
Acceleration of Protein Degradation by 20S Proteasome-Binding Peptides Generated by In Vitro Artificial Evolution
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Generation of 20S-Interacting Peptides Using the cDNA Display Method
2.2. Proteasome Inhibition by 20S-BP1 and Its Analogs


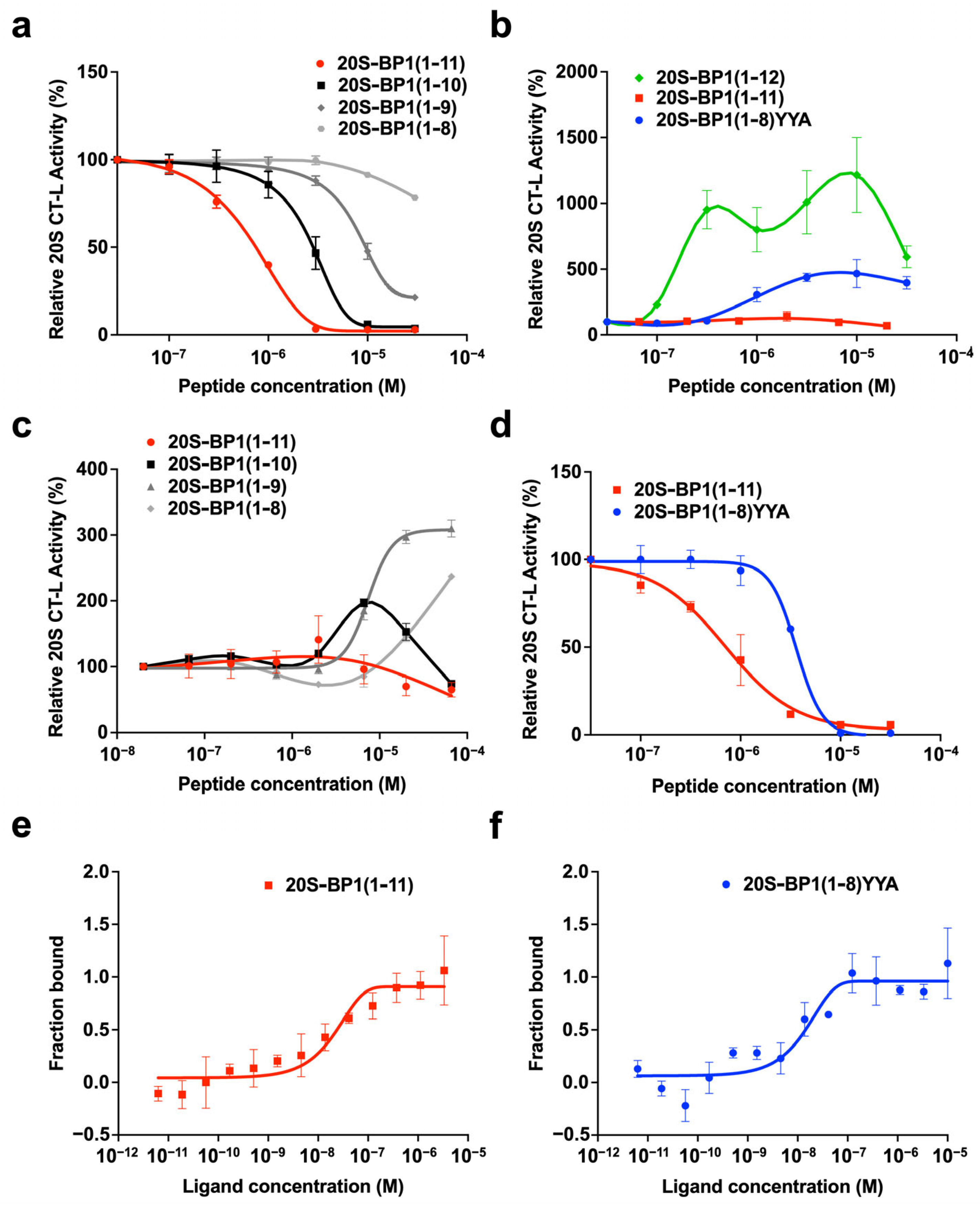
2.3. Proteasome Stimulation by 20S-BP1(1-12) and Its Analogs
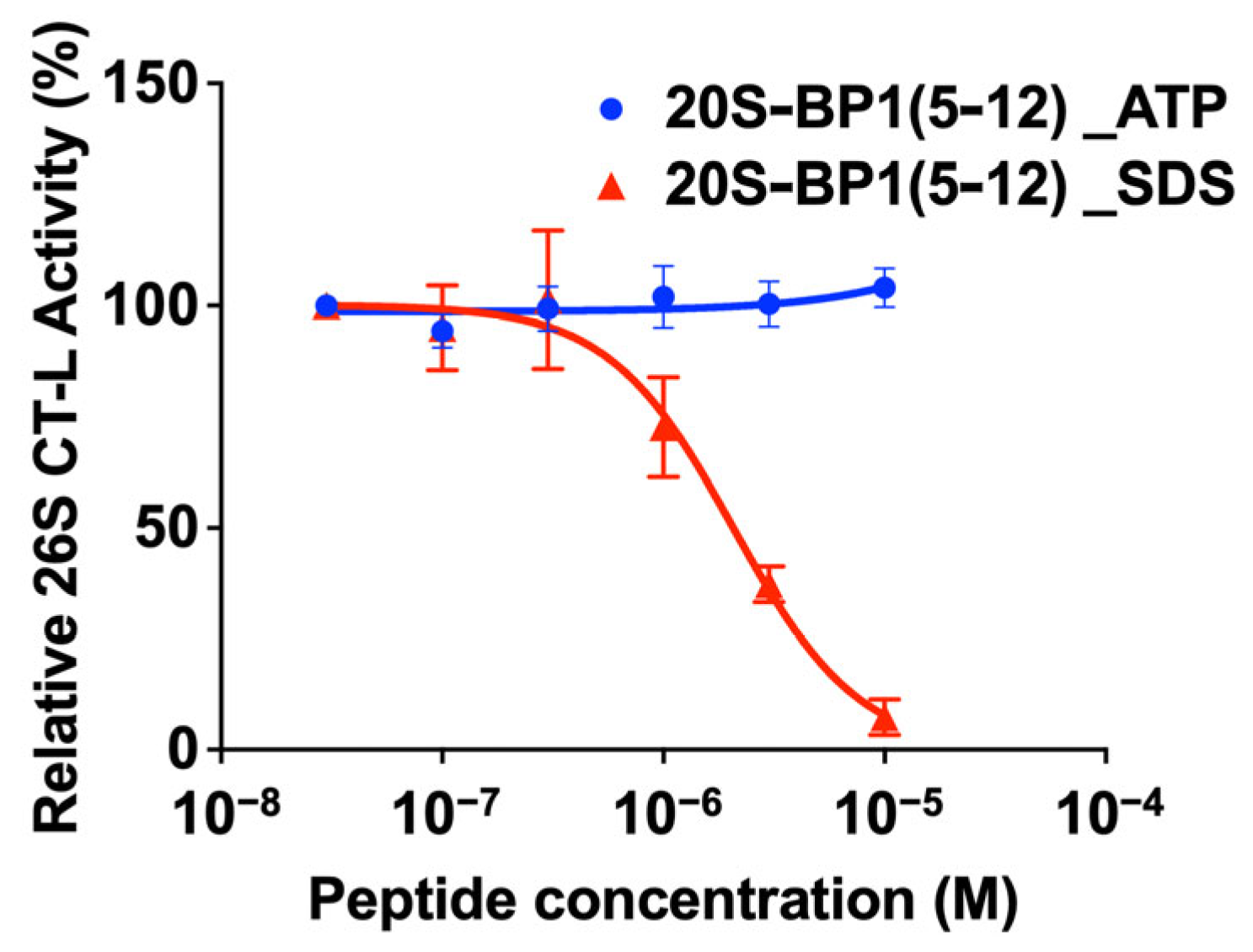
2.4. Effect on the 26S Holoenzyme
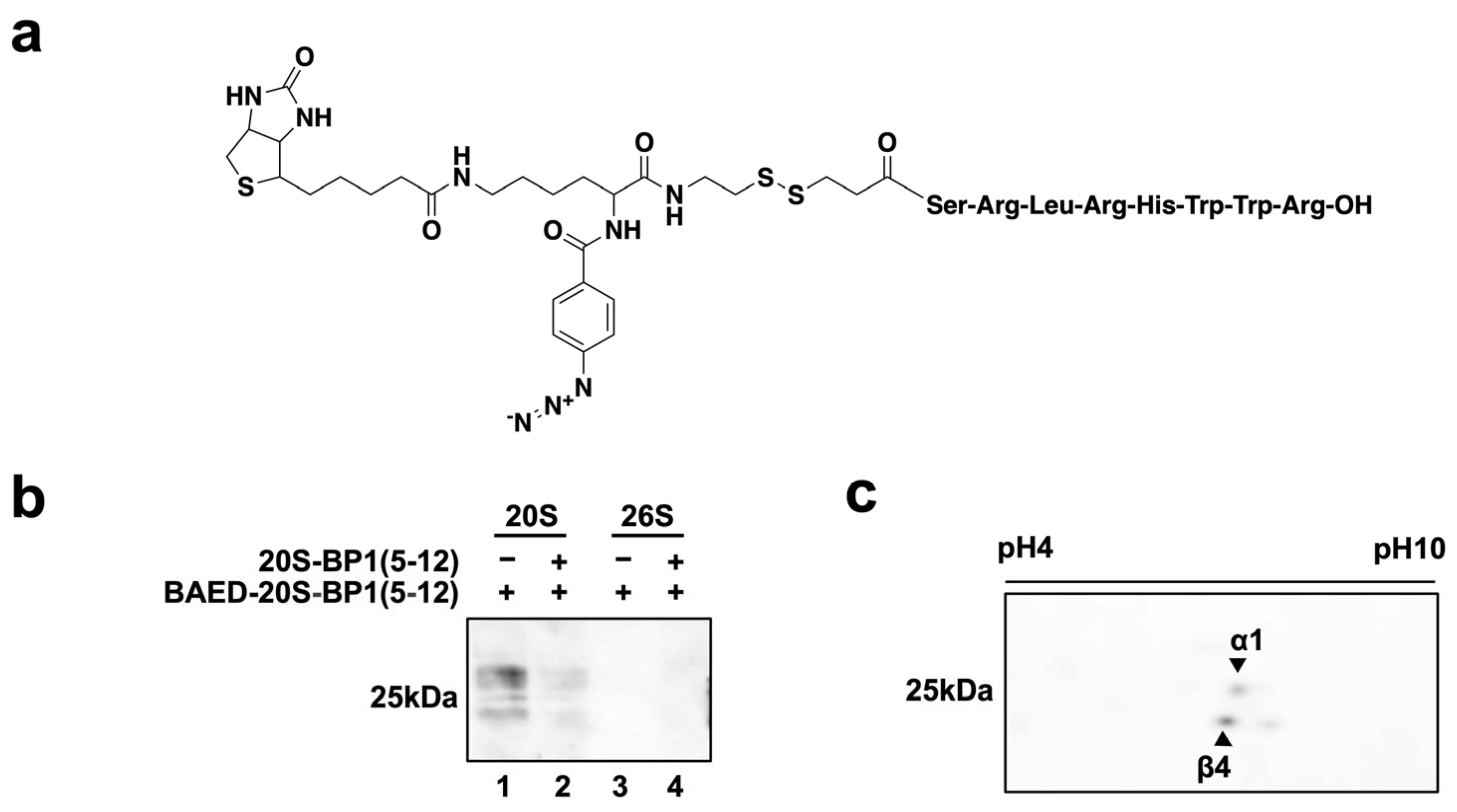
2.5. Photoaffinity Labeling of 20S-BP1-Binding Subunits in the 20S CP
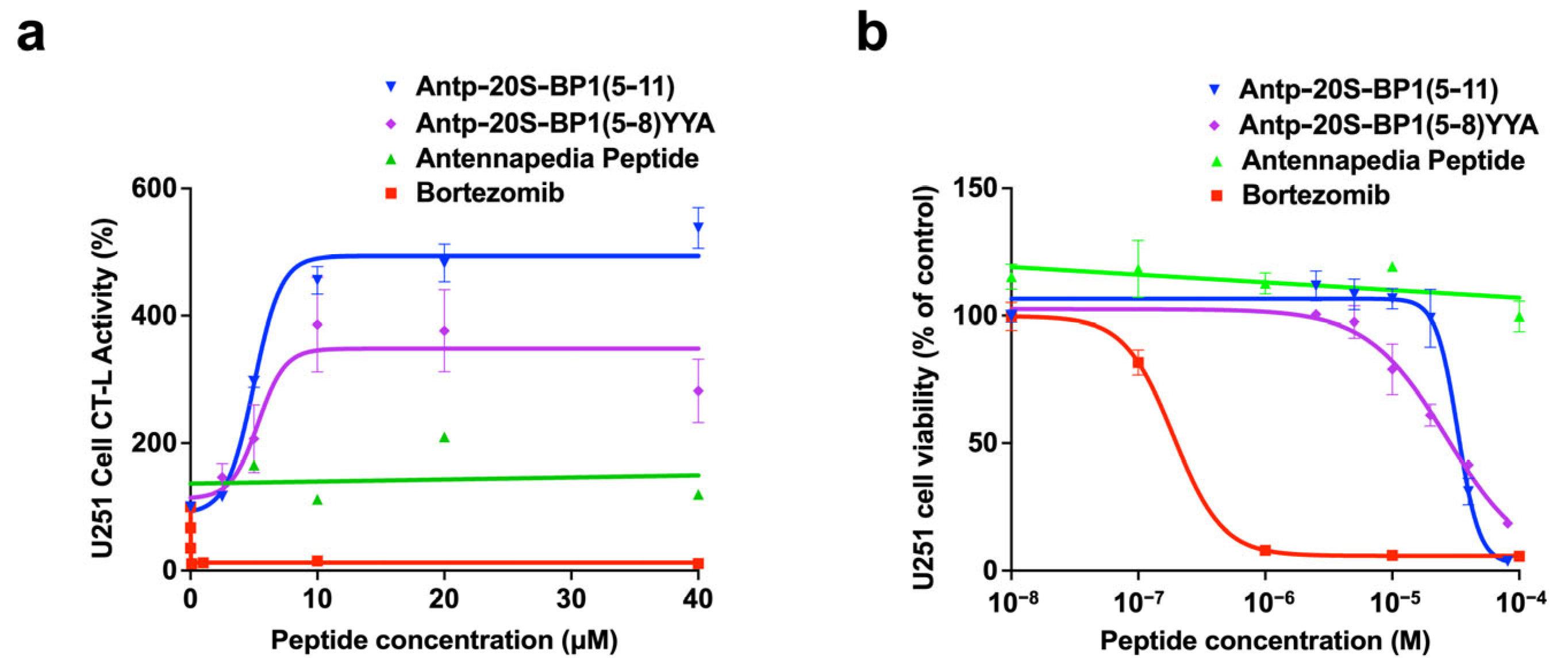
2.6. Effect of 20S-BP1 Analogs with a Cell-Penetrating Peptide on Intracellular Proteasomes
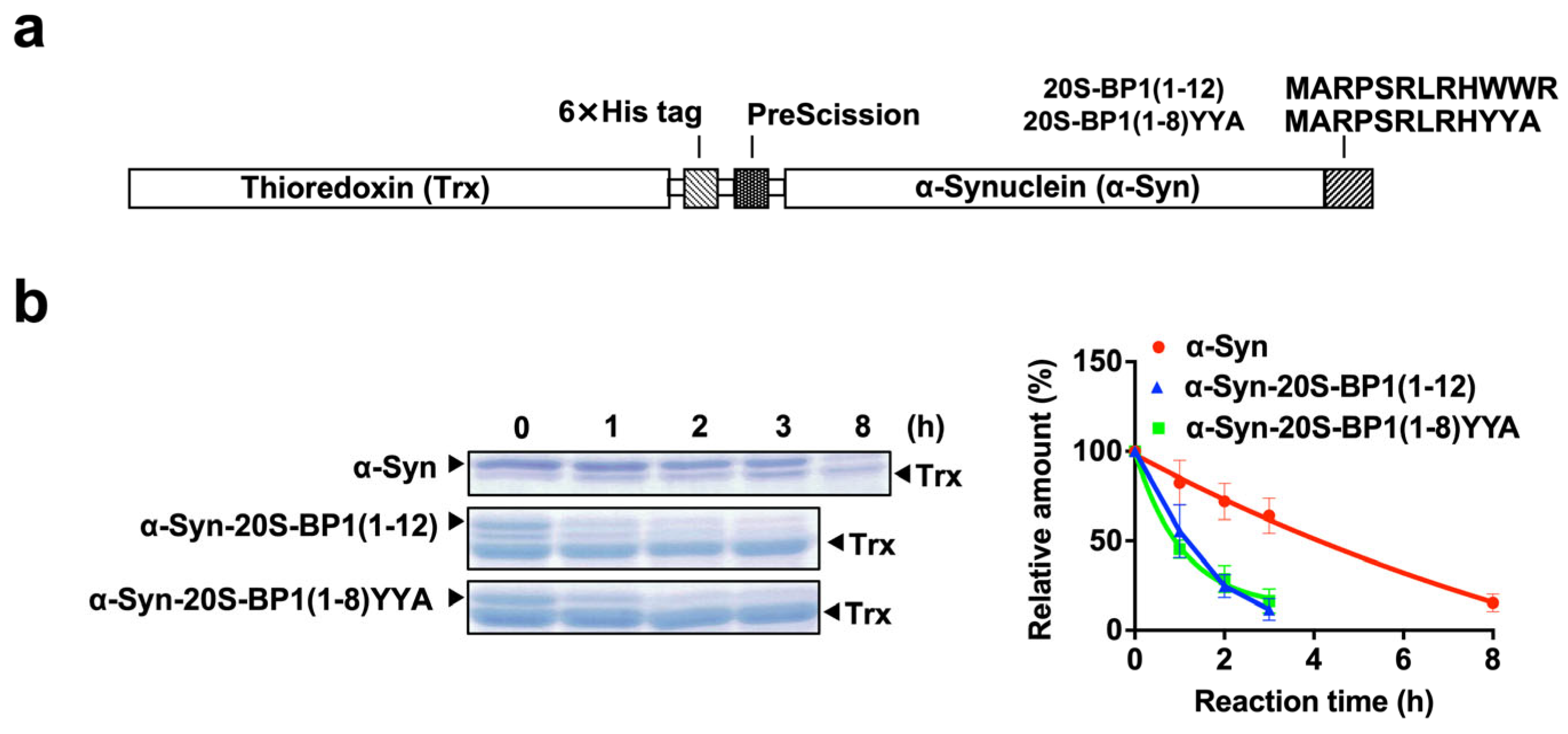
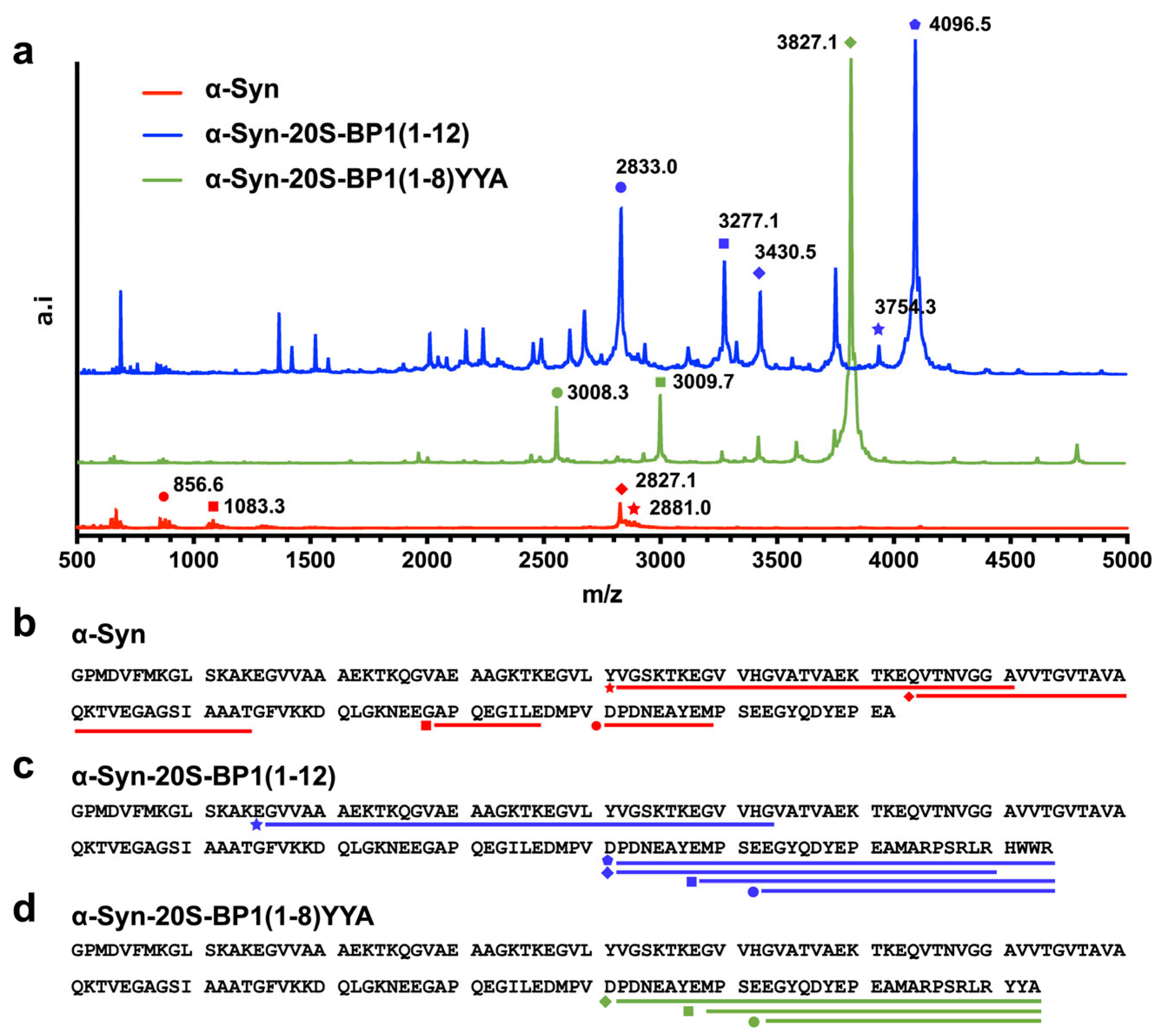
2.7. Enhanced Degradation of 20S CP Substrate Proteins Harboring a 20S-BP1 Sequence
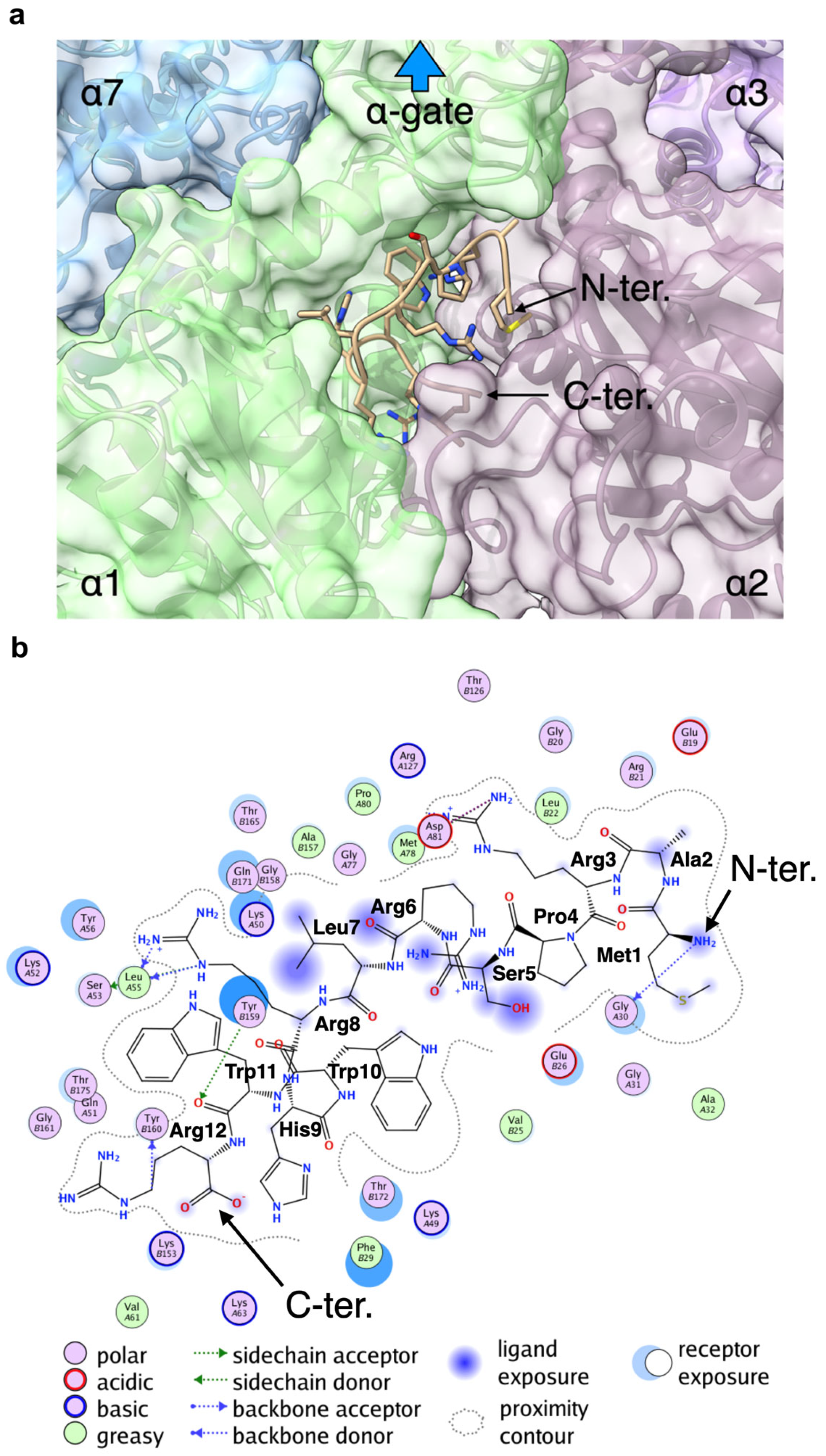
2.8. Docking Study of the 20S-BP1 on the α-Ring of the 20S CP
2.9. Final Considerations
3. Materials and Methods
3.1. The cDNA Display Method
3.2. Peptide Synthesis
3.3. Proteasome Fluorometric Substrate Assay
3.4. Fluorescent Microscale Thermophoresis (MST) Assay
3.5. Photoaffinity Labeling
3.6. Cell Growth Inhibition Assay
3.7. The 20S Proteasome Assay of Substrate Proteins
