
Comparative Application of Terminal Restriction Fragment Analysis Tools to Large-Scale Genomic Assays

 ,
,
Abstract
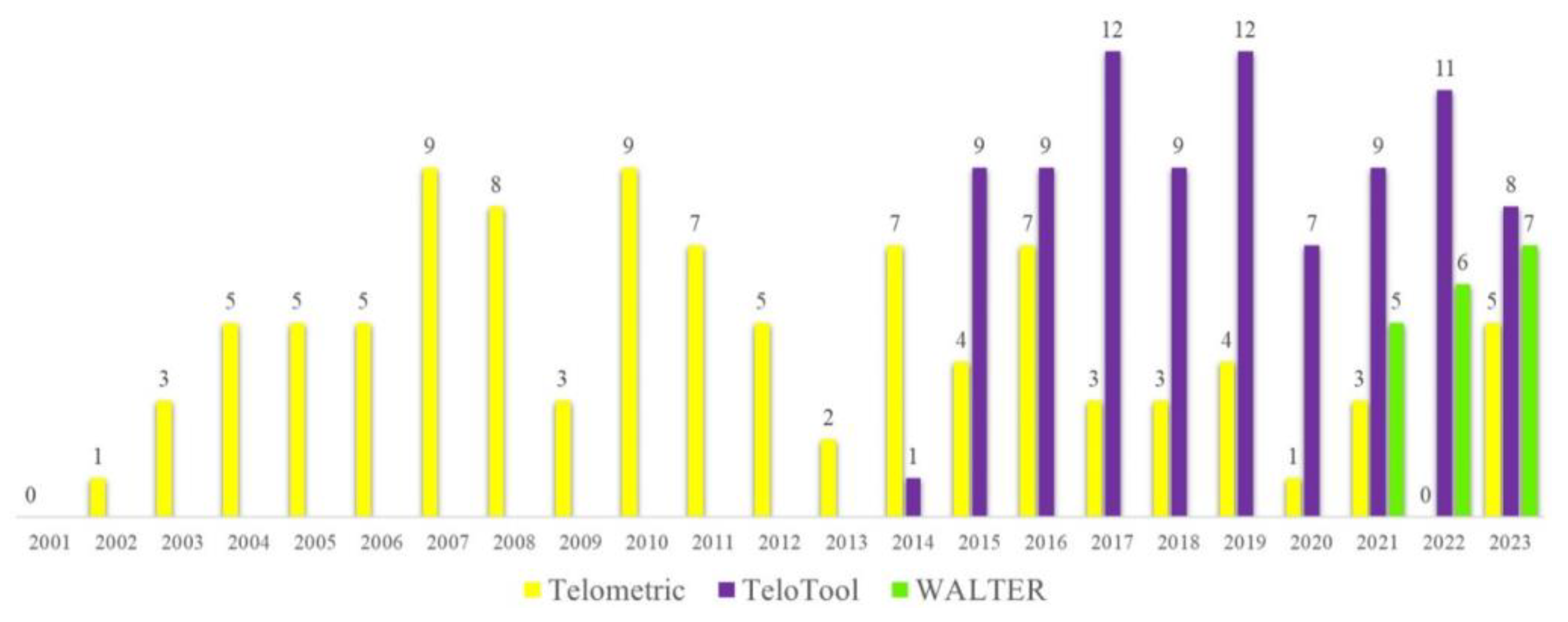
:1. Introduction
2. Results
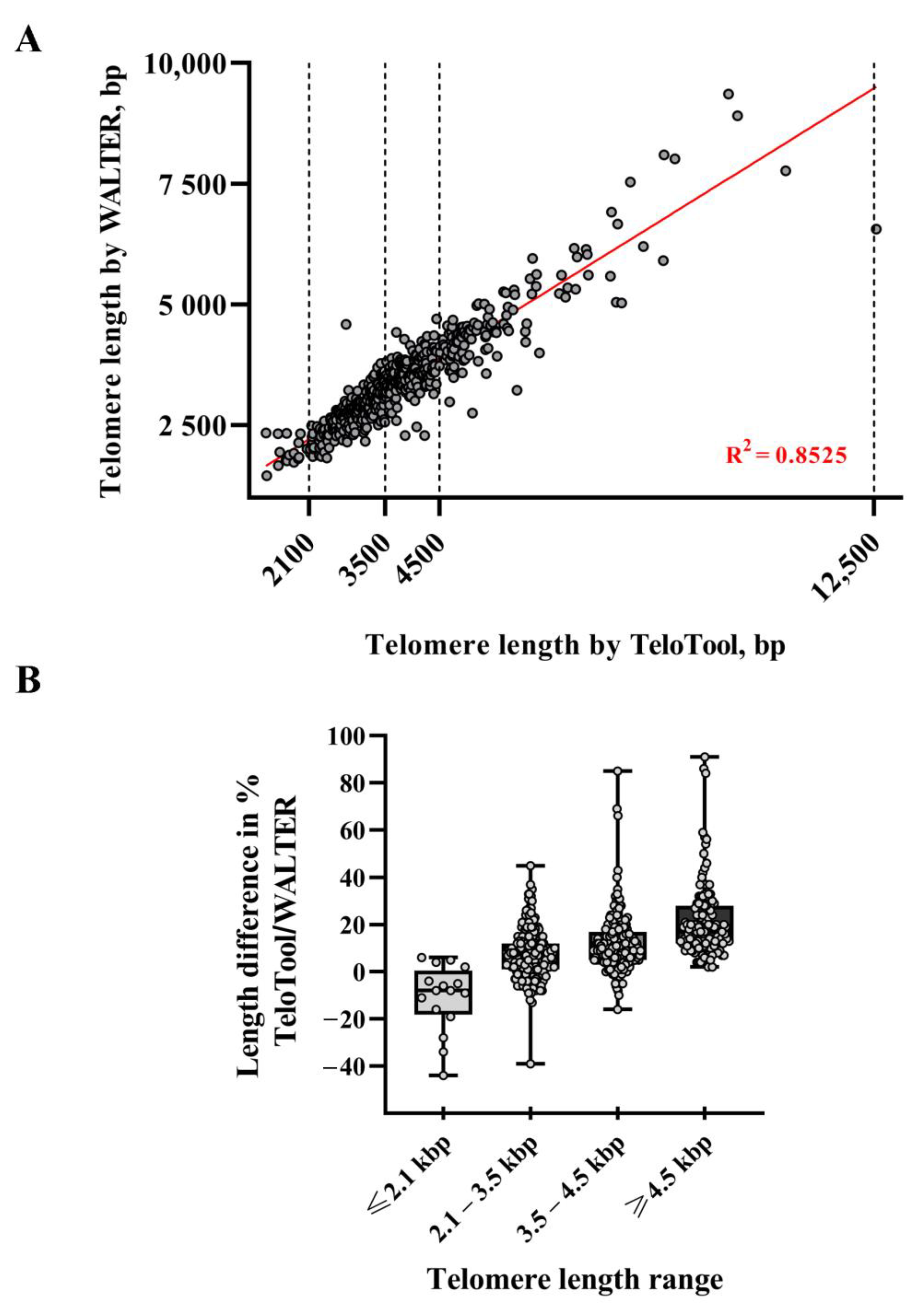
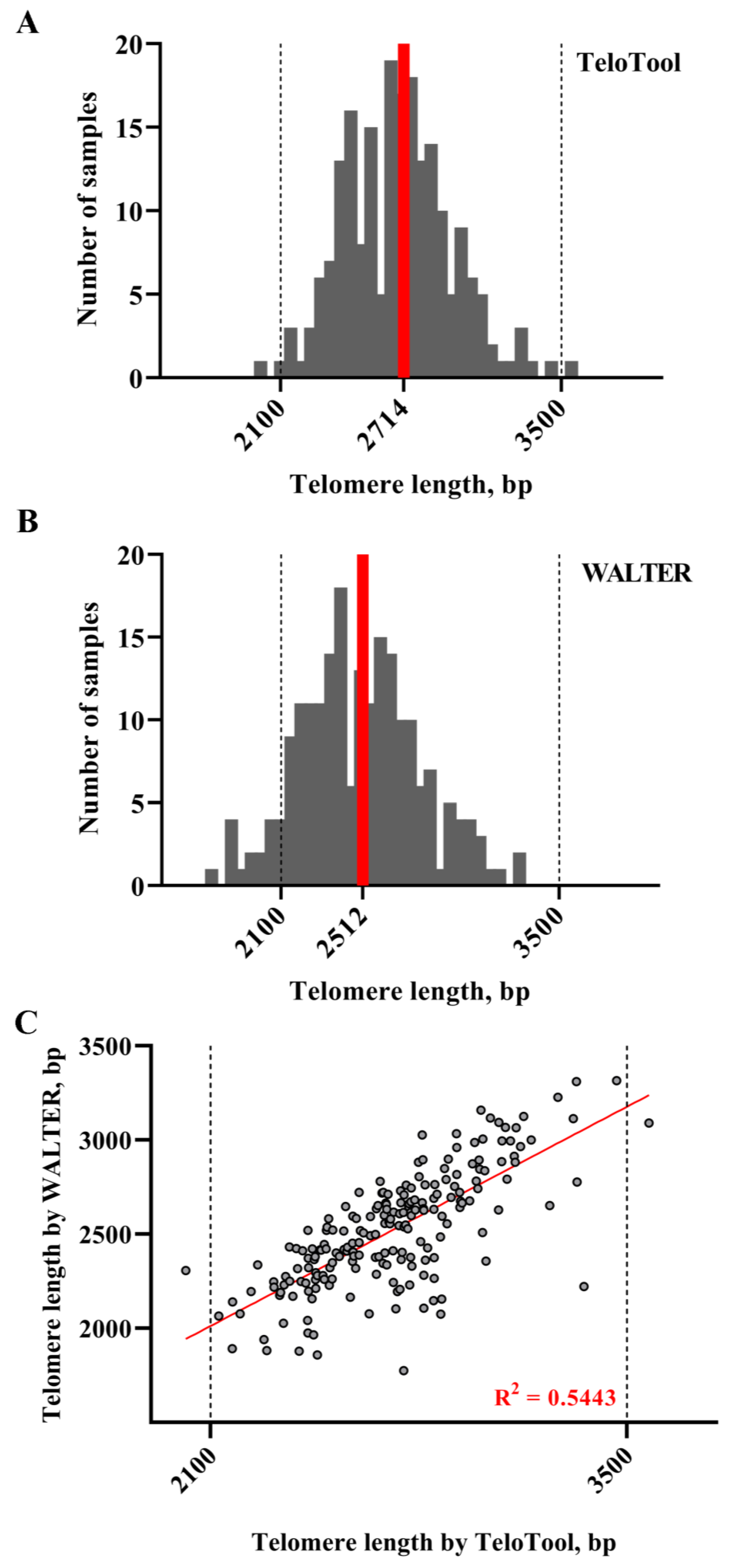
2.1. TeloTool—WALTER Comparison: General Differences and Similarities in Analyzing a Large-Scale Dataset
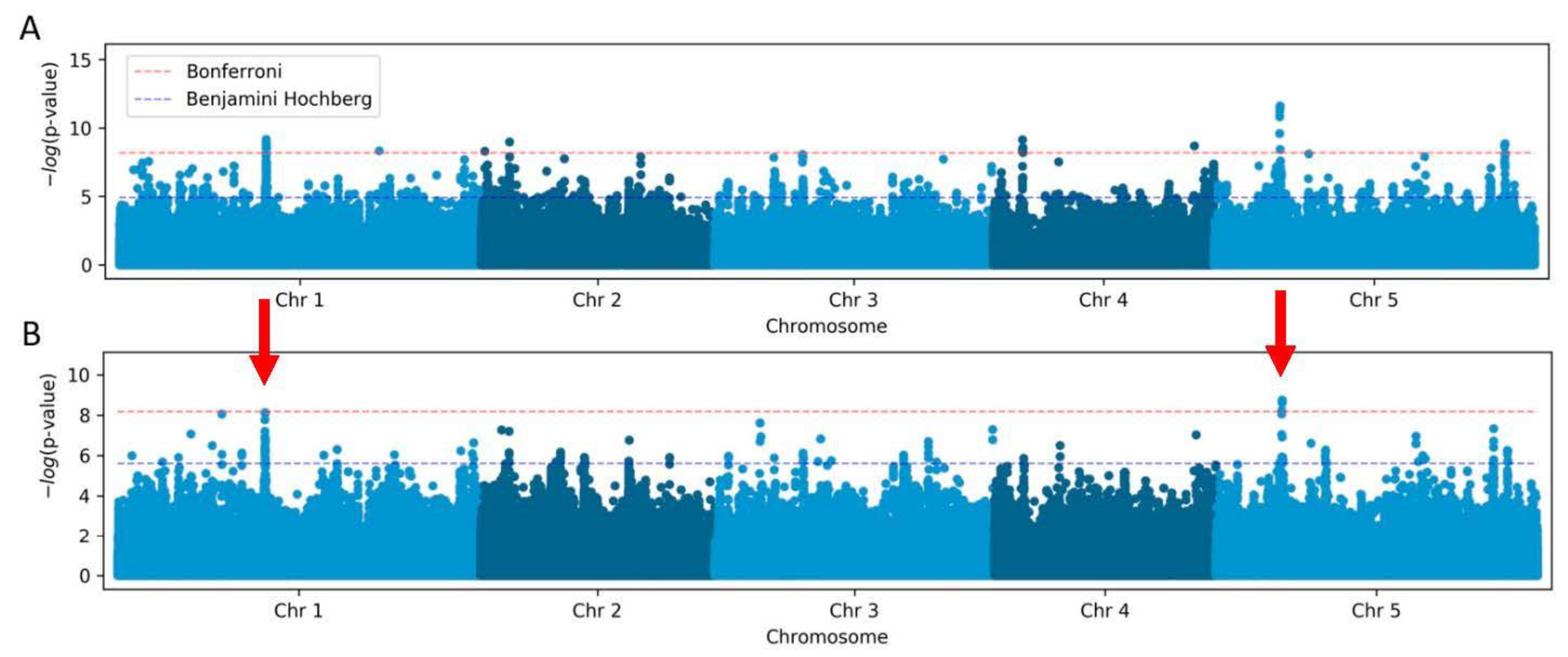
2.2. Telomere Length Estimates by TeloTool or WALTER Methods Can Affect GWAS Outcomes
2.3. Comparison of TeloTool and WALTER Datasets Generated for the Arabidopsis T-DNA Mutant Screen
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Telomere Length Measurement
4.3. Genome-Wide Association Study
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McClintock, B. The behavior in successive nuclear divisions of a chromosome broken at meiosis. Proc. Natl. Acad. Sci. USA 1939, 25, 405–416. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The stability of broken ends of chromosomes in Zea mays. Genetics 1941, 26, 234–282. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Grill, S.; Nandakumar, J. Molecular mechanisms of telomere biology disorders. J. Biol. Chem. 2021, 296, 100064. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The Limited in vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Shampay, J.; Szostak, J.W.; Blackburn, E.H. DNA sequences of telomeres maintained in yeast. Nature 1984, 310, 154–157. [Google Scholar] [CrossRef]
- Fajkus, J.; Kovarik, A.; Kralovics, R.; Bezdek, M. Organization of telomeric and subtelomeric chromatin in the higher plant Nicotiana tabacum. Mol. Gen. Genet. 1995, 247, 633–638. [Google Scholar] [CrossRef]
