
Phenotype–Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Case Presentations
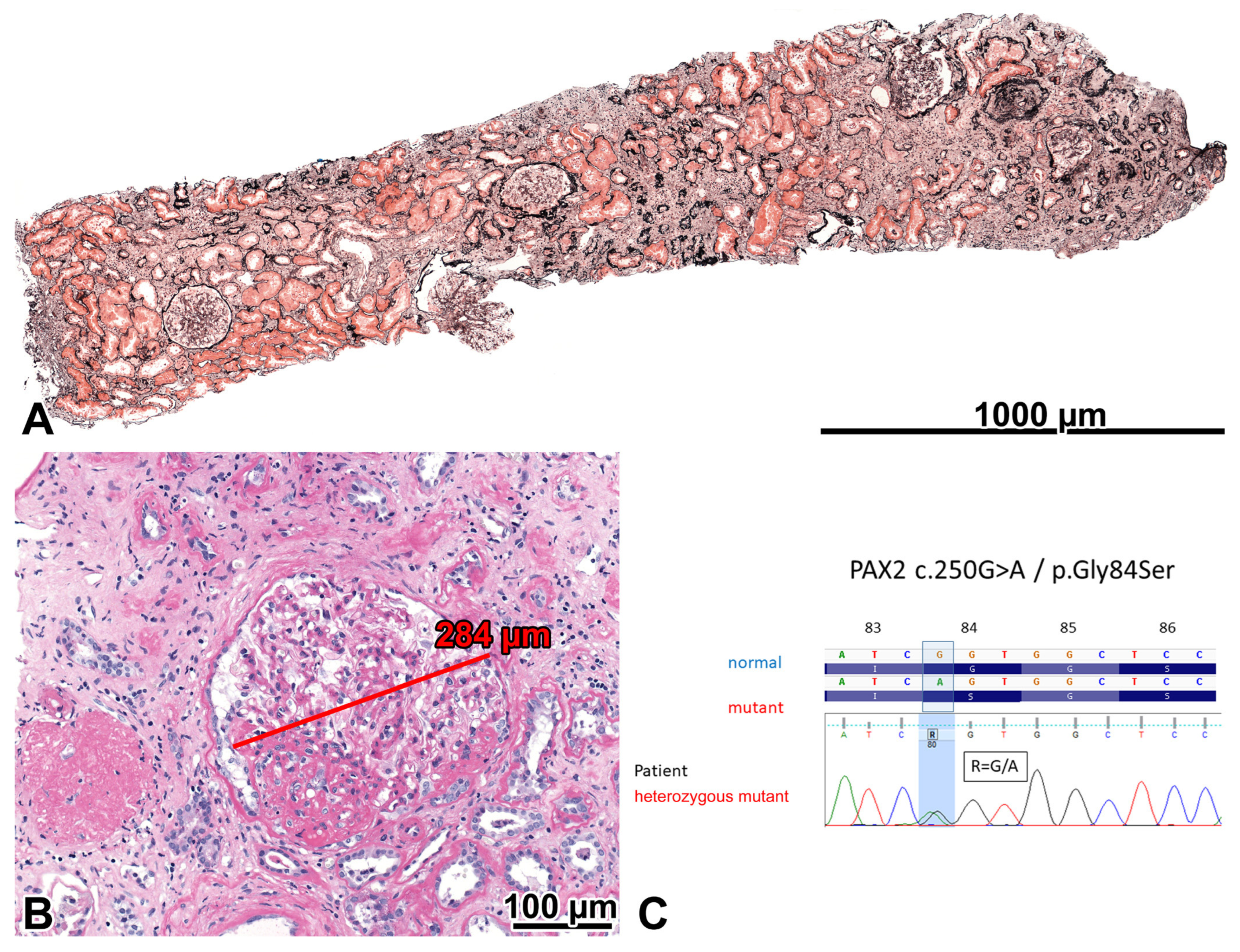
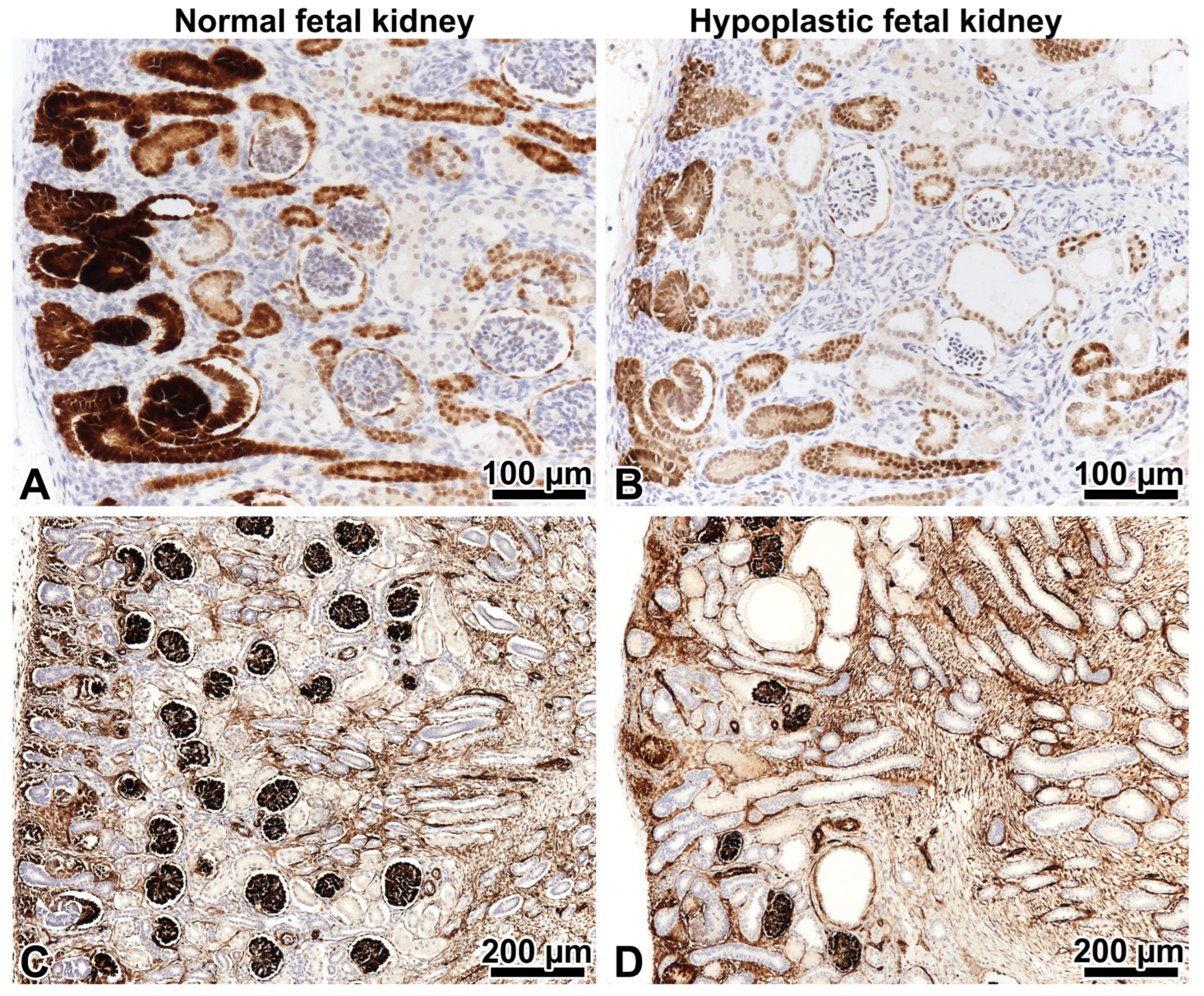
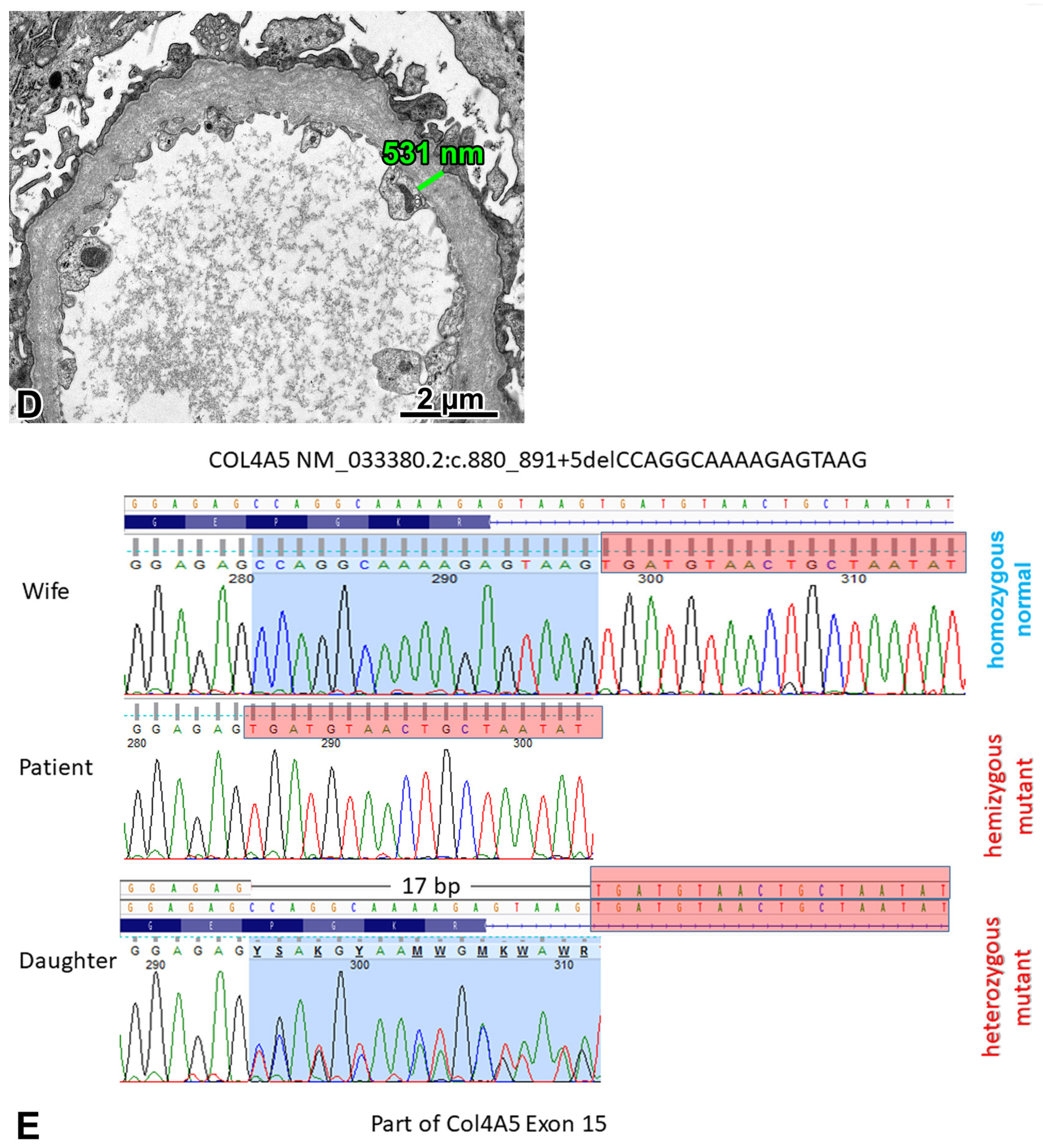
2.2. Patient 1
2.2.1. Renal Biopsy Findings and Genetic Analysis
2.2.2. Investigations Performed after the Genotyping Procedure
2.2.3. Comments
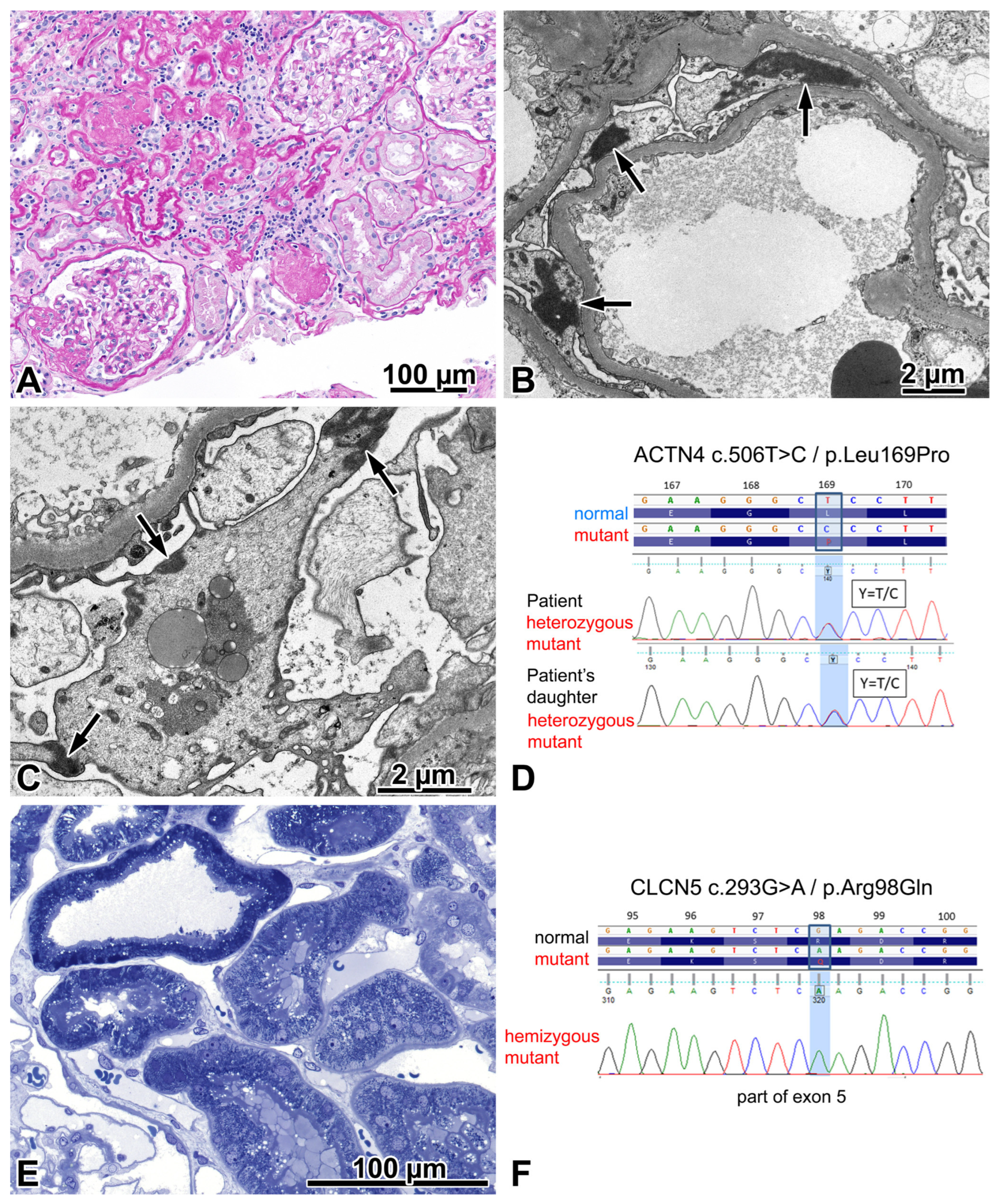
2.3. Patient 2
2.3.1. Renal Biopsy Findings and Results of a Genetic Analysis
2.3.2. Investigations Made after Performing the Genotyping Procedure
2.3.3. Comments
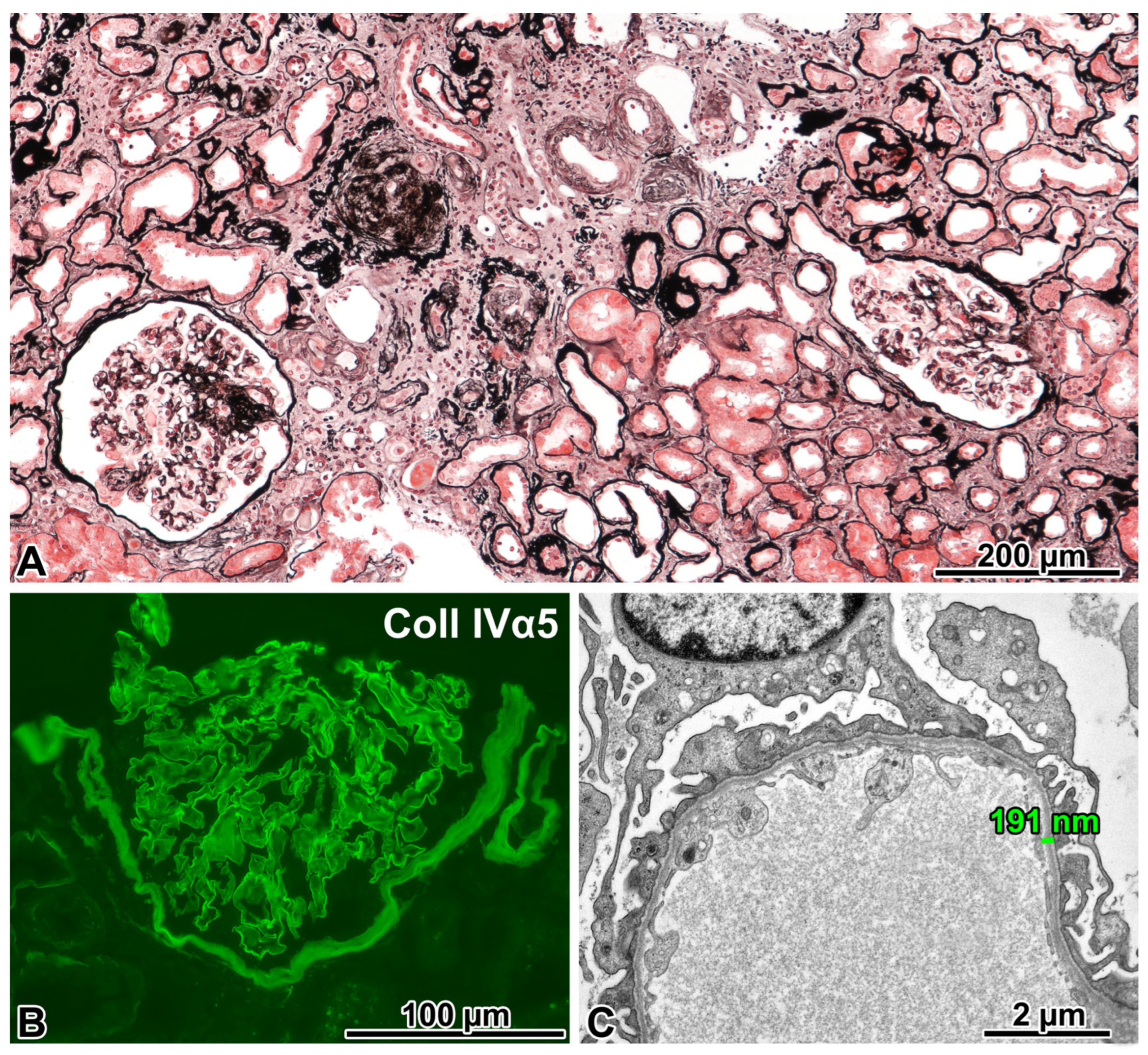
2.4. Patient 3
2.4.1. Renal Biopsy Findings and a Post-Biopsy Evaluation for Alport Syndrome
2.4.2. Genetic Analysis
2.4.3. Comments
3. Discussion
Lessons Learned from the Case Presentations
4. Materials and Methods
4.1. Evaluation of Renal Biopsy Samples
4.2. Additional Immunostainings and Measurements
4.3. Genetic Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Vriese, A.S.; Sethi, S.; Nath, K.A.; Glassock, R.J.; Fervenza, F.C. Differentiating primary, genetic, and secondary FSGS in adults: A clinicopathologic approach. J. Am. Soc. Nephrol. 