3.8. Docking Study of 20S-BP1(1-12) and α-Ring of 20S CP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Mayor, T.; Sharon, M.; Glickman, M.H. Tuning the proteasome to brighten the end of the journey. Am. J. Physiol. Cell Physiol. 2016, 311, C793–C804. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef]
- Saeki, Y. Ubiquitin recognition by the proteasome. J. Biochem. 2017, 161, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 2002, 10, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Kunjappu, M.J.; Hochstrasser, M. Assembly of the 20S proteasome. Biochim. Biophys. Acta 2014, 1843, 2–12. [Google Scholar] [CrossRef]
- Sahu, I.; Glickman, M.H. Proteasome in action: Substrate degradation by the 26S proteasome. Biochem. Soc. Trans. 2021, 49, 629–644. [Google Scholar] [CrossRef]
- Rabl, J.; Smith, D.M.; Yu, Y.; Chang, S.C.; Goldberg, A.L.; Cheng, Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell 2008, 30, 360–368. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef]
- Hohn, T.J.; Grune, T. The proteasome and the degradation of oxidized proteins: Part III-Redox regulation of the proteasomal system. Redox. Biol. 2014, 2, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.P. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Lambour, T.; Garrigues, L.; Amalric, F.; Vigneron, N.; Menneteau, T.; Stella, A.; Monsarrat, B.; Van den Eynde, B.; Burlet-Schiltz, O.; et al. Deciphering preferential interactions within supramolecular protein complexes: The proteasome case. Mol. Syst. Biol. 2015, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S proteasome complex during oxidative stress. Sci. Signal. 2010, 3, ra88. [Google Scholar] [CrossRef]
- Pickering, A.M.; Koop, A.L.; Teoh, C.Y.; Ermak, G.; Grune, T.; Davies, K.J. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010, 432, 585–594. [Google Scholar] [CrossRef]
- Sahu, I.; Mali, S.M.; Sulkshane, P.; Xu, C.; Rozenberg, A.; Morag, R.; Sahoo, M.P.; Singh, S.K.; Ding, Z.; Wang, Y.; et al. The 20S as a stand-alone proteasome in cells can degrade the ubiquitin tag. Nat. Commun. 2021, 12, 6173. [Google Scholar] [CrossRef]
- Suskiewicz, M.J.; Sussman, J.L.; Silman, I.; Shaul, Y. Context-dependent resistance to proteolysis of intrinsically disordered proteins. Protein Sci. 2011, 20, 1285–1297. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Karpowicz, P.; Jankowska, E.; Bohmann, J.; Pickering, A.M.; Gaczynska, M. New Peptide-Based Pharmacophore Activates 20S Proteasome. Molecules 2020, 25, 1439. [Google Scholar] [CrossRef]