- Burr, B.; Burr, F.A.; Matz, E.C.; Romero-Severson, J. Pinning down loose ends: Mapping telomeres and factors affecting their length. Plant Cell 1992, 4, 953–960. [Google Scholar] [CrossRef]
- Lai, T.P.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos. Trans. R Soc. Lond B Biol. Sci. 2018, 373, 20160451. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Challoner, P.B. Identification of a telomeric DNA sequence in Trypanosoma brucei. Cell 1984, 36, 447–457. [Google Scholar] [CrossRef]
- Ausubel, M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Current Protocols in Molecular Biology; John Wiley & Sons, Inc.: Media, PA, USA, 1989; pp. 1–146. [Google Scholar]
- Lansdorp, P.M.; Verwoerd, N.P.; van de Rijke, F.M.; Dragowska, V.; Little, M.T.; Dirks, R.W.; Raap, A.K.; Tanke, H.J. Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet. 1996, 5, 685–691. [Google Scholar] [CrossRef]
- Fojtová, M.; Fajkus, P.; Polanská, P.; Fajkus, J. Terminal Restriction Fragments (TRF) Method to Analyze Telomere Lengths. Bio-Protocol 2015, 5, e1671. [Google Scholar] [CrossRef]
- Mender, I.; Shay, J.W. Telomere Restriction Fragment (TRF) Analysis. Bio-Protocol 2015, 5, e1658. [Google Scholar] [CrossRef]
- Jain, M.; Madeka, S.; Khattar, E. Optimization of Performance Parameters of the TAGGG Telomere Length Assay. J. Vis. Exp. 2023, 194, e65288. [Google Scholar] [CrossRef] [PubMed]
- Nigmatullina, L.R.; Sharipova, M.R.; Shakirov, E.V. Non-radioactive TRF assay modifications to improve telomeric DNA detection efficiency in plants. BioNanoScience 2016, 6, 325–328. [Google Scholar] [CrossRef]
