem. J. 2012, 441, 275–283. [Google Scholar] [CrossRef]
- Oláhová, M.; Hardy, S.A.; Hall, J.; Yarham, J.W.; Haack, T.B.; Wilson, W.C.; Alston, C.L.; He, L.; Aznauryan, E.; Brown, R.M.; et al. LRPPRC mutations cause early-onset multisystem mitochondrial disease outside of the French-Canadian population. Brain 2015, 138 Pt 12, 3503–3519. [Google Scholar] [CrossRef]
- Fleck, D.; Phu, L.; Verschueren, E.; Hinkle, T.; Reichelt, M.; Bhangale, T.; Haley, B.; Wang, Y.; Graham, R.; Kirkpatrick, D.S.; et al. PTCD1 Is Required for Mitochondrial Oxidative-Phosphorylation: Possible Genetic Association with Alzheimer’s Disease. J. Neurosci. 2019, 39, 4636–4656. [Google Scholar] [CrossRef]
- Rackham, O.; Davies, S.M.K.; Shearwood, A.-M.J.; Hamilton, K.L.; Whelan, J.; Filipovska, A. Pentatricopeptide repeat domain protein 1 lowers the levels of mitochondrial leucine tRNAs in cells. Nucleic Acids Res. 2009, 37, 5859–5867. [Google Scholar] [CrossRef]
- Perks, K.L.; Rossetti, G.; Kuznetsova, I.; Hughes, L.A.; Ermer, J.A.; Ferreira, N.; Busch, J.D.; Rudler, D.L.; Spahr, H.; Schöndorf, T.; et al. PTCD1 Is Required for 16S rRNA Maturation Complex Stability and Mitochondrial Ribosome Assembly. Cell Rep. 2018, 23, 127–142. [Google Scholar] [CrossRef]
- Newman, W.G.; Friedman, T.B.; Conway, G.S.; Demain, L.A.M. Perrault Syndrome. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Shen, Y.; Wang, X.; Shen, X.; Wang, Y.; Wang, S.; Zhang, Y.; Yao, X.; Xu, Y.; Sang, M.; Pan, J.; et al. Geniposide Possesses the Protective Effect on Myocardial Injury by Inhibiting Oxidative Stress and Ferroptosis via Activation of the Grsf1/GPx4 Axis. Front. Pharmacol. 2022, 13, 879870. [Google Scholar] [CrossRef]
- Jourdain, A.A.; Koppen, M.; Wydro, M.; Rodley, C.D.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Martinou, J.-C. GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab. 2013, 17, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Giardina, G.; Brunotti, P.; Fiascarelli, A.; Cicalini, A.; Costa, M.; Buckle, A.M.; Di Salvo, M.L.; Giorgi, A.; Marani, M.; Paone, A.; et al. How pyridoxal 5’-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 2015, 282, 1225–1241. [Google Scholar] [CrossRef]
- Tramonti, A.; Cuyàs, E.; Encinar, J.A.; Pietzke, M.; Paone, A.; Verdura, S.; Arbusà, A.; Martin-Castillo, B.; Giardina, G.; Joven, J.; et al. Metformin Is a Pyridoxal-5′-phosphate (PLP)-Competitive Inhibitor of SHMT2. Cancers 2021, 13, 4009. [Google Scholar] [CrossRef]
- Morscher, R.J.; Ducker, G.S.; Li, S.H.-J.; Mayer, J.A.; Gitai, Z.; Sperl, W.; Rabinowitz, J.D. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018, 554, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.B.; Moss, T.; Mokashi, S.; Srikanth, S.; Jones, J.R.; Foley, J.R.; Skinner, C.; Lichty, A.; Kocur, A.; Wood, T.; et al. Functional Assessment of Homozygous ALDH18A1 Variants Reveals Alterations in Amino Acid and Antioxidant Metabolism. Hum. Mol. Genet. 2023, 32, 732–744. [Google Scholar] [CrossRef]
- Saito, Y.; Nakagawa, T.; Kakihana, A.; Nakamura, Y.; Nabika, T.; Kasai, M.; Takamori, M.; Yamagishi, N.; Kuga, T.; Hatayama, T.; et al. Yeast Two-Hybrid and One-Hybrid Screenings Identify Regulators of hsp70 Gene Expression. J. Cell Biochem. 2016, 117, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Dores-Silva, P.R.; Kiraly, V.T.R.; Moritz, M.N.d.O.; Serrão, V.H.B.; dos Passos, P.M.S.; Spagnol, V.; Teixeira, F.R.; Gava, L.M.; Cauvi, D.M.; Ramos, C.H.I.; et al. New insights on human Hsp70-escort protein 1: Chaperone activity, interaction with liposomes, cellular localizations and HSPA’s self-assemblies remodeling. Int. J. Biol. Macromol. 2021, 182, 772–784. [Google Scholar] [CrossRef]
- Srivastava, S.; Savanur, M.A.; Sinha, D.; Birje, A.; Vigneshwaran, R.; Saha, P.P.; D’Silva, P. Regulation of mitochondrial protein import by the nucleotide exchange factors GrpEL1 and GrpEL2 in human cells. J. Biol. Chem. 2017, 292, 18075–18090. [Google Scholar] [CrossRef]
- Jackson, T.D.; Crameri, J.J.; Muellner-Wong, L.; Frazier, A.E.; Palmer, C.S.; Formosa, L.E.; Hock, D.H.; Fujihara, K.M.; Stait, T.; Sharpe, A.J.; et al. Sideroflexin 4 is a complex I assembly factor that interacts with the MCIA complex and is required for the assembly of the ND2 module. Proc. Natl. Acad. Sci. USA 2022, 119, e2115566119. [Google Scholar] [CrossRef]
- Muster, B.; Kohl, W.; Wittig, I.; Strecker, V.; Joos, F.; Haase, W.; Bereiter-Hahn, J.; Busch, K. Respiratory chain complexes in dynamic mitochondria display a patchy distribution in life cells. PLoS ONE 2010, 5, e11910. [Google Scholar] [CrossRef]
