
De Novo Potent Peptide Nucleic Acid Antisense Oligomer Inhibitors Targeting SARS-CoV-2 RNA-Dependent RNA Polymerase via Structure-Guided Drug Design


Abstract
:
1. Introduction
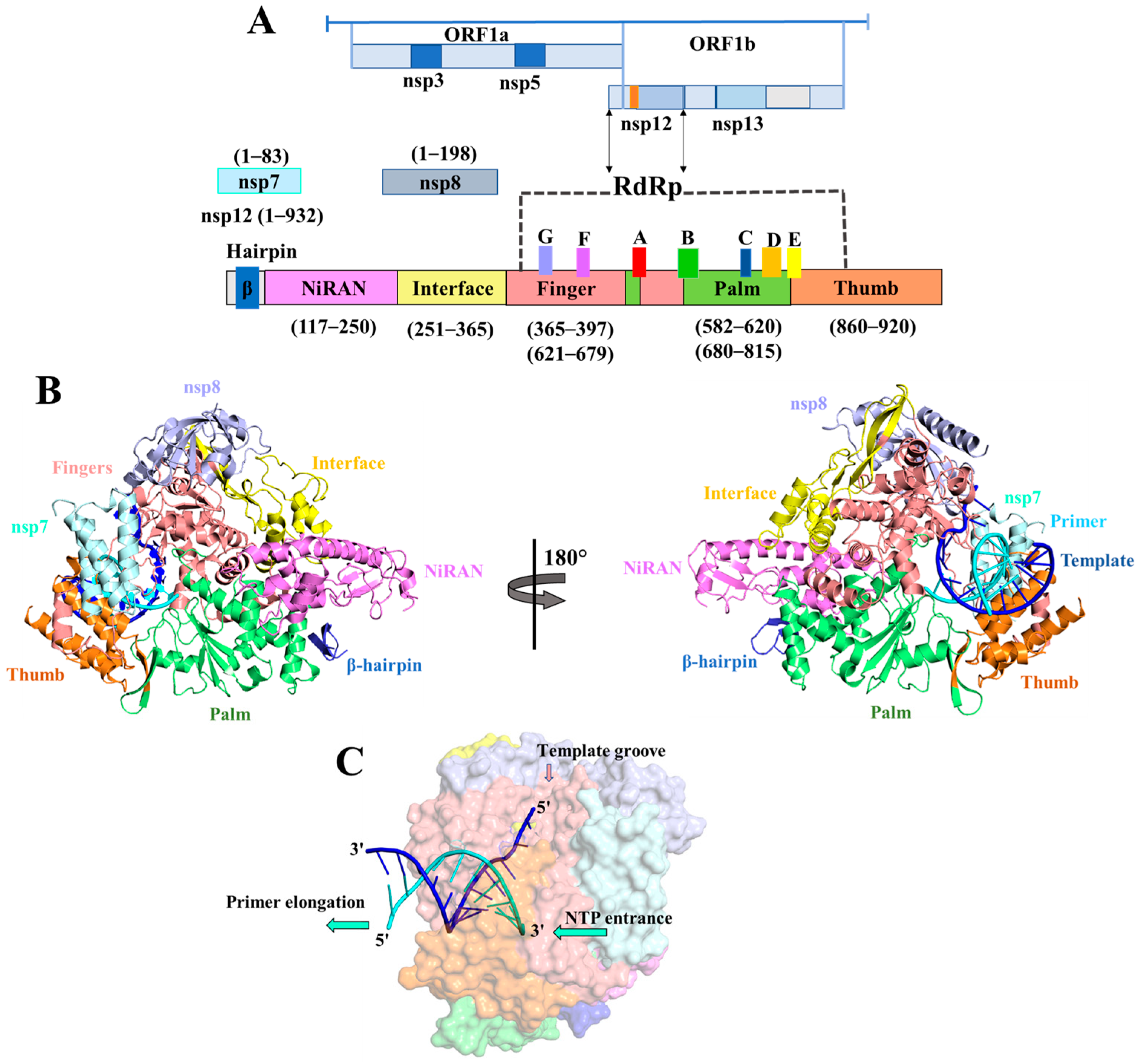
2. Results
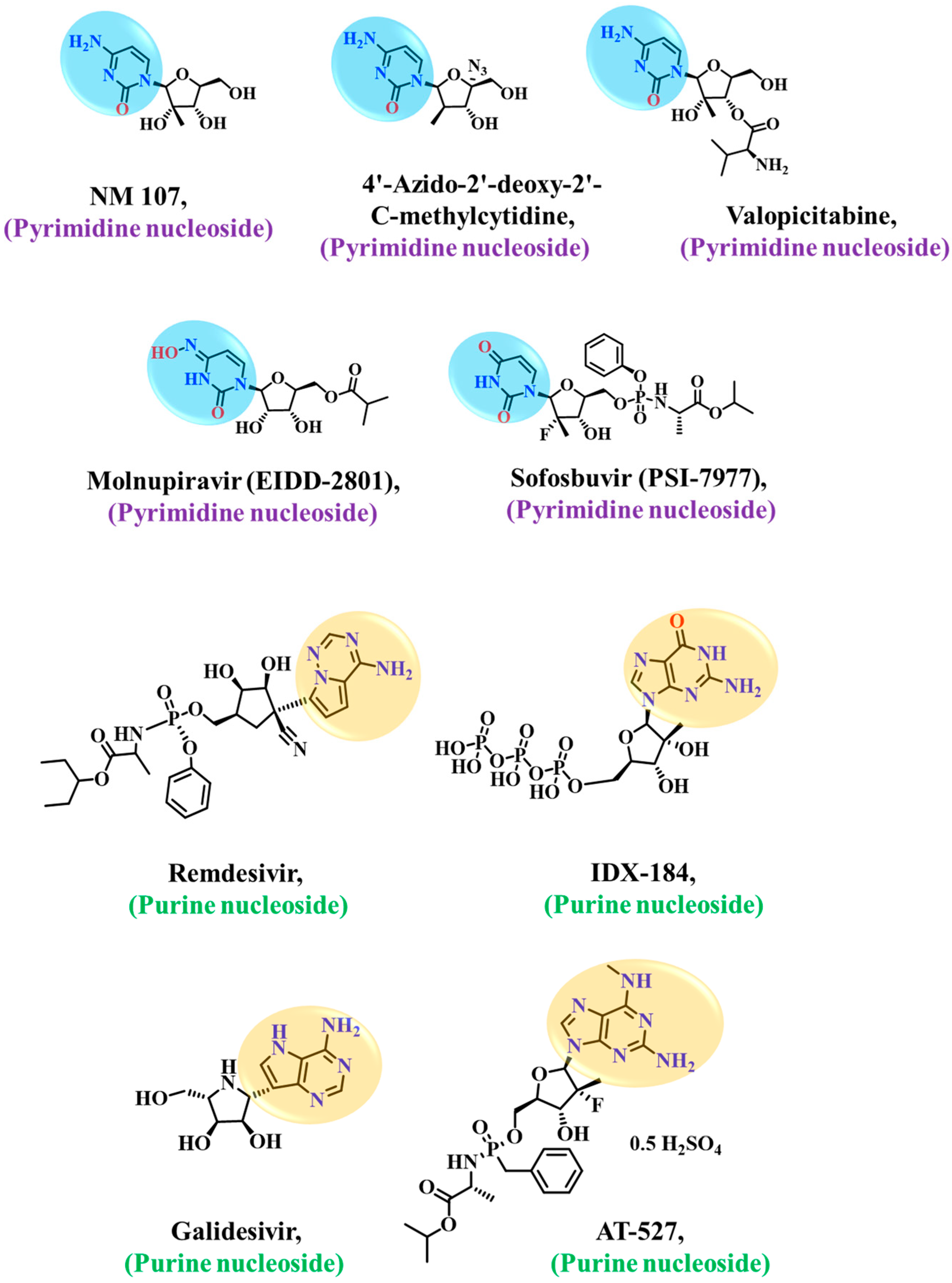
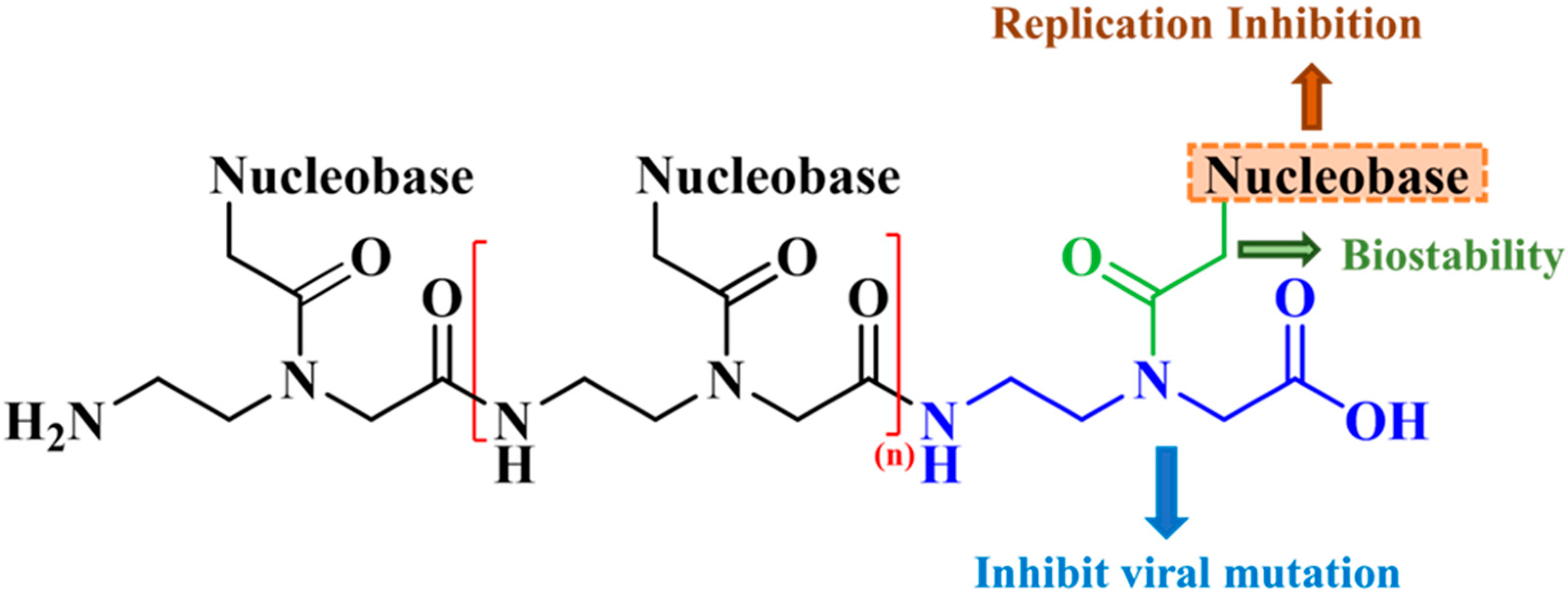
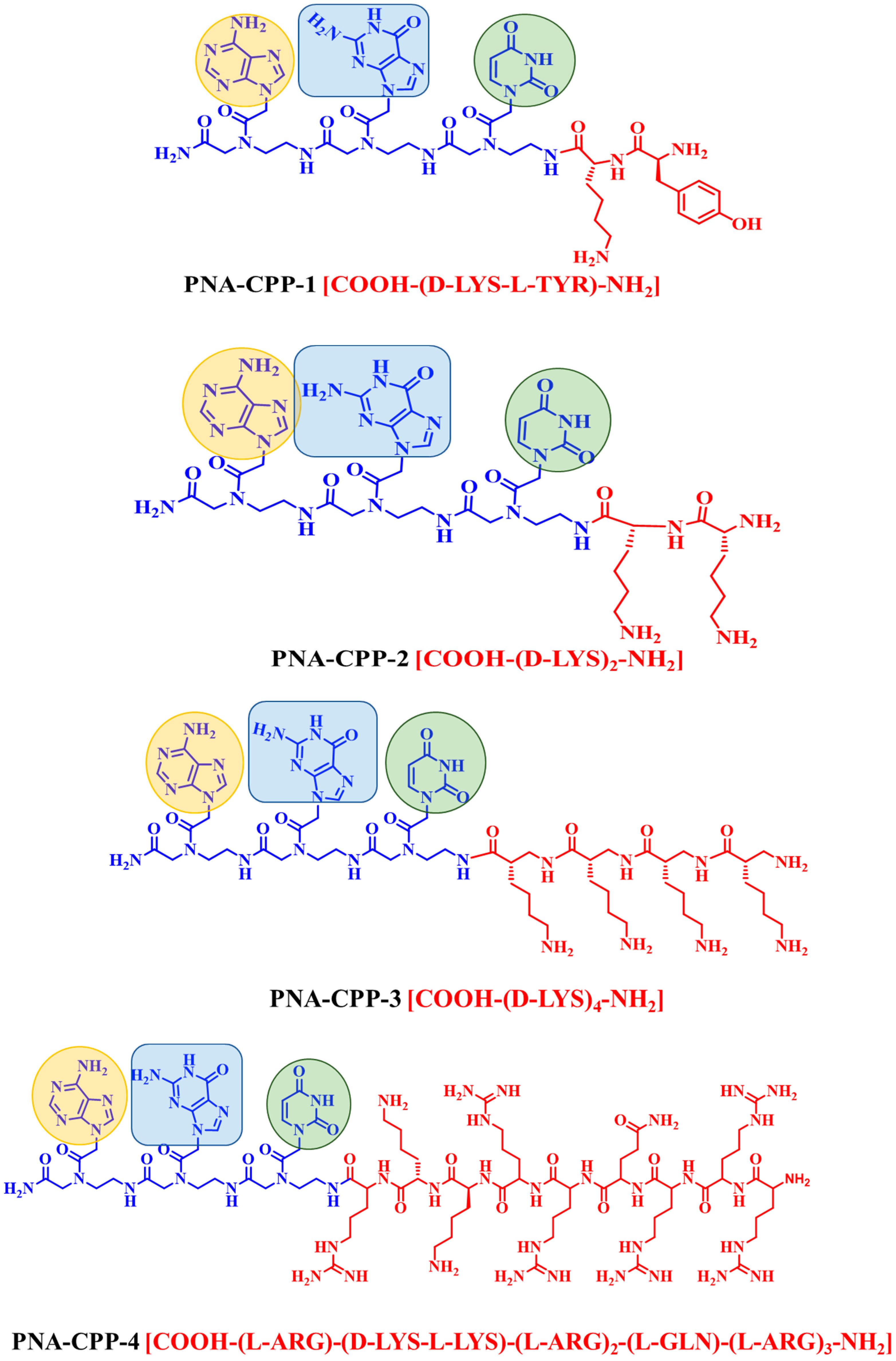
2.1. Design Strategy of PNA Analogs