- Grant, J.D.; Broccoli, D.; Muquit, M.; Manion, F.J.; Tisdall, J.; Ochs, M.F. Telometric: A tool providing simplified, reproducible measurements of telomeric DNA from constant field agarose gels. Biotechniques 2001, 31, 1314–1316, 1318. [Google Scholar] [CrossRef]
- Göhring, J.; Fulcher, N.; Jacak, J.; Riha, K. TeloTool: A new tool for telomere length measurement from terminal restriction fragment analysis with improved probe intensity correction. Nucleic Acids Res. 2014, 42, e21. [Google Scholar] [CrossRef]
- Michaeli, J.; Smoom, R.; Serruya, N.; El Ayoubi, H.; Rotshenker-Olshinka, K.; Srebnik, N.; Michaeli, O.; Eldar-Geva, T.; Tzfati, Y. Leukocyte Telomere Length Correlates with Extended Female Fertility. Cells 2022, 11, 513. [Google Scholar] [CrossRef]
- Kusuma, F.K.; Prabhu, A.; Tieo, G.; Ahmed, S.M.; Dakle, P.; Yong, W.K.; Pathak, E.; Madan, V.; Jiang, Y.Y.; Tam, W.L.; et al. Signalling inhibition by ponatinib disrupts productive alternative lengthening of telomeres (ALT). Nat. Commun. 2023, 14, 1919. [Google Scholar] [CrossRef] [PubMed]
- Lyčka, M.; Peska, V.; Demko, M.; Spyroglou, I.; Kilar, A.; Fajkus, J.; Fojtová, M. WALTER: An easy way to online evaluate telomere lengths from terminal restriction fragment analysis. BMC Bioinform. 2021, 22, 145. [Google Scholar] [CrossRef] [PubMed]
- Mannherz, W.; Agarwal, S. Thymidine nucleotide metabolism controls human telomere length. Nat. Genet. 2023, 55, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Schreglmann, S.R.; Goncalves, T.; Grant-Peters, M.; Kia, D.A.; Soreq, L.; Ryten, M.; Wood, N.W.; Bhatia, K.P.; Tomita, K. Age-related telomere attrition in the human putamen. Aging Cell 2023, 22, e13861. [Google Scholar] [CrossRef]