- Babot, M.; Labarbuta, P.; Birch, A.; Kee, S.; Fuszard, M.; Botting, C.H.; Wittig, I.; Heide, H.; Galkin, A. ND3, ND1 and 39kDa subunits are more exposed in the de-active form of bovine mitochondrial complex I. Biochim. Biophys. Acta 2014, 1837, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Lobo-Jarne, T.; Pérez-Pérez, R.; Fontanesi, F.; Timón-Gómez, A.; Wittig, I.; Peñas, A.; Serrano-Lorenzo, P.; García-Consuegra, I.; Arenas, J.; Martín, M.A.; et al. Multiple pathways coordinate assembly of human mitochondrial complex IV and stabilization of respiratory supercomplexes. EMBO J. 2020, 39, e103912. [Google Scholar] [CrossRef]
- Swenson, S.; Cannon, A.; Harris, N.J.; Taylor, N.G.; Fox, J.L.; Khalimonchuk, O. Analysis of Oligomerization Properties of Heme a Synthase Provides Insights into Its Function in Eukaryotes. J. Biol. Chem. 2016, 291, 10411–10425. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.G.; Swenson, S.; Harris, N.J.; Germany, E.M.; Fox, J.L.; Khalimonchuk, O. The Assembly Factor Pet117 Couples Heme a Synthase Activity to Cytochrome Oxidase Assembly. J. Biol. Chem. 2017, 292, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- International Mouse Phenotyping Consortium. Available online: https://www.mousephenotype.org/data/genes/MGI:1858213 (accessed on 9 November 2023).
- Bareth, B.; Dennerlein, S.; Mick, D.U.; Nikolov, M.; Urlaub, H.; Rehling, P. The heme a synthase Cox15 associates with cytochrome c oxidase assembly intermediates during Cox1 maturation. Mol. Cell Biol. 2013, 33, 4128–4137. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, C.; Yang, Z.; Brown, C.E.; Hollebone, B.P.; Stout, S.A. Petroleum biomarker fingerprinting for oil spill characterization and source identification. In Standard Handbook Oil Spill Environmental Forensics, 2nd ed.; Stout, S.A., Wang, Z., Eds.; Academic Press: Cambridge, MA, USA, 2016; Chapter 4. [Google Scholar]
- Labbé, R.F.; Vreman, H.J.; Stevenson, D.K. Zinc protoporphyrin: A metabolite with a mission. Clin. Chem. 1999, 45, 2060–2072. [Google Scholar] [CrossRef]
- Spasojević, I.; Chen, Y.; Noel, T.J.; Yu, Y.; Cole, M.P.; Zhang, L.; Zhao, Y.; Clair, D.K.S.; Batinić-Haberle, I. Mn porphyrin-based superoxide dismutase (SOD) mimic, MnIIITE-2-PyP5+, targets mouse heart mitochondria. Free Radic. Biol. Med. 2007, 42, 1193–1200. [Google Scholar] [CrossRef]
- Crow, J.P.; Calingasan, N.Y.; Chen, J.; Hill, J.L.; Beal, M.F. Manganese porphyrin given at symptom onset markedly extends survival of ALS mice. Ann. Neurol. 2005, 58, 258–265. [Google Scholar] [CrossRef]
- Watkins, S.; Baron, J.; Tephly, T.R. Identification of cobalt protoporphyrin IX formation in vivo following cobalt administration to rats. Biochem. Pharmacol. 1980, 29, 2319–2323. [Google Scholar] [CrossRef]
- Maitra, D.; Cunha, J.B.; Elenbaas, J.S.; Bonkovsky, H.L.; Shavit, J.A.; Omary, M.B. Porphyrin-Induced Protein Oxidation and Aggregation as a Mechanism of Porphyria-Associated Cell Injury. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 535–548. [Google Scholar] [CrossRef]
- Mayr, S.J.; Mendel, R.R.; Schwarz, G. Molybdenum cofactor biology, evolution and deficiency. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118883. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Gonzales, K.K. Manganese-Induced Parkinsonism Is Not Idiopathic Parkinson’s Disease: Environmental and Genetic Evidence. Toxicol. Sci. 2015, 146, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Vyskočil, A.; Viau, C. Assessment of molybdenum toxicity in humans. J. Appl. Toxicol. 1999, 19, 185–192. [Google Scholar] [CrossRef]
- Ohsawa, H.; Gualerzi, C. Chemical modification in situ of Escherichia coli 30 S ribosomal proteins by the site-specific reagent pyridoxal phosphate. Inactivation of the aminoacyl-tRNA and mRNA binding sites. J. Biol. Chem. 1983, 258, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Montioli, R.; Bellezza, I.; Desbats, M.A.; Voltattorni, C.B.; Salviati, L.; Cellini, B. Deficit of human ornithine aminotransferase in gyrate atrophy: Molecular, cellular, and clinical aspects. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140555. [Google Scholar] [CrossRef] [PubMed]
- Pfanzelt, M.; Maher, T.E.; Absmeier, R.M.; Schwarz, M.; Sieber, S.A. Tailored Pyridoxal Probes Unravel Novel Cofactor-Dependent Targets and Antibiotic Hits in Critical Bacterial Pathogens. Angew. Chem. Int. Ed. Engl. 2022, 61, e202117724. [Google Scholar] [CrossRef] [PubMed]
