
A Transient π–π or Cation–π Interaction between Degron and Degrader Dual Residues: A Key Step for the Substrate Recognition and Discrimination in the Processive Degradation of SulA by ClpYQ (HslUV) Protease in Escherichia coli

Abstract
:
1. Introduction
2. Results
2.1. The Intracellular Degradation of Chromosomal RcsA and SulA by ClpYQ, ClpYY91FQ, ClpYY91WQ and ClpYY91HQ
2.2. The Distinct In Vitro MBP-SulA Degradation by ClpY, ClpYY91F, ClpYY91H or ClpYY91W in the Presence of ClpQ and ATP
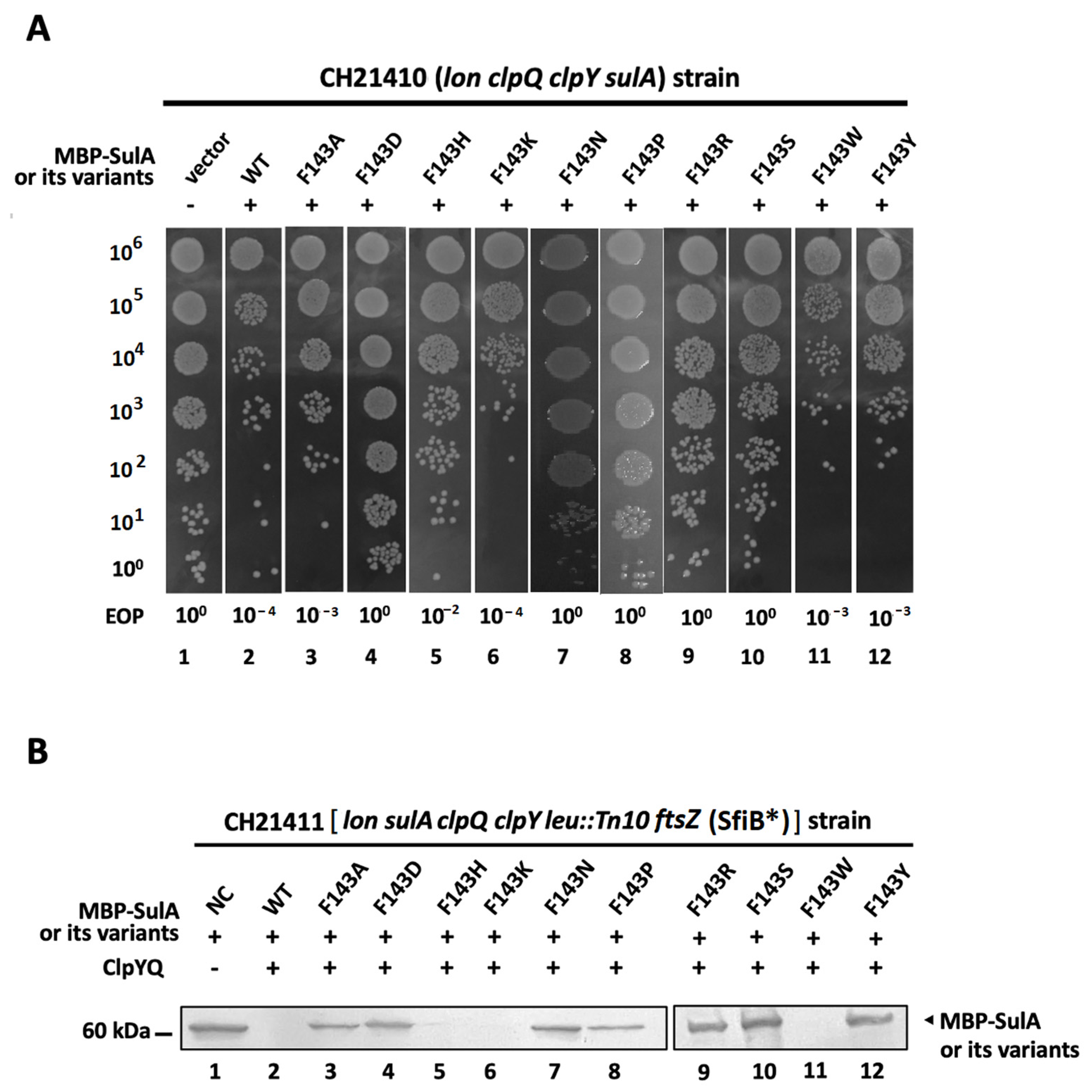
2.3. An Intracellular Degradation of MBP-SulA, SulAF143H, SulAF143K and SulAF143W by ClpYQ Protease
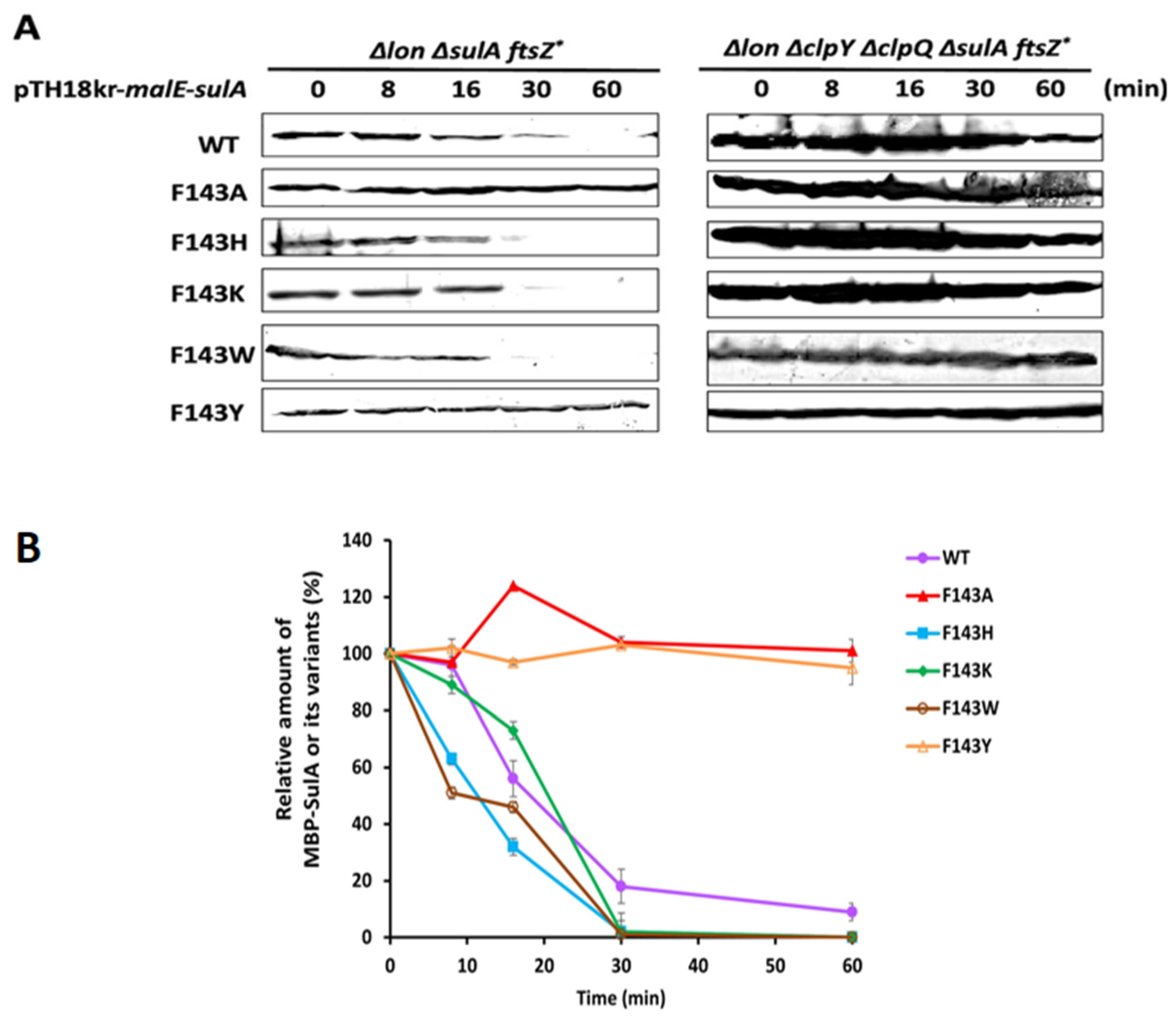
2.4. In Vivo Shorter Half-Life of MBP- SulA, SulAF143H, SulAF143K and SulAF143W Targeted by the Chromosomal ClpYQ Protease at the Higher Temperature
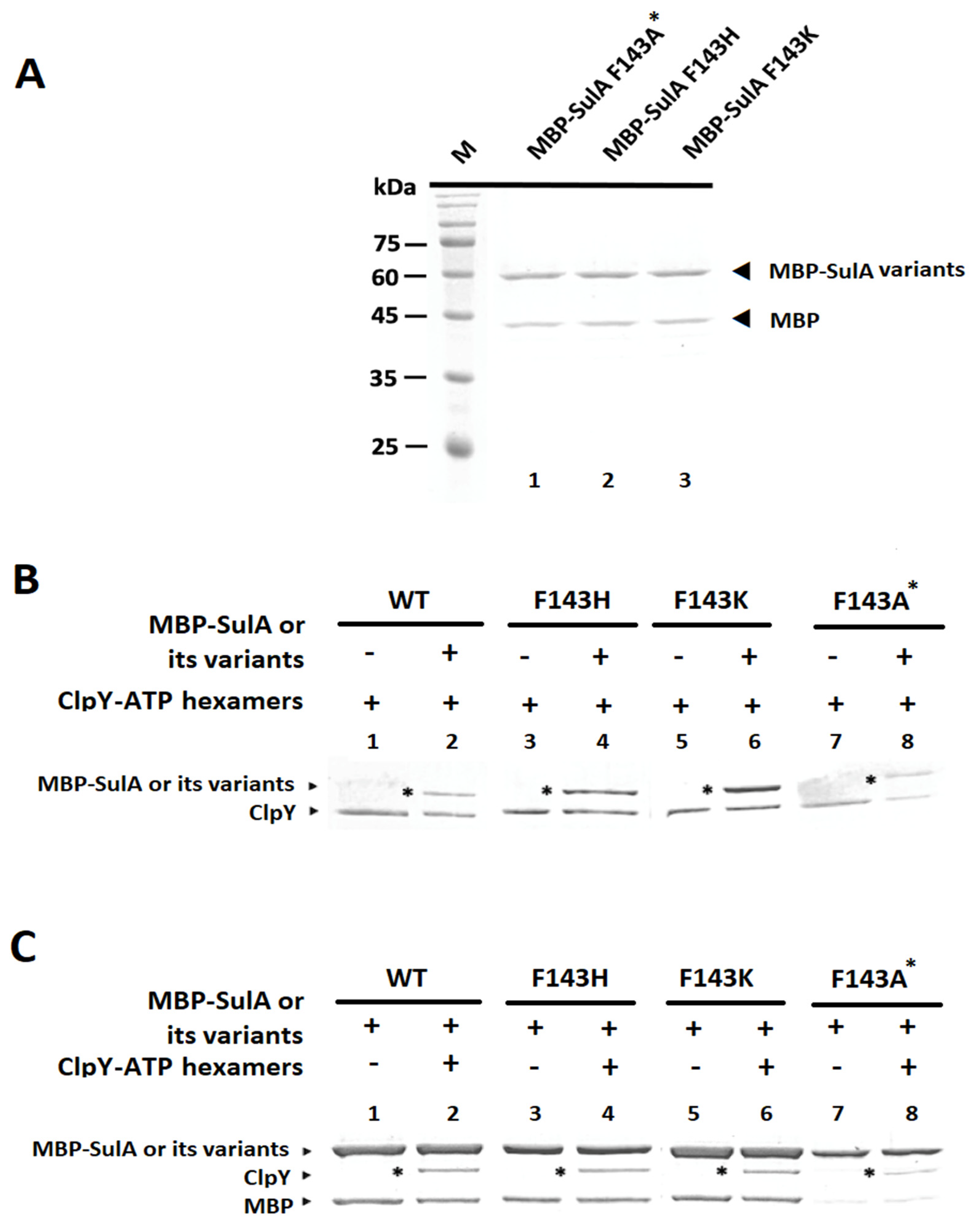
2.5. In Vitro Pull-Down Analyses between ClpY with MBP-SulA and Its Related Derivatives
2.6. In Vitro Degradation of MBP-SulA, MBP-SulAF143H and MBP-SulAF143K by ClpYQ, ClpYY91FQ and ClpYY91HQ
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids and Culture Media
4.2. Site-Directed Mutagenesis for ClpY and MBP-SulA Derivative Mutants
4.3. In Vivo Degradation Assays of MBP-SulA and Its Derivative Mutants
4.4. Protein Expression and Purification
4.5. In Vitro Degradation Assays of MBP-SulA and Its Derivatives by ClpY or ClpY Mutants in the Presence of ClpQ with ATP
4.6. In Vitro Pull-Down Assays of ClpY and MBP-SulA or SulA Variants
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gottesman, S. Proteolysis in bacterial regulatory circuits. Annu. Rev. Cell Dev. Biol. 2003, 19, 565–587. [Google Scholar] [CrossRef] [PubMed]
- Izert, M.A.; Klimecka, M.M.; Górna, M.W. Applications of bacterial degrons and degraders—Toward targeted protein degradation in bacteria. Front. Mol. Biosci. 2021, 8, 669762. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.E.; Burland, V.; Plunkett, G., 3rd; Daniels, D.L.; Blattner, F.R. Sequence analysis of four new heat-shock genes constituting the hslTS/ibpAB and hslVU operons in Escherichia coli. Gene 1993, 134, 1–6. [Google Scholar] [CrossRef]