- Choi, J.Y.; Abdulkina, L.R.; Yin, J.; Chastukhina, I.B.; Lovell, J.T.; Agabekian, I.A.; Young, P.G.; Razzaque, S.; Shippen, D.E.; Juenger, T.E.; et al. Natural variation in plant telomere length is associated with flowering time. Plant Cell 2021, 33, 1118–1134. [Google Scholar] [CrossRef]
- Seren, Ü. GWA-Portal: Genome-wide association studies made easy. Methods Mol. Biol. 2018, 1761, 303–319. [Google Scholar] [CrossRef]
- Abdulkina, L.R.; Kobayashi, C.; Lovell, J.T.; Chastukhina, I.B.; Aklilu, B.; Agabekyan, I.A.; Valeeva, L.R.; Nyamsuren, C.; Aglyamova, G.V.; Sharipova, M.R.; et al. Components of the ribosome biogenesis pathway underlie establishment of telomere length set point in Arabidopsis. Nat. Commun. 2019, 10, 5479. [Google Scholar] [CrossRef]
- Fulcher, N.; Teubenbacher, A.; Kerdaffrec, E.; Farlow, A.; Nordborg, M.; Riha, K. Genetic architecture of natural variation of telomere length in Arabidopsis thaliana. Genetics 2015, 199, 625–635. [Google Scholar] [CrossRef]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef]
- Alonso, J.M.; Stepanova, A.N.; Leisse, T.J.; Kim, C.J.; Chen, H.; Shinn, P.; Stevenson, D.K.; Zimmerman, J.; Barajas, P.; Cheuk, R.; et al. Genome-Wide Insertional Mutagenesis of Arabidopsis thaliana. Science 2003, 301, 653–657. [Google Scholar] [CrossRef]