- Montioli, R.; Desbats, M.A.; Grottelli, S.; Doimo, M.; Bellezza, I.; Voltattorni, C.B.; Salviati, L.; Cellini, B. Molecular and cellular basis of ornithine delta-aminotransferase deficiency caused by the V332M mutation associated with gyrate atrophy of the choroid and retina. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3629–3638. [Google Scholar] [CrossRef]
- Levchenko, I.; Seidel, M.; Sauer, R.T.; Baker, T.A. A specificity-enhancing factor for the ClpXP degradation machine. Science 2000, 289, 2354–2356. [Google Scholar] [CrossRef]
- Fei, X.; Bell, T.A.; Barkow, S.R.; Baker, T.A.; Sauer, R.T. Structural basis of ClpXP recognition and unfolding of ssrA-tagged substrates. Elife 2020, 9, e61496. [Google Scholar] [CrossRef]
- Lytvynenko, I.; Paternoga, H.; Thrun, A.; Balke, A.; Müller, T.A.; Chiang, C.H.; Nagler, K.; Tsaprailis, G.; Anders, S.; Bischofs, I.; et al. Alanine Tails Signal Proteolysis in Bacterial Ribosome-Associated Quality Control. Cell 2019, 178, 76–90.e22. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.W.; Hennig, M.; Hohenester, E.; Jansonius, J.N.; Schirmer, T. Crystal structure of human recombinant ornithine aminotransferase. J. Mol. Biol. 1998, 277, 81–102. [Google Scholar] [CrossRef] [PubMed]
- LaBreck, C.J.; May, S.; Viola, M.G.; Conti, J.; Camberg, J.L. The Protein Chaperone ClpX Targets Native and Non-native Aggregated Substrates for Remodeling, Disassembly, and Degradation with ClpP. Front. Mol. Biosci. 2017, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Truscott, K.N.; Bezawork-Geleta, A.; Dougan, D.A. Unfolded protein responses in bacteria and mitochondria: A central role for the ClpXP machine. IUBMB Life 2011, 63, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef]
- Snider, J.; Houry, W.A. MoxR AAA+ ATPases: A novel family of molecular chaperones? J. Struct. Biol. 2006, 156, 200–209. [Google Scholar] [CrossRef]
- Wong, K.S.; Houry, W.A. Novel structural and functional insights into the MoxR family of AAA+ ATPases. J. Struct. Biol. 2012, 179, 211–221. [Google Scholar] [CrossRef]
- Snider, J.; Thibault, G.; Houry, W.A. The AAA+ superfamily of functionally diverse proteins. Genome Biol. 2008, 9, 216. [Google Scholar] [CrossRef]
- Felix, J.; Bumba, L.; Liesche, C.; Fraudeau, A.; Rébeillé, F.; El Khoury, J.Y.; Huard, K.; Gallet, B.; Moriscot, C.; Kleman, J.-P.; et al. The AAA+ ATPase RavA and its binding partner ViaA modulate E. coli aminoglycoside sensitivity through interaction with the inner membrane. Nat. Commun. 2022, 13, 5502. [Google Scholar] [CrossRef]
- Wong, K.S.; Snider, J.D.; Graham, C.; Greenblatt, J.F.; Emili, A.; Babu, M.; Houry, W.A. The MoxR ATPase RavA and its cofactor ViaA interact with the NADH:ubiquinone oxidoreductase I in Escherichia coli. PLoS ONE 2014, 9, e85529. [Google Scholar] [CrossRef]
- Wong, K.S.; Bhandari, V.; Janga, S.C.; Houry, W.A. The RavA-ViaA Chaperone-Like System Interacts with and Modulates the Activity of the Fumarate Reductase Respiratory Complex. J. Mol. Biol. 2017, 429, 324–344. [Google Scholar] [CrossRef] [PubMed]
- El Bakkouri, M.; Gutsche, I.; Kanjee, U.; Zhao, B.; Yu, M.; Goret, G.; Schoehn, G.; Burmeister, W.P.; Houry, W.A. Structure of RavA MoxR AAA+ protein reveals the design principles of a molecular cage modulating the inducible lysine decarboxylase activity. Proc. Natl. Acad. Sci. USA 2010, 107, 22499–22504. [Google Scholar] [CrossRef] [PubMed]
- Tomar, P.C.; Lakra, N.; Mishra, S.N. Cadaverine: A lysine catabolite involved in plant growth and development. Plant Signal Behav. 2013, 8, e25850. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Hall, J. Endogenous polyamine function—The RNA perspective. Nucleic Acids Res. 2014, 42, 11275–11290. [Google Scholar] [CrossRef]
- Michael, A.J. Biosynthesis of polyamines and polyamine-containing molecules. Biochem. J. 2016, 473, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Jessop, M.; Arragain, B.; Miras, R.; Fraudeau, A.; Huard, K.; Bacia-Verloop, M.; Catty, P.; Felix, J.; Malet, H.; Gutsche, I. Structural insights into ATP hydrolysis by the MoxR ATPase RavA and the LdcI-RavA cage-like complex. Commun. Biol. 2020, 3, 46. [Google Scholar] [CrossRef]
- Ramírez, P.; Guiliani, N.; Valenzuela, L.; Beard, S.; Jerez, C.A. Differential protein expression during growth of Acidithiobacillus ferrooxidans on ferrous iron, sulfur compounds, or metal sulfides. Appl. Environ. Microbiol. 2004, 70, 4491–4498. [Google Scholar] [CrossRef]