- Missiakas, D.; Schwager, F.; Betton, J.M.; Georgopoulos, C.; Raina, S. Identification and characterization of HsIVHsIU (ClpQ ClpY) proteins involved in overall proteolysis of misfolded proteins in Escherichia coli. EMBO J. 1996, 15, 6899–6909. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.C.; Trame, C.B.; Tsuruta, H.; Wilbanks, S.M.; Reddy, V.S.; McKay, D.B. Crystal and solution structures of an HslUV protease-chaperone complex. Cell 2009, 103, 633–643. [Google Scholar] [CrossRef]
- Lien, H.Y.; Shy, R.S.; Peng, S.S.; Wu, Y.L.; Weng, Y.T.; Chen, H.H.; Su, P.C.; Ng, W.F.; Chen, Y.C.; Chang, P.Y.; et al. Characterization of the Escherichia coli ClpY (HslU) substrate recognition site in the ClpYQ (HslUV) protease using the yeast two-hybrid system. J. Bacteriol. 2009, 191, 4218–4231. [Google Scholar] [CrossRef]
- Ruiz-Gonzalez, M.X.; Marin, I. Proteasome-related HslU and HslV genes typical of eubacteria are widespread in eukaryotes. J. Mol. Evol. 2006, 63, 504–512. [Google Scholar] [CrossRef]
- Dong, S.; Chen, H.; Zhou, Q.; Liao, N. Protein degradation control and regulation of bacterial survival and pathogenicity: The role of protein degradation systems in bacteria. Mol. Biol. Rep. 2021, 48, 7575–7585. [Google Scholar] [CrossRef]
- Rohrwild, M.; Coux, O.; Huang, H.C.; Moerschell, R.P.; Yoo, S.J.; Seol, J.H.; Chung, C.H.; Goldberg, A.L. HslV-HslU: A novel ATP-dependent protease complex in Escherichia coli related to the eukaryotic proteasome. Proc. Natl. Acad. Sci. USA 1996, 93, 5808–5813. [Google Scholar] [CrossRef]
- Kessel, M.; Wu, W.F.; Gottesman, S.; Kocsis, E.; Steven, A.C.; Maurizi, M.R. Six-fold rotational symmetry of ClpQ, the E. coli homolog of the 20S proteasome, and its ATP-dependent activator, ClpY. FEBS Lett. 1996, 398, 274–278. [Google Scholar] [CrossRef]