- Ungar, L.; Yosef, N.; Sela, Y.; Sharan, R.; Ruppin, E.; Kupiec, M. A genome-wide screen for essential yeast genes that affect telomere length maintenance. Nucleic Acids Res. 2009, 37, 3840–3849. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.N.; Han, T.X.; Du, L.L.; Zhou, J.Q. A genome-wide screen for Schizosaccharomyces pombe deletion mutants that affect telomere length. Cell Res. 2010, 20, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.S.; Riha, K.; Gao, F.; Ren, S.; McKnight, T.D.; Shippen, D.E. Disruption of the telomerase catalytic subunit gene from Arabidopsis inactivates telomerase and leads to a slow loss of telomeric DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 14813–14818. [Google Scholar] [CrossRef]
- Gutiérrez, M.L.; Rodríguez-González, R.; Pascual-Díaz, J.P.; Fuentes, I.; Garcia, S. Online Resources Useful for Plant Cytogenetics and Cytogenomics Research. In Plant Cytogenetics and Cytogenomics. Methods in Molecular Biology; Heitkam, T., Garcia, S., Eds.; Humana: New York, NY, USA, 2023; Volume 2672, pp. 549–560. [Google Scholar]
- Kato, K.; Kawaguchi, A.; Nagata, K. Template activating factor-I epigenetically regulates the TERT transcription in human cancer cells. Sci. Rep. 2021, 11, 17726. [Google Scholar] [CrossRef]
- Fajkus, P.; Kilar, A.; Nelson, A.D.L.; Holá, M.; Peška, V.; Goffová, I.; Fojtová, M.; Zachová, D.; Fulnečková, J.; Fajkus, J. Evolution of plant telomerase RNAs: Farther to the past, deeper to the roots. Nucleic Acids Res. 2021, 49, 7680–7694. [Google Scholar] [CrossRef]
- Poláková, E.; Albanaz, A.T.S.; Zakharova, A.; Novozhilova, T.S.; Gerasimov, E.S.; Yurchenko, V. Ku80 is involved in telomere maintenance but dispensable for genomic stability in Leishmania mexicana. PLoS Negl. Trop. Dis. 2021, 15, e0010041. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Telomere Length Range * | ≤2100 bp | 2101–3500 bp | 3501–4500 bp | ≥4501 bp |