2018, 29, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Pinto e Vairo, F.; Hogan, M.C.; Erickson, S.B.; El Ters, M.; Bentall, A.J.; Kukla, A.; Greene, E.L.; Hernandez, L.H.; Sethi, S.; et al. Identification of genetic causes of focal segmental glomerulosclerosis increases with proper patient selection. Mayo Clin. Proc. 2021, 96, 2342–2353. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Wetzels, J.F.; Glassock, R.J.; Sethi, S.; Fervenza, F.C. Therapeutic trials in adult FSGS: Lessons learned and the road forward. Nat. Rev. Nephrol. 2021, 17, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integrationof genetic testing and pathology for the diagnosis of adults with FSGS. Clin. J. Am. Soc. Nephrol. 2019, 14, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chun, J.; Genovese, G.; Knob, A.U.; Benjamin, A.; Wilkins, M.S.; Friedman, D.J.; Appel, G.B.; Lifton, R.P.; Mane, S.; et al. Contributions of rare gene variants to familial and sporadic FSGS. J. Am. Soc. Nephrol. 2019, 30, 1625–1640. [Google Scholar] [CrossRef]
- Bitó, L.; Kalmár, T.; Maróti, Z.; Turkevi-Nagy, S.; Bereczki, C.; Iványi, B. PAX2 mutation-related oligomeganephronia in a young adult patient. Case Rep. Nephrol. Dial. 2020, 10, 163–173. [Google Scholar] [CrossRef]