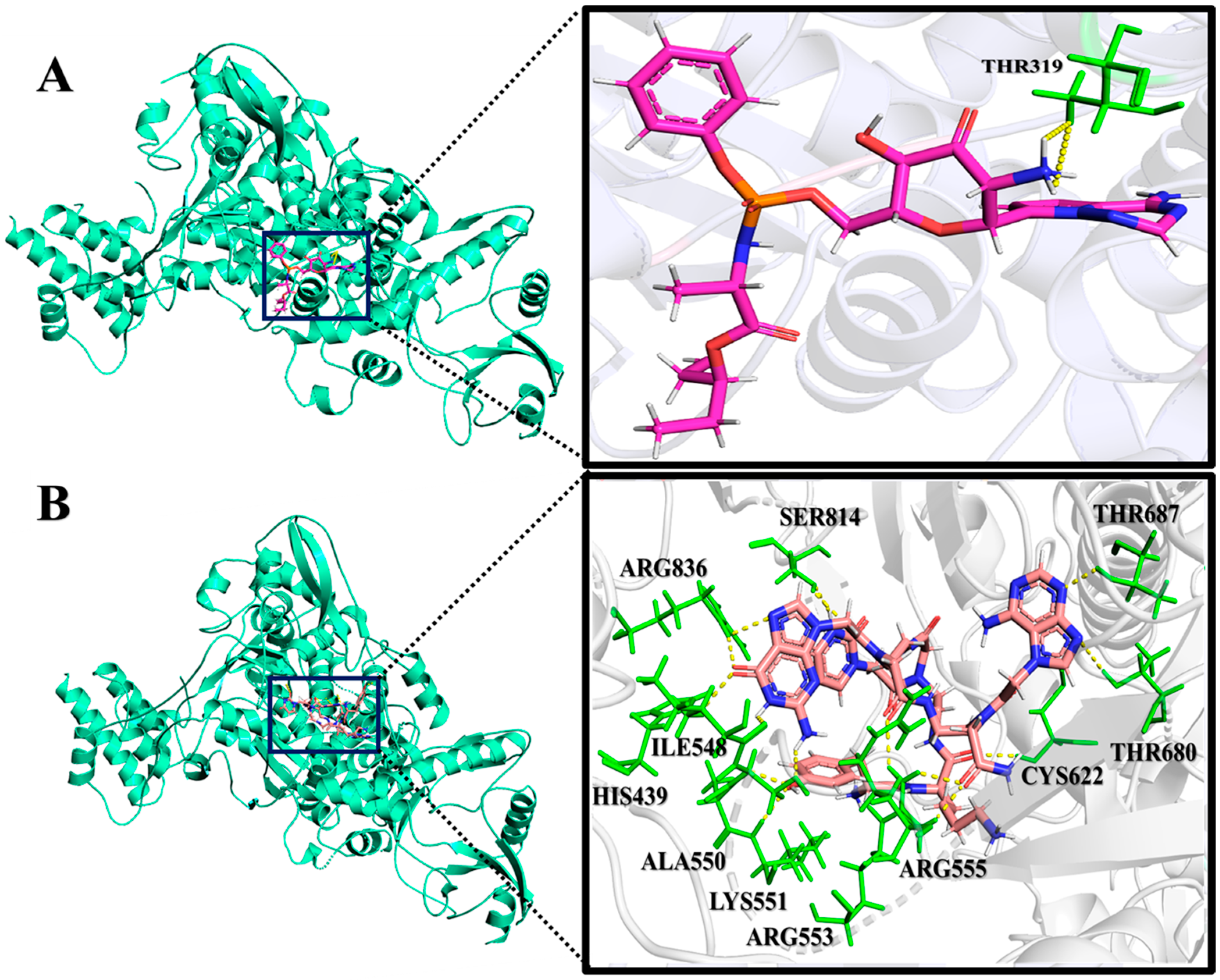
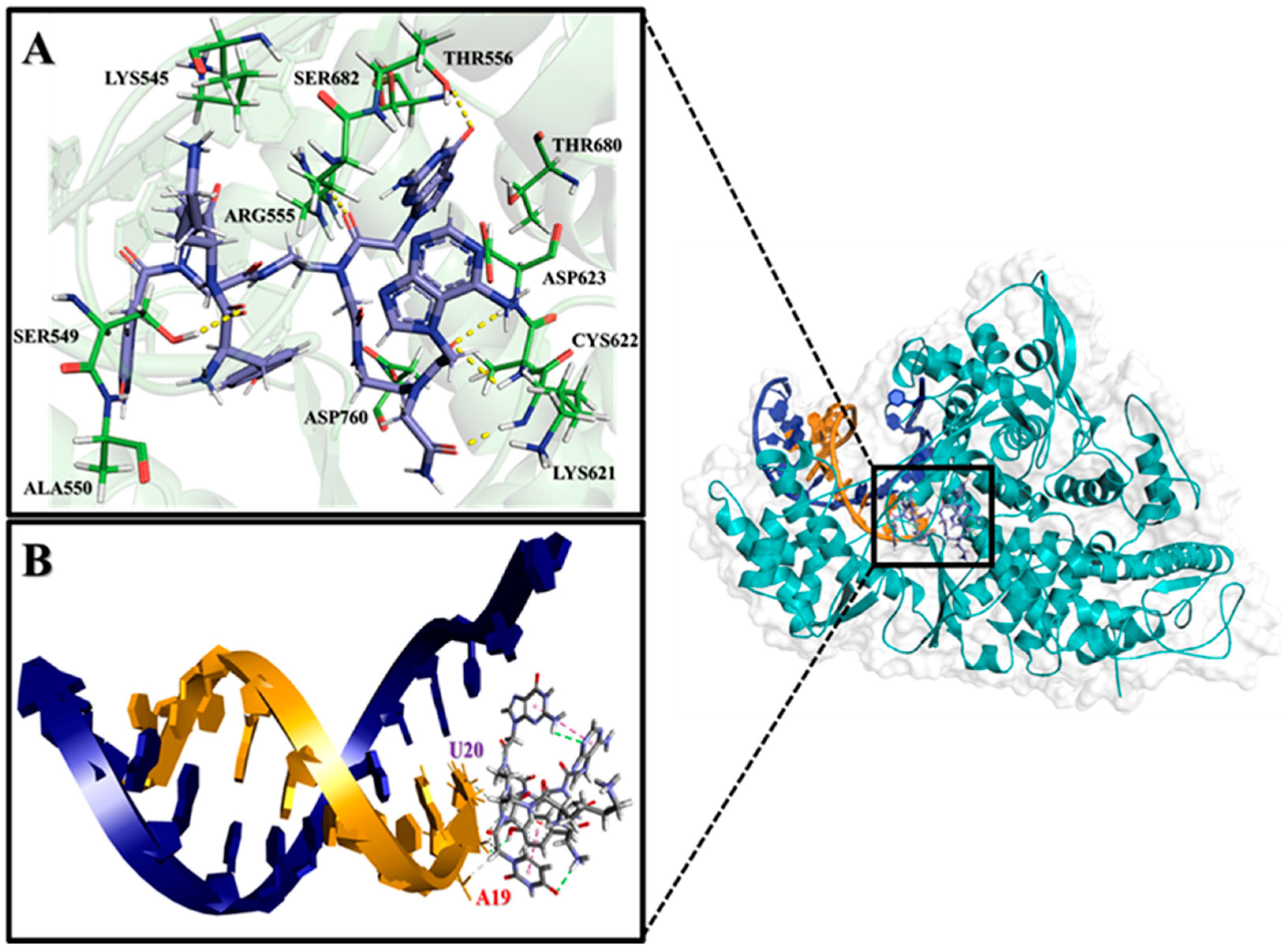
2.2. Analysis of Molecular Interactions by Molecular Docking
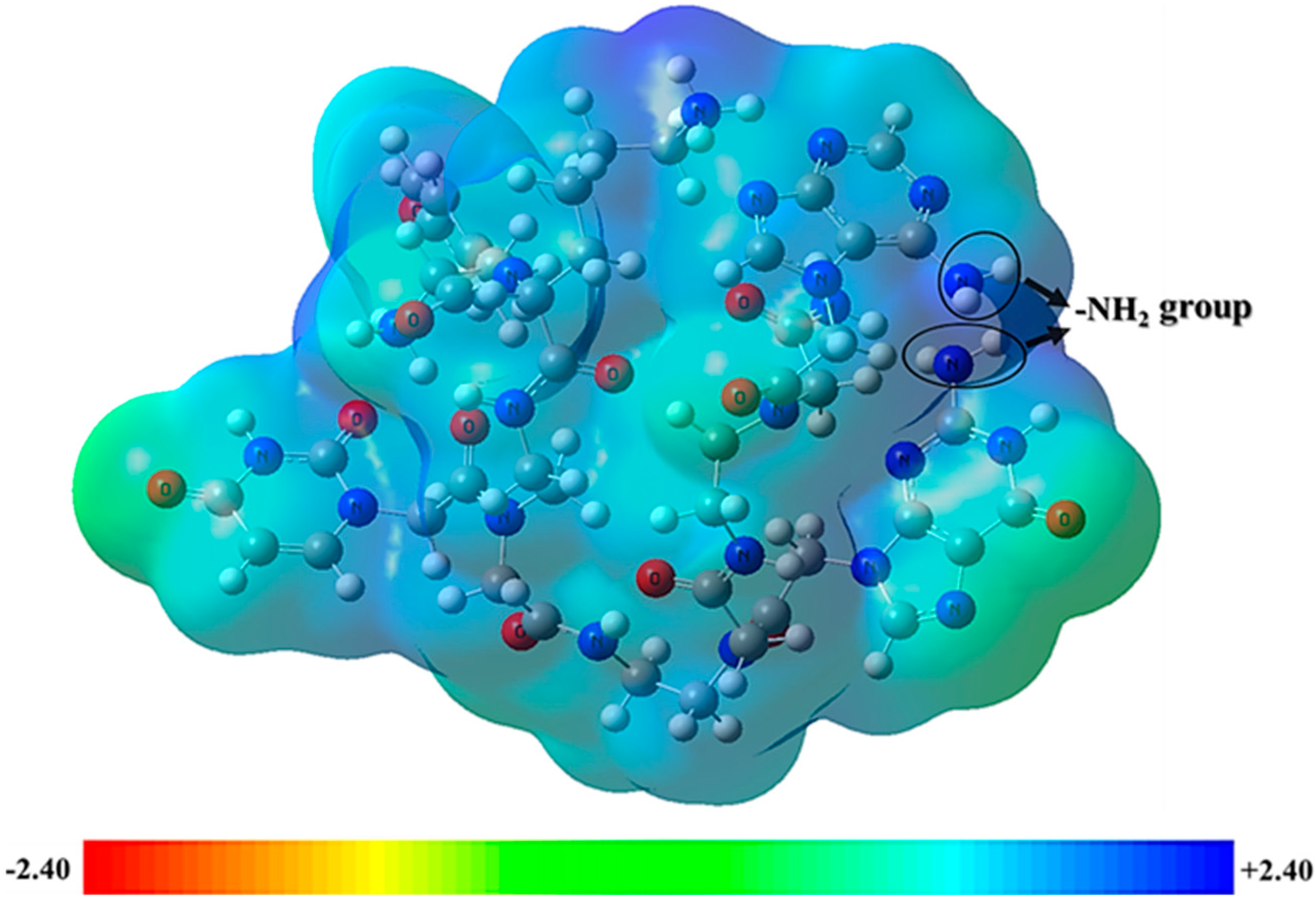
2.3. Frontier Molecular Orbitals and Molecular Electrostatic Potential Analysis
2.4. Global Chemical Descriptors (GCDs)
2.5. Analysis of Molecular Dynamics Simulation Trajectory