- Rohrwild, M.; Pfeifer, G.; Santarius, U.; Muller, S.A.; Huang, H.C.; Engel, A.; Baumeister, W.; Goldberg, A.L. The ATP-dependent HslVU protease from Escherichia coli is a four-ring structure resembling the proteasome. Nat. Struct. Biol. 1997, 4, 133–139. [Google Scholar] [CrossRef]
- Bochtler, M.; Hartmann, C.; Song, H.K.; Bourenkov, G.P.; Bartunik, H.D.; Huber, R. The structure of HslU and the ATP-dependent protease HslU-HslV. Nature 2000, 403, 800–805. [Google Scholar] [CrossRef]
- Yoo, S.J.; Seol, J.H.; Shin, D.H.; Rohrwild, M.; Kang, M.S.; Tanaka, K.; Goldberg, A.L.; Chung, C.H. Purification and characterization of the heat shock proteins HslV and HslU that form a new ATP-dependent protease in Escherichia coli. J. Biol. Chem. 1996, 271, 14035–14040. [Google Scholar] [CrossRef]
- Burton, R.E.; Baker, T.A.; Sauer, R.T. Nucleotide-dependent substrate recognition by the AAA+ HslUV protease. Nat. Struct. Mol. Biol. 2005, 12, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Rho, Y.M.; Koh, O.J.; Ahn, S.W.; Seong, I.S.; Song, J.J.; Bang, O.; Seol, J.H.; Wang, J.; Eom, S.H.; et al. Role of the GYVG pore motif of HslU ATPase in protein unfolding and translocation for degradation by HslV peptidase. J. Biol. Chem. 2005, 280, 22892–22898. [Google Scholar] [CrossRef]
- Hsieh, F.C.; Chen, C.T.; Weng, Y.T.; Peng, S.S.; Chen, Y.C.; Huang, L.Y.; Hu, H.T.; Wu, Y.L.; Lin, N.C.; Wu, W.F. Stepwise activity of ClpY (HslU) mutants in the processive degradation of Escherichia coli ClpYQ (HslUV) protease substrates. J. Bacteriol. 2011, 193, 5465–5476. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, F.C.; Chang, L.K.; Tsai, C.H.; Kuan, J.E.; Wu, K.F.; Wu, C.; Wu, W.F. Roles of double-loop (130~159 aa and 175~209 aa) in ClpY(HslU)-I domain for SulA substrate degradation by ClpYQ(HslUV) protease in Escherichia coli. J. Gen. Appl. Microbiol. 2021, 66, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Koga, N.; Kameda, T.; Okazaki, K.; Takada, S. Paddling mechanism for the substrate translocation by AAA+ motor revealed by multiscale molecular simulations. Proc. Natl. Acad. Sci. USA 2009, 106, 18237–18242. [Google Scholar] [CrossRef]