- Ren, Y.L.; Li, Y.; Gao, J.; Zhou, X.J.; Yang, L.; Wang, S.X. Pathological and clinical characteristics of late-onset oligomeganephronia based on a histomorphometric study. BMC Nephrol. 2023, 24, 54–64. [Google Scholar] [CrossRef]
- Solomon, R.; Tellier, A.L.; Attie-Bittach, T.; Amiel, J.; Vekemans, M.; Lyonnet, S.; Dureau, P.; Niaudet, P.; Gubler, M.C.; Broyer, M. PAX2 mutations in oligomeganephronia. Kidney Int. 2001, 59, 457–462. [Google Scholar] [CrossRef]
- Kambham, N.; Markowitz, G.S.; Valeri, A.M.; Lin, J.; D’Agati, V.D. Obesity-related glomerulopathy: An emerging epidemic. Kidney Int. 2001, 59, 1498–1509. [Google Scholar] [CrossRef]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef]
- Porteous, S.; Torban, E.; Cho, N.P.; Cunliffe, H.; Chua, L.; McNoe, L.; Ward, T.; Souza, E.; Gus, P.; Giugliani, R.; et al. Primary renal hypoplasia in humans and mice with PAX2 mutations: Evidence of increased apoptosis in fetal kidneys of Pax21Neu +/− mutant mice. Hum. Mol. Genet. 2000, 9, 1–11. [Google Scholar] [CrossRef]