- Tsai, Y.-C.C.; Ye, F.; Liew, L.; Liu, D.; Bhushan, S.; Gao, Y.-G.; Mueller-Cajar, O. Insights into the mechanism and regulation of the CbbQO-type Rubisco activase, a MoxR AAA+ ATPase. Proc. Natl. Acad. Sci. USA 2020, 117, 381–387. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Y.; Wang, X.; Liu, L. Hexameric structure of the ATPase motor subunit of magnesium chelatase in chlorophyll biosynthesis. Protein Sci. 2020, 29, 1040–1046. [Google Scholar] [CrossRef]
- Farmer, D.A.; Brindley, A.A.; Hitchcock, A.; Jackson, P.J.; Johnson, B.; Dickman, M.J.; Hunter, C.N.; Reid, J.D.; Adams, N.B.P. The ChlD subunit links the motor and porphyrin binding subunits of magnesium chelatase. Biochem. J. 2019, 476, 1875–1887. [Google Scholar] [CrossRef]
- Jensen, P.E.; Gibson, L.C.D.; Henningsen, K.W.; Hunter, C.N. Expression of the chlI, chlD, and chlH genes from the Cyanobacterium synechocystis PCC6803 in Escherichia coli and demonstration that the three cognate proteins are required for magnesium-protoporphyrin chelatase activity. J. Biol. Chem. 1996, 271, 16662–16667. [Google Scholar] [CrossRef]
- Walker, C.J.; Willows, R.D. Mechanism and regulation of Mg-chelatase. Biochem. J. 1997, 327 Pt 2, 321–333. [Google Scholar] [CrossRef]
- Papenbrock, J.; Mock, H.-P.; Tanaka, R.; Kruse, E.; Grimm, B. Role of magnesium chelatase activity in the early steps of the tetrapyrrole biosynthetic pathway. Plant Physiol. 2000, 122, 1161–1169. [Google Scholar] [CrossRef]
- Fodje, M.; Hansson, A.; Hansson, M.; Olsen, J.; Gough, S.; Willows, R.; Al-Karadaghi, S. Interplay between an AAA module and an integrin I domain may regulate the function of magnesium chelatase. J. Mol. Biol. 2001, 311, 111–122. [Google Scholar] [CrossRef]
- Kuznetsov, S.; Milenkin, A.; Antonov, I. Translational Frameshifting in the chlD Gene Gives a Clue to the Coevolution of the Chlorophyll and Cobalamin Biosyntheses. Microorganisms 2022, 10, 1200. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.A.; Hunter, C.N.; Warren, M.J. Biosynthesis of the modified tetrapyrroles-the pigments of life. J. Biol. Chem. 2020, 295, 6888–6925. [Google Scholar] [CrossRef] [PubMed]
- Cammarano, P.; Pons, S.; Romeo, A.; Galdieri, M.; Gualerzi, C. Characterization of unfolded and compact ribosomal subunits from plants and their relationship to those of lower and higher animals: Evidence for physicochemical heterogeneity among eucaryotic ribosomes. Biochim. Biophys. Acta 1972, 281, 571–596. [Google Scholar] [CrossRef] [PubMed]
- Sreedhara, A.; Cowan, J. Structural and catalytic roles for divalent magnesium in nucleic acid biochemistry. Biometals 2002, 15, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Yamagami, R.; Sieg, J.P.; Bevilacqua, P.C. Functional Roles of Chelated Magnesium Ions in RNA Folding and Function. Biochemistry 2021, 60, 2374–2386. [Google Scholar] [CrossRef] [PubMed]
- Welty, R.; Rau, M.; Pabit, S.; Dunstan, M.S.; Conn, G.L.; Pollack, L.; Hall, K.B. Ribosomal Protein L11 Selectively Stabilizes a Tertiary Structure of the GTPase Center rRNA Domain. J. Mol. Biol. 2020, 432, 991–1007. [Google Scholar] [CrossRef]
- Akanuma, G.; Kobayashi, A.; Suzuki, S.; Kawamura, F.; Shiwa, Y.; Watanabe, S.; Yoshikawa, H.; Hanai, R.; Ishizuka, M. Defect in the formation of 70S ribosomes caused by lack of ribosomal protein L34 can be suppressed by magnesium. J. Bacteriol. 2014, 196, 3820–3830. [Google Scholar] [CrossRef] [PubMed]
- Welty, R.; Hall, K.B. Nucleobases Undergo Dynamic Rearrangements during RNA Tertiary Folding. J. Mol. Biol. 2016, 428, 4490–4502. [Google Scholar] [CrossRef] [PubMed]
- Kehrein, K.; Schilling, R.; Möller-Hergt, B.V.; Wurm, C.A.; Jakobs, S.; Lamkemeyer, T.; Langer, T.; Ott, M. Organization of Mitochondrial Gene Expression in Two Distinct Ribosome-Containing Assemblies. Cell Rep. 2015, 10, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Réblová, K.; Špačková, N.; Štefl, R.; Csaszar, K.; Koča, J.; Leontis, N.B.; Šponer, J. Non-Watson-Crick basepairing and hydration in RNA motifs: Molecular dynamics of 5S rRNA loop E. Biophys. J. 2003, 84, 3564–3582. [Google Scholar] [CrossRef] [PubMed]
- Uno, T.; Aoki, K.; Shikimi, T.; Hiranuma, Y.; Tomisugi, Y.; Ishikawa, Y. Copper insertion facilitates water-soluble porphyrin binding to rA.rU and rA.dT base pairs in duplex RNA and RNA.DNA hybrids. Biochemistry 2002, 41, 13059–13066. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Han, G.; Jia, G.; Zhou, J.; Li, C. Study on the interaction of porphyrin with G-quadruplex DNAs. Biophys. Chem. 2008, 137, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Havlová, K.; Fajkus, J. G4 Structures in Control of Replication and Transcription of rRNA Genes. Front. Plant Sci. 2020, 11, 593692. [Google Scholar] [CrossRef]