- Kravats, A.; Jayasinghe, M.; Stan, G. Unfolding and translocation pathway of substrate protein controlled by structure in repetitive allosteric cycles of the ClpY ATPase. Proc. Natl. Acad. Sci. USA 2011, 108, 2234–2239. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Chang, C.F.; Kuo, C.L.; Chen, M.C.; Yu, C.H.; Lin, P.I.; Wu, W.F. Subunit oligomerization and substrate recognition of the Escherichia coli ClpYQ (HslUV) protease implicated by in vivo protein-protein interactions in the yeast two-hybrid system. J. Bacteriol. 2003, 185, 2393–2401. [Google Scholar] [CrossRef]
- Ramachandran, R.; Hartmann, C.; Song, H.K.; Huber, R.; Bochtler, M. Functional interactions of HslV (ClpQ) with the ATPase HslU (ClpY). Proc. Natl. Acad. Sci. USA 2002, 99, 7396–7401. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Lee, J.W.; Eom, S.H.; Seol, J.H.; Chung, C.H. Binding of MG132 or deletion of the Thr active sites in HslV subunits increases the affinity of HslV protease for HslU ATPase and makes this interaction nucleotide-independent. J. Biol. Chem. 2008, 283, 33258–33266. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, E.; Jeong, M.S.; Jeon, Y.J.; Eom, S.H.; Seol, J.H.; Chung, C.H. HslVU ATP-dependent protease utilizes maximally six among twelve threonine active sites during proteolysis. J. Biol. Chem. 2009, 284, 33475–33484. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Kay, L.E. Tracing an allosteric pathway regulating the activity of the HslV protease. Proc. Natl. Acad. Sci. USA 2014, 111, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Baytshtok, V.; Fei, X.; Shih, T.T.; Grant, R.A.; Santos, J.C.; Baker, T.A.; Sauer, R.T. Heat activates the AAA+ HslUV protease by melting an axial autoinhibitory plug. Cell Rep. 2021, 34, 108639. [Google Scholar] [CrossRef]
- Chang, C.Y.; Hu, H.T.; Tsai, C.H.; Wu, W.F. The degradation of RcsA by ClpYQ(HslUV) protease in Escherichia coli. Microbiol. Res. 2016, 184, 42–50. [Google Scholar] [CrossRef]