- Barua, M.; Stellaci, E.; Stella, L.; Weins, A.; Genovese, G.; Muto, V.; Caputo, V.; Toka, H.R.; Charoonratana, V.T.; Tartaglia, M.; et al. Mutations in PAX2 associate with adult-onset FSGS. J. Am. Soc. Nephrol. 2014, 25, 1942–1953. [Google Scholar] [CrossRef] [PubMed]
- Madariaga, L.; Moriniere, V.; Jeanpierre, C.; Bouvier, R.; Loget, P.; Martinovic, J.; Dechelotte, P.; Leporrier, N.; Thauvin-Robinet, C.; Jensen, U.B.; et al. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin. J. Am. Soc. Nephrol. 2013, 8, 1179–1187. [Google Scholar] [CrossRef]
- Barua, M.; Brown, E.J.; Charoonratana, V.T.; Genovese, G.; Sun, H.; Pollak, M.R. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013, 83, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; DuMontier, C.; Pollak, M.R. The role of alpha-actinin-4 in human kidney disease. Cell Biosci. 2015, 5, 44–51. [Google Scholar] [CrossRef]
- Henderson, J.M.; Alexander, M.P.; Pollak, M.R. Patients with ACTN4 mutations demonstrate distinctive features of glomerular injury. J. Am. Soc. Nephrol. 