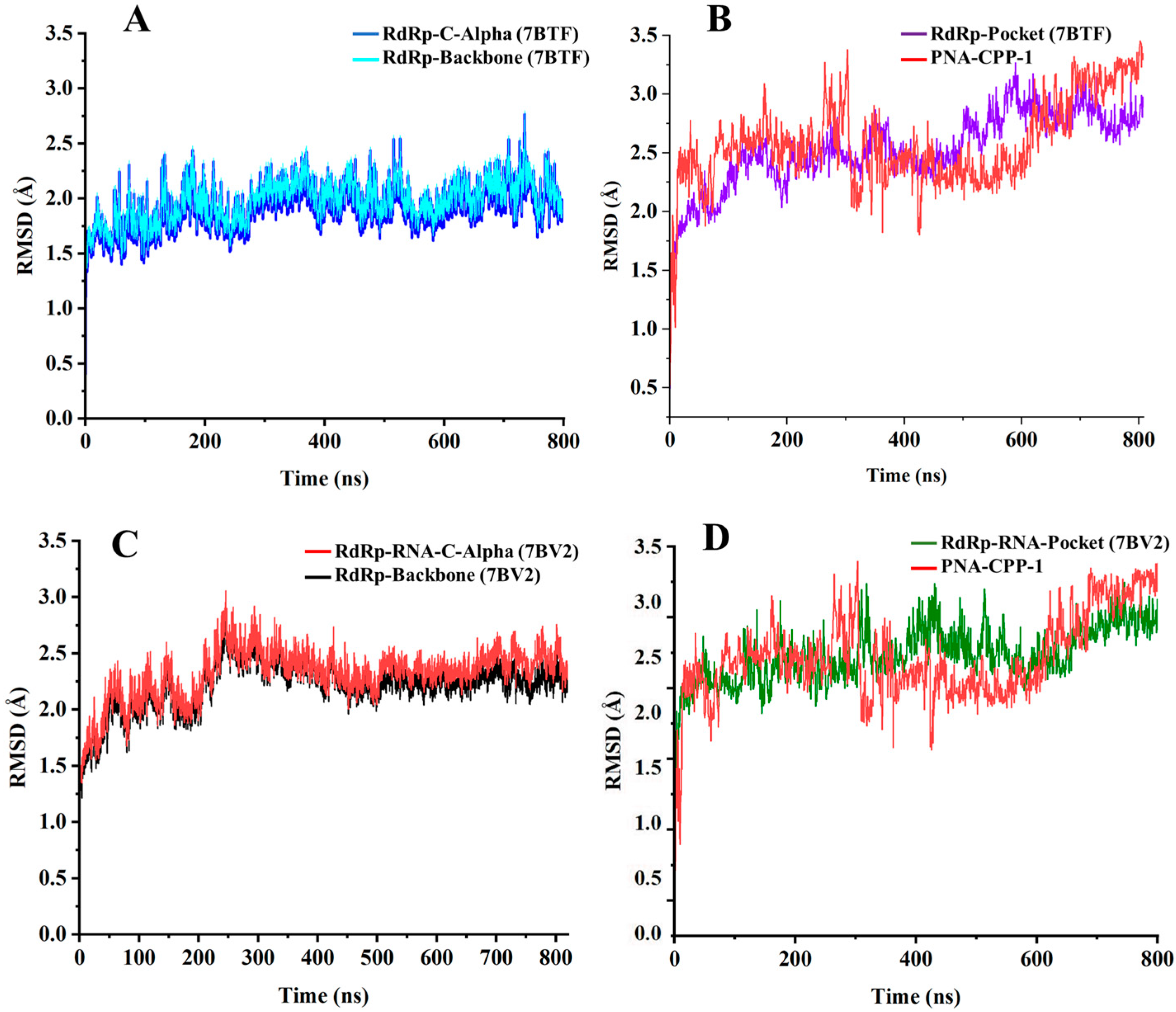
2.6. Stability Analysis by RMSD
2.7. Steered Molecular Dynamics Simulations
2.8. Radius of Gyration Analysis
2.9. Solvent-Accessible Surface Area (SASA) Analysis
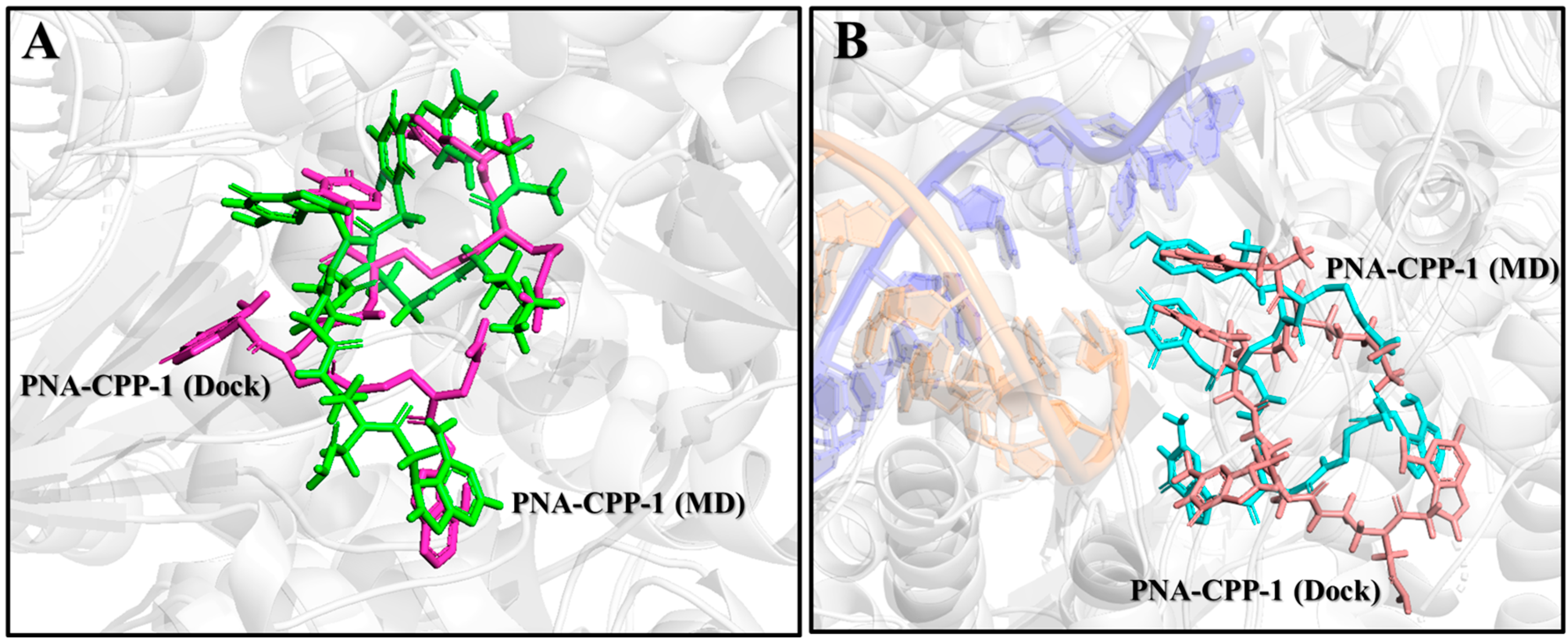
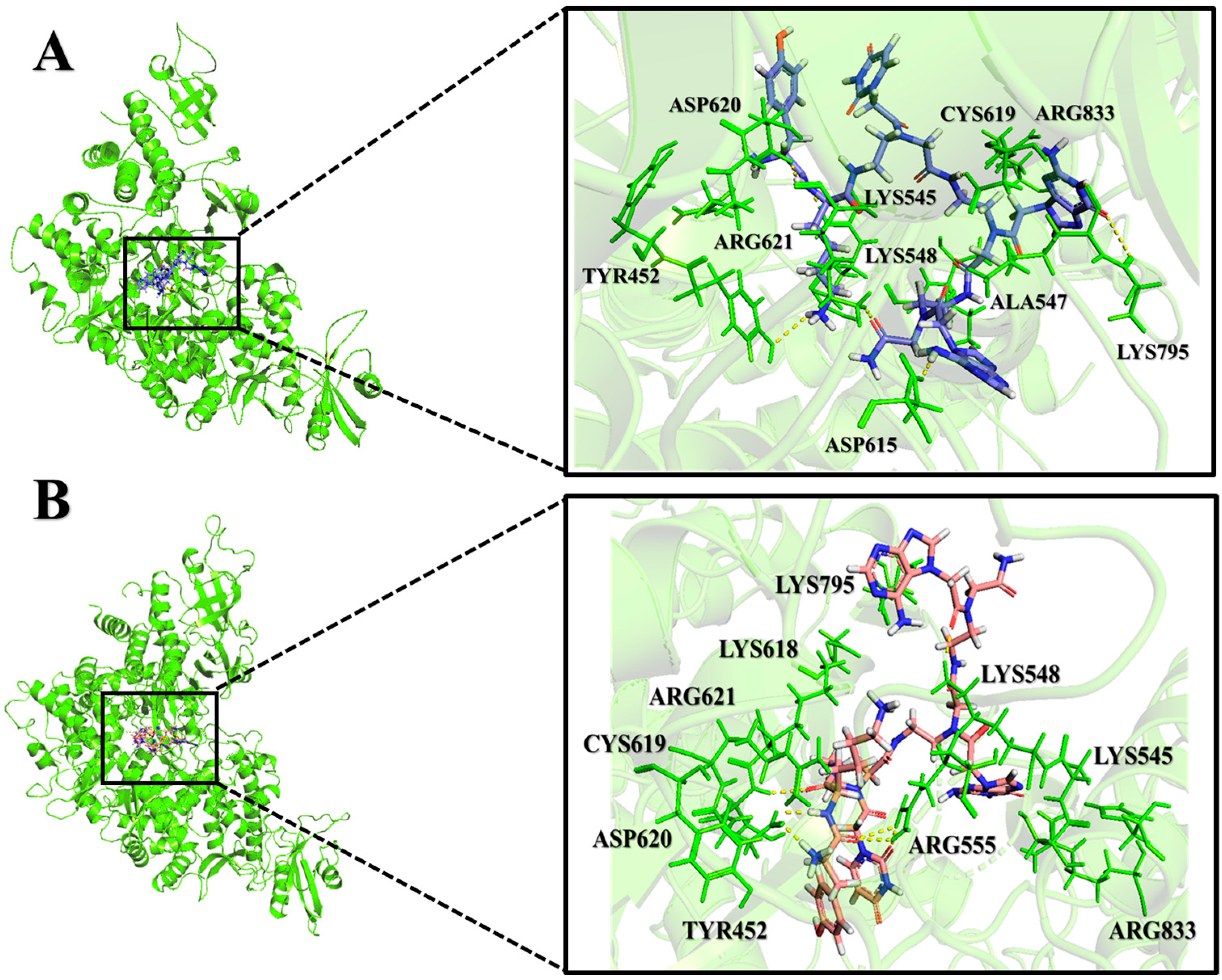
2.10. Comparison of PNA-CPP-1 Complex before and after MD Simulation
2.11. In-Silico Pharmacokinetic Analysis
2.12. Proposed Synthetic Route for PNA-CPP-1
3. Discussion
4. Materials and Methods
4.1. Preparation of Receptors and Ligands
4.2. Prediction of the Binding Site of the Receptor
4.3. Molecular Docking
4.4. Interaction Analysis and Visualization of the Receptor–Ligand Complex
4.5. Quantum Chemical Analysis
4.6. Molecular Dynamics Simulations
4.7. Steered Molecular Dynamics Simulations
4.8. Binding Free Energy Analysis
- ∆Gligand = total energy of the ligand in the solvent.
- ∆Gprotein = total energy of the protein in solvent.
- ∆GMM = molecular mechanics interaction energy, where the ∆Gelec. and ∆GVdW are the electrostatic and Van der Waals interactions, respectively.
- ∆GPB and ∆GSA represent polar solvation and non-polar solvation energy, respectively.