- Kanemori, M.; Nishihara, K.; Yanagi, H.; Yura, T. Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of σ32 and abnormal proteins in Escherichia coli. J. Bacteriol. 1997, 179, 7219–7225. [Google Scholar] [CrossRef]
- Kanemori, M.; Yanagi, H.; Yura, T. The ATP-dependent HslVU/ClpQY protease participates in turnover of cell division inhibitor SulA in Escherichia coli. J. Bacteriol. 1999, 181, 3674–3680. [Google Scholar] [CrossRef]
- Khattar, M.M. Overexpression of the hslVU operon suppresses SOS-mediated inhibition of cell division in Escherichia coli. FEBS Lett. 1997, 414, 402–404. [Google Scholar]
- Meng, J.; Young, G.; Chen, J. The Rcs system in Enterobacteriaceae: Envelope stress responses and virulence regulation. Front. Microbiol. 2021, 12, 627104. [Google Scholar] [CrossRef]
- Lau-Wong, I.C.; Locke, T.; Ellison, M.J.; Raivio, T.L.; Frost, L.S. Activation of the Cpx regulon destabilizes the F plasmid transfer activator, TraJ, via the HslVU protease in Escherichia coli. Mol. Microbiol. 2008, 67, 516–527. [Google Scholar] [CrossRef]
- Liang, W.; Deutscher, M.P. Transfer-messenger RNA-SmpB protein regulates ribonuclease R turnover by promoting binding of HslUV and Lon proteases. J. Biol. Chem. 2012, 287, 33472–33479. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Ho, Y.H.; Sung, T.C.; Wu, W.F.; Chen, C.S. Escherichia coli proteome microarrays identified the substrates of ClpYQ protease. Mol. Cell. Proteom. 2017, 16, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-F.; Zhou, Y.N.; Gottesman, S. Redundant in vivo proteolytic activities of Escherichia coli Lon and the ClpYQ (HslUV) protease. J. Bacteriol. 1999, 181, 3681–3687. [Google Scholar] [CrossRef] [PubMed]
- Huisman, O.; D’Ari, R. An inducible DNA-replication-cell division coupling mechanism in E. coli. Nature 1981, 290, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Huisman, O.; D’Ari, R.; Gottesman, S. Cell division control in Escherichia coli: Specific induction of the SOS SfiA protein is sufficient to block septation. Proc. Natl. Acad. Sci. USA 1984, 81, 4490–4494. [Google Scholar] [CrossRef] [PubMed]