2009, 20, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.K.; Arif, E.; Morinelli, T.; Wilson, R.C.; Hardiman, G.; Deng, P.; Arthur, J.M.; Velez, J.C.Q.; Nihalani, D.; Janech, M.G. A novel CLCN5 mutationassociated with focal segmental glomerulosclerosis and podocyte injury. Kidney Int. Rep. 2018, 3, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Nozu, K.; Nakanishi, K.; Abe, Y.; Udagawa, T.; Okada, S.; Okamoto, T.; Kaito, H.; Kanemoto, K.; Kobayashi, A.; Tanaka, E.; et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 2019, 23, 158–168. [Google Scholar] [CrossRef]
- Hashimura, Y.; Nozu, K.; Kaito, H.; Nakanishi, K.; Fu, X.J.; Ohtsubo, H.; Hashimoto, F.; Oka, M.; Ninchoji, T.; Ishimori, S.; et al. Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV α5 chain. Kidney Int. 2014, 85, 1208–1213. [Google Scholar] [CrossRef]
- Nagano, C.; Hara, S.; Yoshikawa, N.; Takeda, A.; Gotoh, Y.; Hamada, R.; Matsuoka, K.; Yamamoto, M.; Fujinaga, S.; Sakuraya, K.; et al. Clinical, pathological, and genetic characteristics in patients with focal segmental glomerulosclerosis. Kidney360 2022, 3, 1384–1393. [Google Scholar] [CrossRef]