- Gizynska, M.; Witkowska, J.; Karpowicz, P.; Rostankowski, R.; Chocron, E.S.; Pickering, A.M.; Osmulski, P.; Gaczynska, M.; Jankowska, E. Proline- and Arginine-Rich Peptides as Flexible Allosteric Modulators of Human Proteasome Activity. J. Med. Chem. 2019, 62, 359–370. [Google Scholar] [CrossRef]
- Dal Vechio, F.H.; Cerqueira, F.; Augusto, O.; Lopes, R.; Demasi, M. Peptides that activate the 20S proteasome by gate opening increased oxidized protein removal and reduced protein aggregation. Free Radic. Biol. Med. 2014, 67, 304–313. [Google Scholar] [CrossRef]
- Smith, D.M.; Chang, S.C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A.L. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol. Cell 2007, 27, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Kloss, A.; Henklein, P.; Siele, D.; Schmolke, M.; Apcher, S.; Kuehn, L.; Sheppard, P.W.; Dahlmann, B. The cell-penetrating peptide octa-arginine is a potent inhibitor of proteasome activities. Eur. J. Pharm. Biopharm. 2009, 72, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Yoshimura, T.; Ichihara, A. Role of substrate in reversible activation of proteasomes (multi-protease complexes) by sodium dodecyl sulfate. J. Biochem. 1989, 106, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Long, M.J.; Rosenberg, M.M.; Li, S.; Kobjack, A.; Lessans, P.; Coffey, R.T.; Hedstrom, L. Boc(3)Arg-Linked Ligands Induce Degradation by Localizing Target Proteins to the 20S Proteasome. ACS Chem. Biol. 2016, 11, 3328–3337. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Naimuddin, M.; Biyani, M.; Sasaki, T.; Machida, M.; Kubo, T.; Funatsu, T.; Husimi, Y.; Nemoto, N. cDNA display: A novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions. Nucleic Acids Res. 2009, 37, e108. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Komatsu, M.; Biyani, M.; Futakami, M.; Kawakubo, T.; Yamamoto, K.; Nishigaki, K. Proven in vitro evolution of protease cathepsin E-inhibitors and -activators at pH 4.5 using a paired peptide method. J. Pept. Sci. 2012, 18, 711–719. [Google Scholar] [CrossRef]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharmacol. 2005, 145, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lanzas, R.; Castano, J.G. Proteins directly interacting with mammalian 20S proteasomal subunits and ubiquitin-independent proteasomal degradation. Biomolecules 2014, 4, 1140–1154. [Google Scholar] [CrossRef]
- Alvarez-Castelao, B.; Goethals, M.; Vandekerckhove, J.; Castano, J.G. Mechanism of cleavage of alpha-synuclein by the 20S proteasome and modulation of its degradation by the RedOx state of the N-terminal methionines. Biochim. Biophys. Acta 2014, 1843, 352–365. [Google Scholar] [CrossRef]
- Meszaros, B.; Kumar, M.; Gibson, T.J.; Uyar, B.; Dosztanyi, Z. Degrons in cancer. Sci. Signal. 2017, 10, 1557–1567. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, D.; Lee, C.H.; Chung, K.C.; Kim, J.; Paik, S.R. Calpain-resistant fragment(s) of alpha-synuclein regulates the synuclein-cleaving activity of 20S proteasome. Arch. Biochem. Biophys. 2006, 455, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Pawel Ciemny, M.; Oleniecki, T.; Kuriata, A.; Badaczewska-Dawid, A.E.; Kolinski, A.; Kmiecik, S. CABS-dock standalone: A toolbox for flexible protein-peptide docking. Bioinformatics 2019, 35, 4170–4172. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Grune, T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life 2008, 60, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Myers, N.; Moscovitz, O.; Sharon, M.; Prilusky, J.; Shaul, Y. Thermo-resistant intrinsically disordered proteins are efficient 20S proteasome substrates. Mol. Biosyst. 2012, 8, 368–373. [Google Scholar] [CrossRef]
- Balog, E.M.; Lockamy, E.L.; Thomas, D.D.; Ferrington, D.A. Site-specific methionine oxidation initiates calmodulin degradation by the 20S proteasome. Biochemistry 2009, 48, 3005–3016. [Google Scholar] [CrossRef]
- Available online: https://emboss.bioinformatics.nl/cgi-bin/emboss/epestfind (accessed on 1 August 2023).