- Cordell, S.C.; Robinson, E.J.; Lowe, J. Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proc. Natl. Acad. Sci. USA 2003, 100, 7889–7894. [Google Scholar] [CrossRef]
- Chang, C.Y.; Weng, Y.T.; Hwang, L.Y.; Hu, H.T.; Shih, P.S.; Kuan, J.E.; Wu, K.F.; Wu, W.F. Specific regions of the SulA protein recognized and degraded by the ATP-dependent ClpYQ (HslUV) protease in Escherichia coli. Microbiol. Res. 2019, 220, 21–31. [Google Scholar] [CrossRef]
- Seong, I.S.; Oh, J.Y.; Lee, J.W.; Tanaka, K.; Chung, C.H. The HslU ATPase acts as a molecular chaperone in prevention of aggregation of SulA, an inhibitor of cell division in Escherichia coli. FEBS Lett. 2000, 477, 224–229. [Google Scholar] [CrossRef]
- Paik, D.; Lee, H.; Kim, H.; Choi, J.M. Thermodynamics of π–π interactions of benzene and phenol in water. Int. J. Mol. Sci. 2022, 23, 9811. [Google Scholar] [CrossRef]
- Infield, D.T.; Rasouli, A.; Galles, G.D.; Chipot, C.; Tajkhorshid, E.; Ahern, C.A. Cation-π interactions and their functional roles in membrane proteins. J. Mol. Biol. 2021, 433, 167035. [Google Scholar] [CrossRef] [PubMed]
- Thakuria, R.; Nath, N.K.; Saha, B.K. The nature and applications of π–π interactions: A perspective. Cryst. Growth Des. 2019, 19, 523–528. [Google Scholar] [CrossRef]
- Daze, K.D.; Hof, F. The cation-π interaction at protein-protein interaction interfaces: Developing and learning from synthetic mimics of proteins that bind methylated lysines. Acc. Chem. Res. 2013, 46, 937–945. [Google Scholar] [CrossRef]
- Kumar, K.; Woo, S.M.; Siu, T.; Cortopassi, W.A.; Duarte, F.; Paton, R.S. Cation-π interactions in protein-ligand binding: Theory and data-mining reveal different roles for lysine and arginine. Chem. Sci. 2018, 9, 2655–2665. [Google Scholar] [CrossRef] [PubMed]
- Vernon, R.M.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. eLife 2018, 7, e31486. [Google Scholar] [CrossRef] [PubMed]