- Mestre-Fos, S.; Penev, P.I.; Richards, J.C.; Dean, W.L.; Gray, R.D.; Chaires, J.B.; Williams, L.D. Profusion of G-quadruplexes on both subunits of metazoan ribosomes. PLoS ONE 2019, 14, e0226177. [Google Scholar] [CrossRef]
- Datta, A.; Pollock, K.J.; Kormuth, K.A.; Brosh, R. G-Quadruplex Assembly by Ribosomal DNA: Emerging Roles in Disease Pathogenesis and Cancer Biology. Cytogenet. Genome Res. 2021, 161, 285–296. [Google Scholar] [CrossRef]
- Zamiri, B.; Reddy, K.; Macgregor, R.B., Jr.; Pearson, C.E. TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J. Biol. Chem. 2014, 289, 4653–4659. [Google Scholar] [CrossRef]
- Weisman-Shomer, P.; Cohen, E.; Hershco, I.; Khateb, S.; Wolfovitz-Barchad, O.; Hurley, L.H.; Fry, M. The cationic porphyrin TMPyP4 destabilizes the tetraplex form of the fragile X syndrome expanded sequence d(CGG)n. Nucleic. Acids Res. 2003, 31, 3963–3970. [Google Scholar] [CrossRef]
- Whittaker, C.A.; Hynes, R.O. Distribution and evolution of von Willebrand/integrin A domains: Widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell 2002, 13, 3369–3387. [Google Scholar] [CrossRef]
- Romes, E.M.; Sobhany, M.; Stanley, R.E. The Crystal Structure of the Ubiquitin-like Domain of Ribosome Assembly Factor Ytm1 and Characterization of Its Interaction with the AAA-ATPase Midasin. J. Biol. Chem. 2016, 291, 882–893. [Google Scholar] [CrossRef]
- Chen, Z.; Suzuki, H.; Kobayashi, Y.; Wang, A.C.; DiMaio, F.; Kawashima, S.A.; Walz, T.; Kapoor, T.M. Structural Insights into Mdn1, an Essential AAA Protein Required for Ribosome Biogenesis. Cell 2018, 175, 822–834.e18. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, C.; Diepholz, M.; Baßler, J.; Kressler, D.; Pertschy, B.; Galani, K.; Böttcher, B.; Hurt, E. Mechanochemical removal of ribosome biogenesis factors from nascent 60S ribosomal subunits. Cell 2009, 138, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Inoue, S.; Tanabe, Y.; Sumiya, C.; Koike, S. Significance of mitochondria for porphyrin and heme biosynthesis. Science 1959, 129, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Ma, W.; Sand, Z.; Finlayson, J.; Wang, T.; Brinton, R.D.; Willis, W.T.; Mandarino, L.J. Von Willebrand factor A domain-containing protein 8 (VWA8) localizes to the matrix side of the inner mitochondrial membrane. Biochem. Biophys. Res. Commun. 2020, 521, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.V.; Willoughby, M.M.; Piel, R.B.; Ross, T.A.; Bohovych, I.; Addis, H.G.; Fox, J.L.; Lanzilotta, W.N.; Dailey, H.A.; Wohlschlegel, J.A.; et al. Mitochondrial contact site and cristae organizing system (MICOS) machinery supports heme biosynthesis by enabling optimal performance of ferrochelatase. Redox Biol. 2021, 46, 102125. [Google Scholar] [CrossRef]
- Umair, M.; Khan, M.F.; Aldrees, M.; Nashabat, M.; Alhamoudi, K.M.; Bilal, M.; Alyafee, Y.; Al Tuwaijri, A.; Aldarwish, M.; Al-Rumayyan, A.; et al. Mutated VWA8 Is Associated With Developmental Delay, Microcephaly, and Scoliosis and Plays a Novel Role in Early Development and Skeletal Morphogenesis in Zebrafish. Front. Cell Dev. Biol. 2021, 9, 736960. [Google Scholar] [CrossRef]
- Fujiwara, T.; Harigae, H. Biology of Heme in Mammalian Erythroid Cells and Related Disorders. Biomed. Res. Int. 2015, 2015, 278536. [Google Scholar] [CrossRef]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta 2012, 1823, 1604–1616. [Google Scholar] [CrossRef]
- Diaz, F.; Fukui, H.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol. Cell Biol. 2006, 26, 4872–4881. [Google Scholar] [CrossRef] [PubMed]
- Hiser, L.; Di Valentin, M.; Hamer, A.G.; Hosler, J.P. Cox11p is required for stable formation of the Cu(B) and magnesium centers of cytochrome c oxidase. J. Biol. Chem. 2000, 275, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Bourens, M.; Barrientos, A. A CMC1-knockout reveals translation-independent control of human mitochondrial complex IV biogenesis. EMBO Rep. 2017, 18, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Timón-Gómez, A.; Nývltová, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.A.; Haddad, B.G.; Weis, A.J.; Blackwood, C.S.; Shelton, C.D.; Wuerth, M.E.; Walter, J.D.; Spiegel, P.C. Ribosomal protein L7/L12 is required for GTPase translation factors EF-G, RF3, and IF2 to bind in their GTP state to 70S ribosomes. FEBS J. 2017, 284, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Rodnina, M.V.; Peske, F.; Peng, B.-Z.; Belardinelli, R.; Wintermeyer, W. Converting GTP hydrolysis into motion: Versatile translational elongation factor G. Biol. Chem. 2019, 401, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mestre-Fos, S.; Ito, C.; Moore, C.M.; Reddi, A.R.; Williams, L.D. Human ribosomal G-quadruplexes regulate heme bioavailability. J. Biol. Chem. 2020, 295, 14855–14865. [Google Scholar] [CrossRef]