- T∆S (temperature = T and entropy = S) is the contribution of entropy to the free energy.
4.9. ADMET Study
5. Conclusions and Future Aspects
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PNA | Peptide Nucleic Acid |
| CPP | Cell Penetrating Peptide |
| DIPEA | N, N-Diisopropylethylamine |
| RdRp | RNA-dependent RNA-Polymerase |
| HOBt | Hydroxybenzotriazole |
| HATU | Hexafluorophosphate Azabenzotriazole Tetramethyl Uronium |
| NTP | Nucleotide Triphosphate |
References
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Allan, M.; Lièvre, M.; Laurenson-Schafer, H.; de Barros, S.; Jinnai, Y.; Andrews, S.; Stricker, T.; Formigo, J.P.; Schultz, C.; Perrocheau, A. The World Health Organization COVID-19 surveillance database. Int. J. Equit. Health 2022, 21 (Suppl. S3), 167. [Google Scholar] [CrossRef] [PubMed]
- Hegelund, M.H.; Fjordside, L.; Faurholt-Jepsen, D.; Christensen, D.L.; Bygbjerg, I.C. Opportunistic non-communicable diseases in times of COVID-19. APMIS 2023, 131, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Emergence, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; De Jonghe, S.; Jochmans, D.; et al. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. [Google Scholar] [CrossRef]
- Thakkar, R.; Agarwal, D.K.; Ranaweera, C.B.; Ishiguro, S.; Conda-Sheridan, M.; Gaudreault, N.N.; Comer, J. De novo design of a stapled peptide targeting SARS-CoV-2 spike protein receptor-binding domain. RSC Med. Chem. 2023, 14, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.-J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Gao, S.; Huang, T.; Song, L.; Xu, S.; Cheng, Y.; Cherukupalli, S.; Kang, D.; Zhao, T.; Sun, L.; Zhang, J.; et al. Medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors. Acta Pharm. Sin. B 2022, 12, 581–599. [Google Scholar] [CrossRef]
- Mousavi, S.; Zare, S.; Mirzaei, M.; Feizi, A. Novel drug design for treatment of COVID-19: A systematic review of preclinical studies. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 2044282. [Google Scholar] [CrossRef]
- Pundir, H.; Joshi, T.; Pant, M.; Bhat, S.; Pandey, J.; Chandra, S.; Tamta, S. Identification of SARS-CoV-2 RNA dependent RNA polymerase inhibitors using pharmacophore modelling, molecular docking and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2022, 40, 13366–13377. [Google Scholar] [CrossRef]
- Alanagreh, L.A.; Alzoughool, F.; Atoum, M. The human coronavirus disease COVID-19: Its origin, characteristics, and insights into potential drugs and its mechanisms. Pathogens 2020, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- de Farias, S.T.; Junior, A.P.D.S.; Rêgo, T.G.; José, M.V. Origin and evolution of RNA-dependent RNA polymerase. Front. Genet. 2017, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- White, K.A.; Enjuanes, L.; Berkhout, B. RNA Virus Replication, Transcription and Recombination; Taylor & Francis: Abingdon, UK, 2011; pp. 182–183. [Google Scholar]
- Venkataraman, S.; Prasad, B.V.; Selvarajan, R. RNA dependent RNA polymerases: Insights from structure, function and evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Orta, C.; Ferrero, D.; Verdaguer, N. RNA-dependent RNA polymerases of picornaviruses: From the structure to regulatory mechanisms. Viruses 2015, 7, 4438–4460. [Google Scholar] [CrossRef] [PubMed]
- Bartas, M.; Volná, A.; Beaudoin, C.A.; Poulsen, E.T.; Červeň, J.; Brázda, V.; Pečinka, P. Unheeded SARS-CoV-2 proteins? A deep look into negative-sense RNA. Brief. Bioinform. 2022, 23, bbac045. [Google Scholar] [CrossRef] [PubMed]
- Shehzadi, K.; Saba, A.; Yu, M.; Liang, J. Structure-Based Drug Design of RdRp Inhibitors against SARS-CoV-2. Top. Curr. Chem. 2023, 381, 1–53. [Google Scholar] [CrossRef]
- Te Velthuis, A.J. Common and unique features of viral RNA-dependent polymerases. Cell. Mol. Life Sci. 2014, 71, 4403–4420. [Google Scholar] [CrossRef]
- Raj, K.; Kaur, K.; Gupta, G.; Singh, S. Current understanding on molecular drug targets and emerging treatment strategy for novel coronavirus-19. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 1383–1402. [Google Scholar] [CrossRef]
- Jukič, M.; Janežič, D.; Bren, U. Potential novel thioether-amide or guanidine-linker class of SARS-CoV-2 virus RNA-dependent RNA polymerase inhibitors identified by high-throughput virtual screening coupled to free-energy calculations. Int. J. Mol. Sci. 2021, 22, 11143. [Google Scholar] [CrossRef]
- Tian, L.; Qiang, T.; Liang, C.; Ren, X.; Jia, M.; Zhang, J.; Li, J.; Wan, M.; YuWen, X.; Li, H. RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021, 213, 113201. [Google Scholar] [CrossRef]
- Li, Y.; Cao, L.; Li, G.; Cong, F.; Li, Y.; Sun, J.; Zhang, X. Remdesivir metabolite GS-441524 effectively inhibits SARS-CoV-2 infection in mouse models. J. Med. Chem. 2021, 65, 2785–2793. [Google Scholar] [CrossRef]
- Spinner, C.D.; Gottlieb, R.L.; Criner, G.J.; López, J.R.A.; Cattelan, A.M.; Viladomiu, A.S.; Ogbuagu, O.; Malhotra, P.; Mullane, K.M.; Castagna, A. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: A randomized clinical trial. JAMA 2020, 324, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.D.; Lye, D.C.; Hui, D.S.; Marks, K.M.; Bruno, R.; Montejano, R.; Spinner, C.D.; Galli, M.; Ahn, M.-Y.; Nahass, R.G. Remdesivir for 5 or 10 days in patients with severe COVID-19. N. Engl. J. Med. 2020, 383, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Dallocchio, R.; Dessì, A.; De Vito, A.; Delogu, G.; Serra, P.; Madeddu, G. Early combination treatment with existing HIV antivirals: An effective treatment for COVID-19? Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2435–2448. [Google Scholar] [PubMed]
- Nielsen, P.E. Peptide nucleic acids (PNA) in chemical biology and drug discovery. Chem. Biodivers. 2010, 7, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef]
- Ahn, D.-G.; Lee, W.; Choi, J.-K.; Kim, S.-J.; Plant, E.P.; Almazán, F.; Taylor, D.R.; Enjuanes, L.; Oh, J.-W. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antivir. Res. 2011, 91, 1–10. [Google Scholar] [CrossRef]
- Sahu, B.; Behera, S.K.; Das, R.; Dalvi, T.; Chowdhury, A.; Dewangan, B.; Shard, A. Design and in-silico screening of Peptide Nucleic Acid (PNA) inspired novel pronucleotide scaffolds targeting COVID-19. Curr. Comput.-Aided Drug Des. 2022, 18, 26–40. [Google Scholar] [CrossRef]