- Neel, A.J.; Hilton, M.J.; Sigman, M.S.; Toste, F.D. Exploiting non-covalent π interactions for catalyst design. Nature 2017, 543, 637–646. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef]
- Wilbaux, M.; Mine, N.; Guerout, A.M.; Mazel, D.; Van Melderen, L. Functional interactions between coexisting toxin-antitoxin systems of the ccd family in Escherichia coli O157:H7. J. Bacteriol. 2007, 189, 2712–2719. [Google Scholar] [CrossRef]
- Guzman, L.-M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef]
- Hashimoto-Gotoh, T.; Yamaguchi, M.; Yasojima, K.; Tsujimura, A.; Wakabayashi, Y.; Watanabe, Y. A set of temperature sensitive-replication/-segregation and temperature resistant plasmid vectors with different copy numbers and in an isogenic background (chloramphenicol, kanamycin, lacZ, repA, par, polA). Gene 2000, 241, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1995, 158, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, J.P.; Dougherty, D.A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.M.; Du, Q.S.; Meng, J.Z.; Pang, Z.W.; Huang, R.B. The multiple roles of histidine in protein interactions. Chem. Cent. J. 2013, 7, 44. [Google Scholar] [CrossRef]
- Higashitani, A.; Ishii, Y.; Kato, Y.; Horiuchi, K. Fuctional dissection of a cell-division inhibitor, SulA, of Escherichia coli and its negative regulation by Lon. Mol. Gen. Genet. 1997, 254, 351–357. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ. Available online: https://imagej.nih.gov/ij/ (accessed on 1 July 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Description | Source and Reference |
|---|---|---|
| E. coli strains | ||
| BW25113 | lacIq rrnB3 ΔlacZ4787 hsdR514 Δ(araBAD)567 Δ(rhaBAD)568 rph-1 | [47] |
| JW0941 | BW25113 ΔsulA:Kan | [48] |
| JW3902 | BW25113 ΔclpY:Kan | [48] |
| SG22623 | lon cpsB-lacZ Δara mal::lacIQ | [49] |
| CH21408 | lon cpsB-lacZ Δara mal::lacIQ sulA | This study |
| CH21409 | lon cpsB-lacZ Δara mal::lacIQ sulA leu::Tn10 ftsZ(SfiB*) | This study |
| CH21410 | lon cpsB-lacZ Δara mal::lacIQ clpQ clpY sulA | This study |