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef]
- Njomen, E.; Tepe, J.J. Proteasome Activation as a New Therapeutic Approach to Target Proteotoxic Disorders. J. Med. Chem. 2019, 62, 6469–6481. [Google Scholar] [CrossRef]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files, Version 1.33; 2011. Available online: https://github.com/najoshi/sickle (accessed on 1 December 2016).
- Gordon, A.; Hannon, G.J. Fastx-Toolkit. FASTQ/A Short-Reads Pre-Processing Tools. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 1 December 2016).
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.K.; Chang, J.L.; Burke, D.H. FASTAptamer: A Bioinformatic Toolkit for High-throughput Sequence Analysis of Combinatorial Selections. Mol. Ther. Nucleic Acids 2015, 4, e230. [Google Scholar] [CrossRef] [PubMed]
- Moravec, R.A.; O’Brien, M.A.; Daily, W.J.; Scurria, M.A.; Bernad, L.; Riss, T.L. Cell-based bioluminescent assays for all three proteasome activities in a homogeneous format. Anal. Biochem. 2009, 387, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef]
- Yan, R.; Xu, D.; Yang, J.; Walker, S.; Zhang, Y. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci. Rep. 2013, 3, 2619. [Google Scholar] [CrossRef]
- Sahu, I.; Glickman, M.H. Structural Insights into Substrate Recognition and Processing by the 20S Proteasome. Biomolecules 2021, 11, 148. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | SDS (+) | SDS (−) | |
|---|---|---|---|---|
| IC50 ± SD (μM) | Maximum Active Concentration (μM) | Activity Increase (%) | ||
| 20S-BP1(1-17) | MARPSRLRHWWRLRRRV | 1.35 ± 0.21 | - | - |
| 20S-BP1(1-12) | MARPSRLRHWWR | 0.93 ± 0.30 | 10 | 1215 |
| 20S-BP1(6-17) | RLRHWWRLRRRV | >10 | - | - |
| 20S-BP1(1-11) | MARPSRLRHWW | 0.84 (±0.03) | 0.7 | 149 |
| 20S-BP1(1-10) | MARPSRLRHW | 2.31 (±0.41) | 6.7 | 197 |
| 20S-BP1(1-9) | MARPSRLRH | 7.46 (±1.11) | 66.7 | 310 |
| 20S-BP1(1-8) | MARPSRLR | 8.51 (±0.25) | 66.7 | 237 |
| 20S-BP1(1-7) | MARPSRL | >50 | 66.7 | 179 |
| 20S-BP1(1-6) | MARPSR | >50 | 0.7 | 107 |
| 20S-BP1(2-12) | ARPSRLRHWWR | 4.26 (±1.76) | 0.7 | 172 |
| 20S-BP1(3-12) | RPSRLRHWWR | >50 | 0.7 | 154 |
| 20S-BP1(4-12) | PSRLRHWWR | 0.83 (±0.11) | 2.0 | 163 |
| 20S-BP1(5-12) | SRLRHWWR | 0.82 (±0.11) | 2.0 | 207 |
| 20S-BP1(6-12) | RLRHWWR | 1.64 (±0.20) | 2.0 | 168 |
| 20S-BP1(7-12) | LRHWWR | 1.34 (±0.11) | 6.7 | 157 |
| [Ala1]20S-BP1(1-12) | AARPSRLRHWWR | 3.91 (±0.30) | - | - |
| [Ala3]20S-BP1(1-12) | MAAPSRLRHWWR | 1.10 (±0.01) | - | - |
| [Ala4]20S-BP1(1-12) | MARASRLRHWWR | 3.02 (±0.06) | - | - |
| [Ala5]20S-BP1(1-12) | MARPARLRHWWR | 5.46 (±0.14) | - | - |
| [Ala6]20S-BP1(1-12) | MARPSALRHWWR | 1.59 (±0.06) | - | - |
| [Ala7]20S-BP1(1-12) | MARPSRARHWWR | 5.09 (±0.13) | - | - |
| [Ala8]20S-BP1(1-12) | MARPSRLAHWWR | 1.00 (±0.08) | - | - |
| [Ala9]20S-BP1(1-12) | MARPSRLRAWWR | 2.53 (±0.07) | - | - |
| [Ala10]20S-BP1(1-12) | MARPSRLRHAWR | 2.54 (±0.19) | - | - |
| [Ala11]20S-BP1(1-12) | MARPSRLRHWAR | 2.30 (±0.07) | - | - |
| [Ala12]20S-BP1(1-12) | MARPSRLRHWWA | 0.72 (±0.02) | - | - |
| 20S-BP1(1-8)TYN | MARPSRLRTYN | 9.78 (±1.51) | 30.0 | 260 |