|---|---|---|---|---|
| Number of DNA samples | 16 | 250 | 195 | 123 |
| Number of accessions | 15 | 232 | 179 | 111 |
| Minimum difference, % | −44.00 | −39.00 | −16.00 | 2.00 |
| 25% Percentile | −18.25 | 1.000 | 5.000 | 12.00 |
| Median difference, % | −8.00 | 7.000 | 10.00 | 17.00 |
| 75% Percentile | 0.50 | 12.00 | 17.00 | 28.00 |
| Maximum difference, % | 6.00 | 45.00 | 85.00 | 91.00 |
| Mean difference, % | −10.94 | 7.40 | 12.01 | 21.68 |
| SNP | Chromosome | Position | p-Value | Major Allele | Minor Allele | maf | Effect |
|---|---|---|---|---|---|---|---|
| TeloTool (LM) | |||||||
| 5:5538242 * | 5 | 5538242 | 11.61 | T | C | 0.19 | 3’UTR_3 At5g16850 |
| 1:12379845 * | 1 | 12379845 | 10.46 | G | A | 0.028 | Intergenic |
| 4:2463357 | 4 | 2463357 | 9.13 | C | T | 0.046 | INTRON At4g04870 |
| 2:2422473 | 2 | 2422473 | 8.98 | G | A | 0.059 | Intergenic |
| 5:24491588 | 5 | 24491588 | 8.86 | G | A | 0.055 | Intergenic |
| 4:18303636 | 4 | 18303636 | 8.78 | C | T | 0.028 | Intergenic |
| 4:16935117 | 4 | 16935117 | 8.7 | T | A | 0.04 | MISSENSE aTg/aAg M231K At4g35733 |
| 1:21870431 | 1 | 21870431 | 8.32 | T | G | 0.038 | Intergenic |
| 2:356311 | 2 | 356311 | 8.30 | T | A | 0.036 | Intergenic |
| 3:7407370 | 3 | 7407370 | 8.06 | G | A | 0.104 | MISSENSE Gga/Aga, G349R At3g21120 |
| WALTER (LM) | |||||||
| 5:5538242 * | 5 | 5538242 | 8.73 | T | C | 0.19 | 3’UTR_3 At5g16850 |
| 1:12379845 * | 1 | 12379845 | 10.01 | G | A | 0.028 | Intergenic |
| 1:8743486 | 1 | 8743486 | 8.06 | A | G | 0.04 | INTRON At1g24706 |