| CH21411 | lon cpsB-lacZ Δara mal::lacIQ clpQ clpY sulA leu::Tn10 ftsZ(SfiB*) | This study |
| Plasmids | ||
| pBAD24 | ori(pBR322) araC PBADampr | [50] |
| pBAD33 | ori(pACYC184) araC PBADcmr | [50] |
| pTH18kr | ori(pSC101) Plackanr | [51] |
| pET21a (+) | ori(pBR322) T7 promoter ampr | Novagen |
| pMAL-c2X | ori(pBR322) Ptacampr | NEB |
| pKD4 | Ap Km, kan cassette template | [47] |
| pKD46 | exo, bet, gam ParaB repA101ts oriR101 ampr | [47] |
| pCP20 | FLP+ λI1857+ λPR Repts ampr cmr | [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-H.; Tsai, C.-H.; Chou, C.-C.; Wu, W.-F. A Transient π–π or Cation–π Interaction between Degron and Degrader Dual Residues: A Key Step for the Substrate Recognition and Discrimination in the Processive Degradation of SulA by ClpYQ (HslUV) Protease in Escherichia coli. Int. J. Mol. Sci. 2023, 24, 17353. https://doi.org/10.3390/ijms242417353
Lin C-H, Tsai C-H, Chou C-C, Wu W-F. A Transient π–π or Cation–π Interaction between Degron and Degrader Dual Residues: A Key Step for the Substrate Recognition and Discrimination in the Processive Degradation of SulA by ClpYQ (HslUV) Protease in Escherichia coli. International Journal of Molecular Sciences. 2023; 24(24):17353. https://doi.org/10.3390/ijms242417353
Chicago/Turabian StyleLin, Chu-Hsuan, Chih-Hsuan Tsai, Chun-Chi Chou, and Whei-Fen Wu. 2023. "A Transient π–π or Cation–π Interaction between Degron and Degrader Dual Residues: A Key Step for the Substrate Recognition and Discrimination in the Processive Degradation of SulA by ClpYQ (HslUV) Protease in Escherichia coli" International Journal of Molecular Sciences 24, no. 24: 17353. https://doi.org/10.3390/ijms242417353
APA StyleLin, C.-H., Tsai, C.-H., Chou, C.-C., & Wu, W.-F. (2023). A Transient π–π or Cation–π Interaction between Degron and Degrader Dual Residues: A Key Step for the Substrate Recognition and Discrimination in the Processive Degradation of SulA by ClpYQ (HslUV) Protease in Escherichia coli. International Journal of Molecular Sciences, 24(24), 17353. https://doi.org/10.3390/ijms242417353