- Park, S.; Kim, S.H.; Dezhbord, M.; Lee, E.H.; Jeon, Y.; Jung, D.; Kim, K.H. Cell-permeable peptide nucleic acid antisense oligonucleotide platform targeting human betacoronaviruses. Front. Microbiol. 2023, 14, 1258091. [Google Scholar] [CrossRef]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Sada, M.; Saraya, T.; Ishii, H.; Okayama, K.; Hayashi, Y.; Tsugawa, T.; Nishina, A.; Murakami, K.; Kuroda, M.; Ryo, A. Detailed molecular interactions of favipiravir with SARS-CoV-2, SARS-CoV, MERS-CoV, and influenza virus polymerases in silico. Microorganisms 2020, 8, 1610. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.; Thai, N.Q.; Truong, D.T.; Li, M.S. Remdesivir strongly binds to both RNA-dependent RNA polymerase and main protease of SARS-CoV-2: Evidence from molecular simulations. J. Phys. Chem. B 2020, 124, 11337–11348. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Kang, T.; Ali, H.; Lai, D. Remdesivir strongly binds to RNA-dependent RNA polymerase, membrane protein, and main protease of SARS-CoV-2: Indication from molecular modeling and simulations. Front. Pharmacol. 2021, 12, 710778. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Solanki, K.; Li, Y.; Ma, Z.; Peppelenbosch, M.P.; Baig, M.S.; Pan, Q. Viral polymerase binding and broad-spectrum antiviral activity of molnupiravir against human seasonal coronaviruses. Virology 2021, 564, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Shannon, A.; Fattorini, V.; Sama, B.; Selisko, B.; Feracci, M.; Falcou, C.; Gauffre, P.; El Kazzi, P.; Delpal, A.; Decroly, E. A dual mechanism of action of AT-527 against SARS-CoV-2 polymerase. Nat. Commun. 2022, 13, 621. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Uengwetwanit, T.; Chutiwitoonchai, N.; Wichapong, K.; Karoonuthaisiri, N. Identification of novel SARS-CoV-2 RNA dependent RNA polymerase (RdRp) inhibitors: From in silico screening to experimentally validated inhibitory activity. Comput. Struct. Biotechnol. J. 2022, 20, 882–890. [Google Scholar] [CrossRef]
- Al Sheikh Ali, A.; Khan, D.; Naqvi, A.; Al-Blewi, F.F.; Rezki, N.; Aouad, M.R.; Hagar, M. Design, synthesis, molecular modeling, anticancer studies, and density functional theory calculations of 4-(1, 2, 4-Triazol-3-ylsulfanylmethyl)-1, 2, 3-triazole derivatives. ACS Omega 2020, 6, 301–316. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, M.; Ling, X.; Liu, Y.; Xue, Y.; An, L.; Gu, N.; Jin, M. Design, synthesis, quantum chemical studies and biological activity evaluation of pyrazole–benzimidazole derivatives as potent Aurora A/B kinase inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 3523–3530. [Google Scholar] [CrossRef]
- Kuruvilla, T.K.; Prasana, J.C.; Muthu, S.; George, J.; Mathew, S.A. Quantum mechanical and spectroscopic (FT-IR, FT-Raman) study, NBO analysis, HOMO-LUMO, first order hyperpolarizability and molecular docking study of methyl [(3R)-3-(2-methylphenoxy)-3-phenylpropyl] amine by density functional method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 188, 382–393. [Google Scholar] [CrossRef]
- Shafieyoon, P.; Mehdipour, E.; Mary, Y.S. Synthesis, characterization and biological investigation of glycine-based sulfonamide derivative and its complex: Vibration assignment, HOMO–LUMO analysis, MEP and molecular docking. J. Mol. Struct. 2019, 1181, 244–252. [Google Scholar] [CrossRef]
- Zia, M.; Muhammad, S.; Bibi, S.; Abbasi, S.W.; Al-Sehemi, A.G.; Chaudhary, A.R.; Bai, F.Q. Exploring the potential of novel phenolic compounds as potential therapeutic candidates against SARS-CoV-2, using quantum chemistry, molecular docking and dynamic studies. Bioorganic Med. Chem. Lett. 2021, 43, 128079. [Google Scholar] [CrossRef]
- Adole, V.A. Computational Chemistry Approach for the Investigation of Structural, Electronic, Chemical and Quantum Chemical Facets of Twelve Biginelli Adducts. Organomet. Chem. 2021, 1, 29–40. [Google Scholar]
- Mazola, Y.; Guirola, O.; Palomares, S.; Chinea, G.; Menéndez, C.; Hernández, L.; Musacchio, A. A comparative molecular dynamics study of thermophilic and mesophilic β-fructosidase enzymes. J. Mol. Model. 2015, 21, 228. [Google Scholar] [CrossRef]
- Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2021, 39, 2679–2692. [Google Scholar] [CrossRef]
- Marsh, J.A.; Teichmann, S.A. Relative solvent accessible surface area predicts protein conformational changes upon binding. Structure 2011, 19, 859–867. [Google Scholar] [CrossRef]
- Sahu, S.N.; Mishra, B.; Sahu, R.; Pattanayak, S.K. Molecular dynamics simulation perception study of the binding affinity performance for main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 2444–2459. [Google Scholar] [CrossRef]
- Wakchaure, P.D.; Ghosh, S.; Ganguly, B. Revealing the Inhibition Mechanism of RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2 by Remdesivir and Nucleotide Analogues: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2020, 124, 10641–10652. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, I.; Derr, R.; Zhang, G.; Moelijker, N.; Hendriks, G.; Østerlund, T. (Genotoxicity assessment of potentially mutagenic nucleoside analogues using ToxTracker®. Toxicol. Lett. 2022, 362, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Nabati, M.; Parsaee, H. Potential cardiotoxic effects of remdesivir on cardiovascular system: A literature review. Cardiovasc. Toxicol. 2022, 22, 268–272. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef]
- Aleem, A.; Mahadevaiah, G.; Shariff, N.; Kothadia, J.P. Hepatic manifestations of COVID-19 and effect of remdesivir on liver function in patients with COVID-19 illness. In Baylor University Medical Center Proceedings; Taylor & Francis: Abingdon, UK, 2021; Volume 34, pp. 473–477. [Google Scholar]
- Griffiths, S.K.; Campbell, J.P. Placental structure, function and drug transfer. Contin. Educ. Anaesth. Crit. Care Pain 2015, 15, 84–89. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Cao, D. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006, 34 (Suppl. S1), D302–D305. [Google Scholar] [CrossRef]
- Kumar, S.P.; Patel, C.N.; Rawal, R.M.; Pandya, H.A. Energetic contributions of amino acid residues and its cross-talk to delineate ligand-binding mechanism. Proteins Struct. Funct. Bioinform. 2020, 88, 1207–1225. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Kerwin, S.M. ChemBioOffice Ultra 2010 Suite; ACS Publications: Washington, DC, USA, 2010. [Google Scholar]
- Tian, W.; Chen, C.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins and beyond. Biophys. J. 2018, 114, 50a. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Raya, A.; Barrientos-Salcedo, C.; Rubio-Póo, C.; Soriano-Correa, C. Electronic structure evaluation through quantum chemical descriptors of 17β-aminoestrogens with an anticoagulant effect. Eur. J. Med. Chem. 2011, 46, 2463–2468. [Google Scholar] [CrossRef]