| TeloTool (AMM) | |||||||
| 5:5538242 * | 5 | 5538242 | 11.61 | T | C | 0.19 | 3’UTR_3 At5g16850 |
| 3:23295214 | 3 | 23295214 | 8.66 | T | G | 0.03 | Promoter of At3g63030 |
| 1:21870431 | 1 | 21870431 | 8.08 | T | G | 0.038 | Intergenic |
| WALTER (AMM) | |||||||
| 5:5538242 * | 5 | 5538242 | 9.0 | T | C | 0.19 | 3’UTR_3 At5g16850 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdulkina, L.R.; Agabekian, I.A.; Valeeva, L.R.; Kozlova, O.S.; Sharipova, M.R.; Shakirov, E.V. Comparative Application of Terminal Restriction Fragment Analysis Tools to Large-Scale Genomic Assays. Int. J. Mol. Sci. 2023, 24, 17194. https://doi.org/10.3390/ijms242417194
Abdulkina LR, Agabekian IA, Valeeva LR, Kozlova OS, Sharipova MR, Shakirov EV. Comparative Application of Terminal Restriction Fragment Analysis Tools to Large-Scale Genomic Assays. International Journal of Molecular Sciences. 2023; 24(24):17194. https://doi.org/10.3390/ijms242417194
Chicago/Turabian StyleAbdulkina, Liliia R., Inna A. Agabekian, Liia R. Valeeva, Olga S. Kozlova, Margarita R. Sharipova, and Eugene V. Shakirov. 2023. "Comparative Application of Terminal Restriction Fragment Analysis Tools to Large-Scale Genomic Assays" International Journal of Molecular Sciences 24, no. 24: 17194. https://doi.org/10.3390/ijms242417194
APA StyleAbdulkina, L. R., Agabekian, I. A., Valeeva, L. R., Kozlova, O. S., Sharipova, M. R., & Shakirov, E. V. (2023). Comparative Application of Terminal Restriction Fragment Analysis Tools to Large-Scale Genomic Assays. International Journal of Molecular Sciences, 24(24), 17194. https://doi.org/10.3390/ijms242417194