- Canesin, G.; Muralidharan, A.M.; Swanson, K.D.; Wegiel, B. HO-1 and Heme: G-Quadruplex Interaction Choreograph DNA Damage Responses and Cancer Growth. Cells 2021, 10, 1801. [Google Scholar] [CrossRef]
- Luo, M.; Willis, W.T.; Coletta, D.K.; Langlais, P.R.; Mengos, A.; Ma, W.; Finlayson, J.; Wagner, G.R.; Shi, C.-X.; Mandarino, L.J. Deletion of the Mitochondrial Protein VWA8 Induces Oxidative Stress and an HNF4alpha Compensatory Response in Hepatocytes. Biochemistry 2019, 58, 4983–4996. [Google Scholar] [CrossRef]
- Luo, M.; Ma, W.; Zapata-Bustos, R.; Willis, W.T.; Mandarino, L.J. Deletion of Von Willebrand A Domain Containing Protein (VWA8) raises activity of mitochondrial electron transport chain complexes in hepatocytes. Biochem. Biophys. Rep. 2021, 26, 100928. [Google Scholar] [CrossRef] [PubMed]
- Greber, B.J.; Bieri, P.; Leibundgut, M.; Leitner, A.; Aebersold, R.; Boehringer, D.; Ban, N. The complete structure of the 55S mammalian mitochondrial ribosome. Science 2015, 348, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Öztas, S.; Qiu, W.-Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I.; et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Singh, V.; Khawaja, A.; Naschberger, A.; Nguyen, M.D.; Rorbach, J.; Amunts, A. Structure of the mitoribosomal small subunit with streptomycin reveals Fe-S clusters and physiological molecules. Elife 2022, 11, e77460. [Google Scholar] [CrossRef]
- Amunts, A.; Brown, A.; Toots, J.; Scheres, S.H.W.; Ramakrishnan, V. The structure of the human mitochondrial ribosome. Science 2015, 348, 95–98. [Google Scholar] [CrossRef]
- Jacques, C.; Chevrollier, A.; Loiseau, D.; Lagoutte, L.; Savagner, F.; Malthièry, Y.; Reynier, P. mtDNA controls expression of the Death Associated Protein 3. Exp. Cell Res. 2006, 312, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.; Williams, L.D. A recurrent magnesium-binding motif provides a framework for the ribosomal peptidyl transferase center. Nucleic Acids Res. 2009, 37, 3134–3142. [Google Scholar] [CrossRef]
- Sun, F.-J.; Caetano-Anollés, G. The evolutionary history of the structure of 5S ribosomal RNA. J. Mol. Evol. 2009, 69, 430–443. [Google Scholar] [CrossRef]
- Gongadze, G.M. 5S rRNA and ribosome. Biochemistry 2011, 76, 1450–1464. [Google Scholar] [CrossRef]
- Dontsova, O.A.; Dinman, J.D. 5S rRNA: Structure and Function from Head to Toe. Int. J. Biomed. Sci. 2005, 1, 1–7. [Google Scholar]
- Sloan, K.E.; Bohnsack, M.T.; Watkins, N.J. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013, 5, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, A.; Entelis, N.; Martin, R.P.; Tarassov, I. Biological significance of 5S rRNA import into human mitochondria: Role of ribosomal protein MRP-L18. Genes Dev. 2011, 25, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Entelis, N.S.; Kolesnikova, O.A.; Dogan, S.; Martin, R.P.; Tarassov, I.A. 5 S rRNA and tRNA import into human mitochondria. Comparison of in vitro requirements. J. Biol. Chem. 2001, 276, 45642–45653. [Google Scholar] [CrossRef]
- Smirnov, A.; Tarassov, I.; Mager-Heckel, A.-M.; Letzelter, M.; Martin, R.P.; Krasheninnikov, I.A.; Entelis, N. Two distinct structural elements of 5S rRNA are needed for its import into human mitochondria. RNA 2008, 14, 749–759. [Google Scholar] [CrossRef]
- Tobiasson, V.; Gahura, O.; Aibara, S.; Baradaran, R.; Zíková, A.; Amunts, A. Interconnected assembly factors regulate the biogenesis of mitoribosomal large subunit. EMBO J. 2021, 40, e106292. [Google Scholar] [CrossRef]
- Koper, K.; Han, S.-W.; Pastor, D.C.; Yoshikuni, Y.; Maeda, H.A. Evolutionary origin and functional diversification of aminotransferases. J. Biol. Chem. 2022, 298, 102122. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Han, Q.; Tan, Y.; Ding, H.; Li, J. Current Advances on Structure-Function Relationships of Pyridoxal 5′-Phosphate-Dependent Enzymes. Front. Mol. Biosci. 2019, 6, 4. [Google Scholar] [CrossRef]
- di Salvo, M.L.; Budisa, N.; Contestabile, R. PLP-Dependent Enzymes: A Powerful Tool for Metabolic Synthesis of Non-Canonical Amino Acids. Available online: https://www.beilstein-institut.de/download/65/plp-dependent_enzymes_a_powerful_tool_for_metabolic_synthesis_of_non-canonical_amino_acids_pdf 2012 (accessed on 9 November 2023).