- Frisch, A. Gaussian 09W Reference; Gaussian, Inc.: Wallingford, CT, USA, 2009; Volume 470, 25p. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. Gauss View 6.0; 16; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Sundaraganesan, N.; Ilakiamani, S.; Dominic Joshua, B. FT-Raman and FT-IR spectra, ab initio and density functional studies of 2-amino-4,5-difluorobenzoic acid. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2007, 67, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Izadyar, M.; Khavani, M.; Housaindokht, M.R. Sensing Ability of Hybrid Cyclic Nanopeptides Based on Thiourea Cryptands for Different Ions, A Joint DFT-D3/MD Study. J. Phys. Chem. A 2017, 121, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Jasmine, N.J.; Muthiah, P.T.; Arunagiri, C.; Subashini, A. Vibrational spectra (experimental and theoretical), molecular structure, natural bond orbital, HOMO-LUMO energy, Mulliken charge and thermodynamic analysis of N’-hydroxy-pyrimidine-2-carboximidamide by DFT approach. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 144, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. In Protein Engineering: Methods and Protocols; Springer: New York, NY, USA, 2018; pp. 43–67. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Harrach, M.F.; Drossel, B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef] [PubMed]
- Tomarchio, R.; Patamia, V.; Zagni, C.; Crocetti, L.; Cilibrizzi, A.; Floresta, G.; Rescifina, A. Steered Molecular Dynamics Simulations Study on FABP4 Inhibitors. Molecules 2023, 28, 2731. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Im, W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Shenkin, P.S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The normal-mode entropy in the MM/PBSA method: Effect of system truncation, buffer region, and dielectric constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleobase Sequence | CPP | Compounds | Druggable Cavities | Binding Energy (Kcal/mol) | Ki Values (µmol) |
|---|---|---|---|---|---|
| AGU | COOH-KY-NH2 | PNA-CPP-1 | Cavity 1 | −8.03 | 1.29 |
| Cavity 2 | −7.96 | 1.45 | |||
| Cavity 3 | −8.29 | 0.82 | |||
| Cavity 4 | −9.19 | 0.18 | |||
| AGU | COOH-KK-NH2 | PNA-CPP-2 | Cavity 1 | −7.48 | 3.23 |
| Cavity 2 | −7.59 | 2.69 | |||
| Cavity 3 | −7.80 | 1.91 | |||
| Cavity 4 | −7.22 | 5.06 | |||
| AGU | COOH-dK4-NH2 | PNA-CPP-3 | Cavity 1 | −6.37 | 21.19 |
| Cavity 2 | −7.40 | 3.73 | |||
| Cavity 3 | −7.33 | 4.22 | |||
| Cavity 4 | −6.70 | 12.14 | |||
| AGU | COOH-RRRQRRKKR-NH2 | PNA-CPP-4 | Cavity 1 | −7.59 | 2.73 |
| Cavity 2 | −6.58 | 14.92 | |||
| Cavity 3 | −6.48 | 17.57 | |||
| Cavity 4 | −6.91 | 8.60 | |||
| AGUCG | COOH-KY-NH2 | PNA-CPP-5 | Cavity 1 | −8.70 | 0.41 |
| Cavity 2 | −8.13 | 1.29 | |||
| Cavity 3 | −8.29 | 0.82 | |||
| Cavity 4 | −8.21 | 0.95 | |||
| AGUCG | COOH-KK-NH2 | PNA-CPP-6 | Cavity 1 | −8.23 | 0.92 |
| Cavity 2 | −7.65 | 2.46 | |||
| Cavity 3 | −8.09 | 1.17 | |||
| Cavity 4 | −7.79 | 1.94 | |||
| AGUCG | COOH-RRRQRRKKR-NH2 | PNA-CPP-7 | Cavity 1 | −7.09 | 6.33 |
| Cavity 2 | −7.65 | 2.44 | |||
| Cavity 3 | −8.00 | 1.36 | |||
| Cavity 4 | −8.30 | 0.82 | |||
| AGUCG | COOH-dK4-NH2 | PNA-CPP-8 | Cavity 1 | −7.07 | 6.53 |
| Cavity 2 | −7.14 | 5.83 | |||
| Cavity 3 | −6.67 | 12.86 | |||
| Cavity 4 | −6.39 | 20.63 | |||
| Remdesivir (Reference drug) | Cavity 1 | −8.14 | 1.07 | ||
| Cavity 2 | −7.65 | 2.45 | |||
| Cavity 3 | −7.61 | 2.61 | |||
| Cavity 4 | −8.69 | 0.42 |
| Compound | Binding Energy (Kcal/mol) | Inhibition Constant (µmol) | Residues Involved in H-Bond Interaction |
|---|---|---|---|
| PNA-CPP-1 | −9.94 | 0.051 | SER549, ALA550, ARG555, ARG553, LYS621, CYC622, ASP623, THR680, SER682, ASN691, ASP760, THR556, SER549, LYS545, SER681 |
| Remdesivir | −7.92 | 1.54 | LYS545, SER682, ASN691, ARG553, ARG555 |
| Parameters | PNA-CPP-1 |
|---|---|
| EHOMO (eV) | −6.47 |
| ELUMO (eV) | −1.73 |
| ΔEL-H (eV) | 4.75 |
| Ionization Energy (I) (eV) | 6.47 |
| Electron Affinity (A) | 1.73 |
| Chemical Hardness (η) | 2.37 |
| Chemical potential (μ) | −4.10 |
| Electronegativity index (χ) | 4.10 |
| Electrophilicity index (ω) | 6.68 |
| Softness (S) (e−1V−1) | 0.42 |
| Properties | PNA-CPP-1 | Remdesivir |
|---|---|---|
| Formula | C63H74N22O14 | C27H35N6O8P |
| Fraction Csp3 | 0.35 | 0.48 |
| Num. H-bond acceptors | 20 | 12 |
| Num. H-bond donors | 12 | 4 |
| Molar Refractivity | 353.62 | 150.43 |
| Topological Polar Surface area, TPSA ([Å]2) | 519.07 | 213.36 |
| LogP | −1.81 | 2.06 |
| Water solubility (logS) (mol/L) | −3.87 | −3.05 |
| GI-absorption | moderate | moderate |
| P-gp substrate | Yes | Yes |
| P-glycoprotein I/II inhibitor | Yes | Yes |
| Log VDss (log L/Kg) | 0.76 | 1.86 |
| CYP2C19 inhibitor | No | No |
| CYP2C9 inhibitor | No | No |
| CYP2D6 substrate | No | No |
| CYP3A4 substrate | No | Yes |
| CYP2D6 Inhibitor | No | No |
| CYP3A4 Inhibitor | No | Yes |
| Rat Oral Acute Toxicity | moderate | moderate |
| Renal OCT2 substrate | No | No |
| Acute Toxicity Rule | 0 alert | 0 alert |
| Human hepatotoxicity (H-HT) | No | No |
| Skin sensitivity | No | No |
| Plasma protein binding (PPB) | 79.26% | 65.83% |
| Blood–brain barrier (BBB) permeability | No | No |
| SA score | 5.418 | 4.815 |
| AMES | No | Yes |
| PAINS | 0 alert | 0 alert |
| Brenk | 0 alert | 1 alert: Phosphorus |
| Bioavailability Score | 0.17 | 0.17 |
| Pfizer Rule | Accepted | Accepted |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shehzadi, K.; Yu, M.; Liang, J. De Novo Potent Peptide Nucleic Acid Antisense Oligomer Inhibitors Targeting SARS-CoV-2 RNA-Dependent RNA Polymerase via Structure-Guided Drug Design. Int. J. Mol. Sci. 2023, 24, 17473. https://doi.org/10.3390/ijms242417473
Shehzadi K, Yu M, Liang J. De Novo Potent Peptide Nucleic Acid Antisense Oligomer Inhibitors Targeting SARS-CoV-2 RNA-Dependent RNA Polymerase via Structure-Guided Drug Design. International Journal of Molecular Sciences. 2023; 24(24):17473. https://doi.org/10.3390/ijms242417473
Chicago/Turabian StyleShehzadi, Kiran, Mingjia Yu, and Jianhua Liang. 2023. "De Novo Potent Peptide Nucleic Acid Antisense Oligomer Inhibitors Targeting SARS-CoV-2 RNA-Dependent RNA Polymerase via Structure-Guided Drug Design" International Journal of Molecular Sciences 24, no. 24: 17473. https://doi.org/10.3390/ijms242417473
APA StyleShehzadi, K., Yu, M., & Liang, J. (2023). De Novo Potent Peptide Nucleic Acid Antisense Oligomer Inhibitors Targeting SARS-CoV-2 RNA-Dependent RNA Polymerase via Structure-Guided Drug Design. International Journal of Molecular Sciences, 24(24), 17473. https://doi.org/10.3390/ijms242417473