- Braunisch, M.C.; Riedhammer, K.M.; Herr, P.-M.; Draut, S.; Günthner, R.; Wagner, M.; Weidenbusch, M.; Lungu, A.; Alhaddad, B.; Renders, L.; et al. Identification of disease-causing variants by comprehensive genetic testing with exome sequencing in adults with suspicion of hereditary FSGS. Eur. J. Hum. Gen. 2021, 29, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Schlöndorff, J.S.; Becker, D.J.; Tsukaguchi, H.; Tonna, S.J.; Uscinski, A.L.; Higgs, H.N.; Henderson, J.M.; Pollak, M.R. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Gen. 2010, 42, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; D’Agati, V.D.; Nast, C.C.; Fogo, A.B.; De Vriese, A.S.; Markowitz, G.S.; Glassock, R.J.; Fervenza, F.C.; Seshan, S.V.; Rule, A.; et al. A proposal for standardized grading of chronic changes in native kidney biopsy specimens. Kidney Int. 2017, 91, 787–789. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial manifestation started before age 25 |
| Absence of the nephrotic syndrome |
| Family history of kidney disease |
| Extrarenal manifestations (hearing loss, eye abnormalities, etc.) |
| Undetermined FSGS or secondary FSGS without an identifiable cause |
| Oligomeganephronia on LM or abnormalities in podocytes * or GBM ** on EM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalmár, T.; Turkevi-Nagy, S.; Bitó, L.; Kaiser, L.; Maróti, Z.; Jakab, D.; Letoha, A.; Légrády, P.; Iványi, B. Phenotype–Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis. Int. J. Mol. Sci. 2023, 24, 17489. https://doi.org/10.3390/ijms242417489
Kalmár T, Turkevi-Nagy S, Bitó L, Kaiser L, Maróti Z, Jakab D, Letoha A, Légrády P, Iványi B. Phenotype–Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis. International Journal of Molecular Sciences. 2023; 24(24):17489. https://doi.org/10.3390/ijms242417489
Chicago/Turabian StyleKalmár, Tibor, Sándor Turkevi-Nagy, László Bitó, László Kaiser, Zoltán Maróti, Dániel Jakab, Annamária Letoha, Péter Légrády, and Béla Iványi. 2023. "Phenotype–Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis" International Journal of Molecular Sciences 24, no. 24: 17489. https://doi.org/10.3390/ijms242417489
APA StyleKalmár, T., Turkevi-Nagy, S., Bitó, L., Kaiser, L., Maróti, Z., Jakab, D., Letoha, A., Légrády, P., & Iványi, B. (2023). Phenotype–Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis. International Journal of Molecular Sciences, 24(24), 17489. https://doi.org/10.3390/ijms242417489