- Chen, M.; Liu, C.T.; Tang, Y. Discovery and Biocatalytic Application of a PLP-Dependent Amino Acid gamma-Substitution Enzyme That Catalyzes C-C Bond Formation. J. Am. Chem. Soc. 2020, 142, 10506–10515. [Google Scholar] [CrossRef]
- Ikushiro, H.; Nagami, A.; Takai, T.; Sawai, T.; Shimeno, Y.; Hori, H.; Miyahara, I.; Kamiya, N.; Yano, T. Heme-dependent Inactivation of 5-Aminolevulinate Synthase from Caulobacter crescentus. Sci. Rep. 2018, 8, 14228. [Google Scholar] [CrossRef]
- Deshmukh, D.R.; Srivastava, S.K. Purification and properties of ornithine aminotransferase from rat brain. Experientia 1984, 40, 357–359. [Google Scholar] [CrossRef]
- Kirschning, A. On the evolution of coenzyme biosynthesis. Nat. Prod. Rep. 2022, 39, 2175–2199. [Google Scholar] [CrossRef]
- Böttinger, L.; Mårtensson, C.U.; Song, J.; Zufall, N.; Wiedemann, N.; Becker, T. Respiratory chain supercomplexes associate with the cysteine desulfurase complex of the iron-sulfur cluster assembly machinery. Mol. Biol. Cell 2018, 29, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.S.; Dietz, K.-J. The significance of amino acids and amino acid-derived molecules in plant responses and adaptation to heavy metal stress. J. Exp. Bot. 2006, 57, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, M.M.; Penmatsa, A.; Whittaker, J.W. The Mtm1p carrier and pyridoxal 5′-phosphate cofactor trafficking in yeast mitochondria. Arch. Biochem. Biophys. 2015, 568, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Hartman, H. The evolution of Class II Aminoacyl-tRNA synthetases and the first code. FEBS Lett. 2015, 589, 3499–3507. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Stachelhaus, T.; Marahiel, M.A. Targeted alteration of the substrate specificity of peptide synthetases by rational module swapping. Mol. Gen. Genet. 1998, 257, 308–318. [Google Scholar] [CrossRef]
- Mills, P.B.; Camuzeaux, S.S.; Footitt, E.J.; Mills, K.A.; Gissen, P.; Fisher, L.; Das, K.B.; Varadkar, S.M.; Zuberi, S.; McWilliam, R.; et al. Epilepsy due to PNPO mutations: Genotype, environment and treatment affect presentation and outcome. Brain 2014, 137 Pt 5, 1350–1360. [Google Scholar] [CrossRef]
- Henslee, J.G.; Wakabayashi, Y.; Small, C.; Jones, M.E. Factors influencing pyrroline 5-carboxylate synthesis from glutamate by rat intestinal mucosa mitochondria. Arch. Biochem. Biophys. 1983, 226, 693–703. [Google Scholar] [CrossRef]
- Csonka, L.N.; Leisinger, T. Biosynthesis of Proline. EcoSal Plus 2007, 2. [Google Scholar] [CrossRef]
- Zappa, S.; Li, K.; Bauer, C.E. The tetrapyrrole biosynthetic pathway and its regulation in Rhodobacter capsulatus. Adv. Exp. Med. Biol. 2010, 675, 229–250. [Google Scholar]
- Hoober, J.K.; Kahn, A.; Ash, D.E.; Gough, S.; Kannangara, C.G. Biosynthesis of delta-aminolevulinate in greening barley leaves. IX. Structure of the substrate, mode of gabaculine inhibition, and the catalytic mechanism of glutamate 1-semialdehyde aminotransferase. Carlsberg Res. Commun. 1988, 53, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Eirich, J.; Finkemeier, I.; Grimm, B. Glutamate 1-semialdehyde aminotransferase is connected to GluTR by GluTR-binding protein and contributes to the rate-limiting step of 5-aminolevulinic acid synthesis. Plant Cell 2022, 34, 4623–4640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gao, X.; Coots, R.A.; Conn, C.S.; Liu, B.; Qian, S.-B. Translational control of the cytosolic stress response by mitochondrial ribosomal protein L18. Nat. Struct. Mol. Biol. 2015, 22, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Choi, S.I.; Seong, B.L. 5S rRNA-assisted DnaK refolding. Biochem. Biophys. Res. Commun. 2010, 391, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Brusslan, J.A.; Peterson, M.P. Tetrapyrrole regulation of nuclear gene expression. Photosynth. Res. 2002, 71, 185–194. [Google Scholar] [CrossRef]
- Barros, M.H.; Carlson, C.G.; Glerum, D.; Tzagoloff, A. Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O. FEBS Lett. 2001, 492, 133–138. [Google Scholar] [CrossRef]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef]