| 20S-BP1(1-8)LYL | MARPSRLRLYL | 2.51 (±0.18) | 1.0 | 183 |
| 20S-BP1(1-8)FYK | MARPSRLRFYK | 1.26 (±0.13) | 1.0 | 231 |
| 20S-BP1(1-8)KPV | MARPSRLRKPV | 14.7 (±0.56) | 3.0 | 313 |
| 20S-BP1(1-8)LWK | MARPSRLRLWK | 2.55 (±0.16) | 1.0 | 250 |
| 20S-BP1(1-8)YYA | MARPSRLRYYA | 3.80 (±0.81) | 10.0 | 467 |
| 20S-BP1(2-8)YYA | ARPSRLRYYA | 1.81 (±0.14) | 10.0 | 334 |
| 20S-BP1(3-8)YYA | RPSRLRYYA | 1.59 (±0.00) | 10.0 | 513 |
| 20S-BP1(4-8)YYA | PSRLRYYA | 9.92 (±0.25) | 53.0 | 668 |
| 20S-BP1(5-8)YYA | SRLRYYA | 14.7 (±0.83) | 100.0 | 481 |
| 20S-BP1(6-8)YYA | RLRYYA | 4.44 (±0.23) | 100.0 | 644 |
| 20S-BP1(7-8)YYA | LRYYA | >50 | N.D. | N.D. |
| 20S-BP1(1-8)YY | MARPSRLRYY | 2.64 (±0.28) | 1.2 | 204 |
| 20S-BP1(1-8)Y | MARPSRLRY | 3.71 (±0.80) | N.D. | N.D. |
| Rpt2-C | QEGTPEGLYL | >50 | N.D. | N.D. |
| Rpt5-C | KKKANLQYYA | 28.1 (±3.29) | 100.0 | 1046 |
| Rpt3-C | KDEQEHEFYK | >50 | N.D. | N.D. |
| AntP-20S-BP1(5-11) | RQIKWFQNRRMKWKKSRLRHWW | 1.34 (±0.11) | - | - |
| AntP-20S-BP1(5-8)YYA | RQIKWFQNRRMKWKKSRLRYYA | 0.79 (±0.05) | - | - |
| Antennapedia peptide | RQIKWFQNRRMKWKK | >50 | - | - |
| Name | Range | Target Mass (m/z) | Measured Mass (m/z) |
|---|---|---|---|
| α-Syn | 42–71 | 2881.5 | 2881.0 |
| 64–94 | 2826.5 | 2827.1 | |
| 121–129 | 1082.4 | 1083.3 | |
| 106–116 | 856.4 | 856.6 | |
| α-Syn-20S-BP1(1-12) | 122–142 + 20S-BP1(1-12) | 4095.8 | 4096.5 |
| 16–53 | 3754.0 | 3754.3 | |
| 122–142 + 20S-BP1(1-8) | 3430.5 | 3430.5 | |
| 129–142 + 20S-BP1(1-12) | 3277.5 | 3277.1 | |
| 133–142 + BP1(1-12) | 2833.3 | 2833.0 | |
| α-Syn-20S-BP1(1-8)YYA | 122–142 + 20S-BP1(1-8)YYA | 3826.6 | 3827.1 |
| 129–142 + 20S-BP1(1-8)YYA | 3008.3 | 3009.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Shigeyoshi, K.; Hayakawa, Y.; Fujiwara, S.; Kishida, M.; Ohki, H.; Horibe, T.; Shionyu, M.; Mizukami, T.; Hasegawa, M. Acceleration of Protein Degradation by 20S Proteasome-Binding Peptides Generated by In Vitro Artificial Evolution. Int. J. Mol. Sci. 2023, 24, 17486. https://doi.org/10.3390/ijms242417486
Zhu Y, Shigeyoshi K, Hayakawa Y, Fujiwara S, Kishida M, Ohki H, Horibe T, Shionyu M, Mizukami T, Hasegawa M. Acceleration of Protein Degradation by 20S Proteasome-Binding Peptides Generated by In Vitro Artificial Evolution. International Journal of Molecular Sciences. 2023; 24(24):17486. https://doi.org/10.3390/ijms242417486
Chicago/Turabian StyleZhu, Yunhao, Kaishin Shigeyoshi, Yumiko Hayakawa, Sae Fujiwara, Masamichi Kishida, Hitoshi Ohki, Tomohisa Horibe, Masafumi Shionyu, Tamio Mizukami, and Makoto Hasegawa. 2023. "Acceleration of Protein Degradation by 20S Proteasome-Binding Peptides Generated by In Vitro Artificial Evolution" International Journal of Molecular Sciences 24, no. 24: 17486. https://doi.org/10.3390/ijms242417486
APA StyleZhu, Y., Shigeyoshi, K., Hayakawa, Y., Fujiwara, S., Kishida, M., Ohki, H., Horibe, T., Shionyu, M., Mizukami, T., & Hasegawa, M. (2023). Acceleration of Protein Degradation by 20S Proteasome-Binding Peptides Generated by In Vitro Artificial Evolution. International Journal of Molecular Sciences, 24(24), 17486. https://doi.org/10.3390/ijms242417486