- Antonicka, H.; Pankratz, N.; Nichols, W.C.; Uniacke, S.K.; Halter, C.; Murrell, J.; Rudolph, A.; Shults, C.W.; Conneally, P.M.; Foroud, T. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef]
- Alfadhel, M.; Lillquist, Y.P.; Waters, P.J.; Sinclair, G.; Struys, E.; McFadden, D.; Hendson, G.; Hyams, L.; Shoffner, J.; Vallance, H.D. Infantile cardioencephalopathy due to a COX15 gene defect: Report and review. Am. J. Med. Genet. A 2011, 155A, 840–844. [Google Scholar] [CrossRef]
- Zheng, H.; Ji, C.; Zou, X.; Wu, M.; Jin, Z.; Yin, G.; Li, J.; Feng, C.; Cheng, H.; Gu, S.; et al. Molecular cloning and characterization of a novel human putative transmembrane protein homologous to mouse sideroflexin associated with sideroblastic anemia. DNA Seq. 2003, 14, 369–373. [Google Scholar] [CrossRef]
- Paul, B.T.; Tesfay, L.; Winkler, C.R.; Torti, F.M.; Torti, S.V. Sideroflexin 4 affects Fe-S cluster biogenesis, iron metabolism, mitochondrial respiration and heme biosynthetic enzymes. Sci. Rep. 2019, 9, 19634. [Google Scholar] [CrossRef] [PubMed]
- Hildick-Smith, G.J.; Cooney, J.D.; Garone, C.; Kremer, L.S.; Haack, T.B.; Thon, J.N.; Miyata, N.; Lieber, D.S.; Calvo, S.E.; Akman, H.O.; et al. Macrocytic anemia and mitochondriopathy resulting from a defect in sideroflexin 4. Am. J. Hum. Genet. 2013, 93, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.P.; Schagger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Giese, H.; Ackermann, J.; Heide, H.; Bleier, L.; Dröse, S.; Wittig, I.; Brandt, U.; Koch, I. NOVA: A software to analyze complexome profiling data. Bioinformatics 2015, 31, 440–441. [Google Scholar] [CrossRef] [PubMed]
- Schlattl, M.; Buffler, M.; Windisch, W. Clay Minerals Affect the Solubility of Zn and Other Bivalent Cations in the Digestive Tract of Ruminants In Vitro. Animals 2021, 11, 877. [Google Scholar] [CrossRef]
- Adrain, C.; Creagh, E.M.; Martin, S.J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001, 20, 6627–6636. [Google Scholar] [CrossRef]
- Baghirova, S.; Hughes, B.G.; Hendzel, M.J.; Schulz, R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2015, 2, 440–445. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements, ppm | WT | CLPP-Null | Fold Change | p-Value | Significance |
|---|---|---|---|---|---|
| Iron/Ferrum (Fe) | 83.33 ± 2.82 | 113.85 ± 3.60 | 1.366 | <0.0001 | **** |
| Molybdenum (Mo) | 221.48 ± 9.10 | 299.04 ± 7.04 | 1.350 | <0.0001 | **** |
| Cobalt (Co) | 37.04 ± 1.74 | 44.98 ± 2.46 | 1.214 | 0.0178 | * |
| Manganese (Mn) | 1.47 ± 0.09 | 1.76 ± 0.07 | 1.193 | 0.0235 | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Key, J.; Gispert, S.; Koepf, G.; Steinhoff-Wagner, J.; Reichlmeir, M.; Auburger, G. Translation Fidelity and Respiration Deficits in CLPP-Deficient Tissues: Mechanistic Insights from Mitochondrial Complexome Profiling. Int. J. Mol. Sci. 2023, 24, 17503. https://doi.org/10.3390/ijms242417503
Key J, Gispert S, Koepf G, Steinhoff-Wagner J, Reichlmeir M, Auburger G. Translation Fidelity and Respiration Deficits in CLPP-Deficient Tissues: Mechanistic Insights from Mitochondrial Complexome Profiling. International Journal of Molecular Sciences. 2023; 24(24):17503. https://doi.org/10.3390/ijms242417503
Chicago/Turabian StyleKey, Jana, Suzana Gispert, Gabriele Koepf, Julia Steinhoff-Wagner, Marina Reichlmeir, and Georg Auburger. 2023. "Translation Fidelity and Respiration Deficits in CLPP-Deficient Tissues: Mechanistic Insights from Mitochondrial Complexome Profiling" International Journal of Molecular Sciences 24, no. 24: 17503. https://doi.org/10.3390/ijms242417503
APA StyleKey, J., Gispert, S., Koepf, G., Steinhoff-Wagner, J., Reichlmeir, M., & Auburger, G. (2023). Translation Fidelity and Respiration Deficits in CLPP-Deficient Tissues: Mechanistic Insights from Mitochondrial Complexome Profiling. International Journal of Molecular Sciences, 24(24), 17503. https://doi.org/10.3390/ijms242417503