
Lovastatin Treatment Inducing Apoptosis in Human Pancreatic Cancer Cells by Inhibiting Cholesterol Rafts in Plasma Membrane and Mitochondria

Abstract
:1. Introduction
2. Results
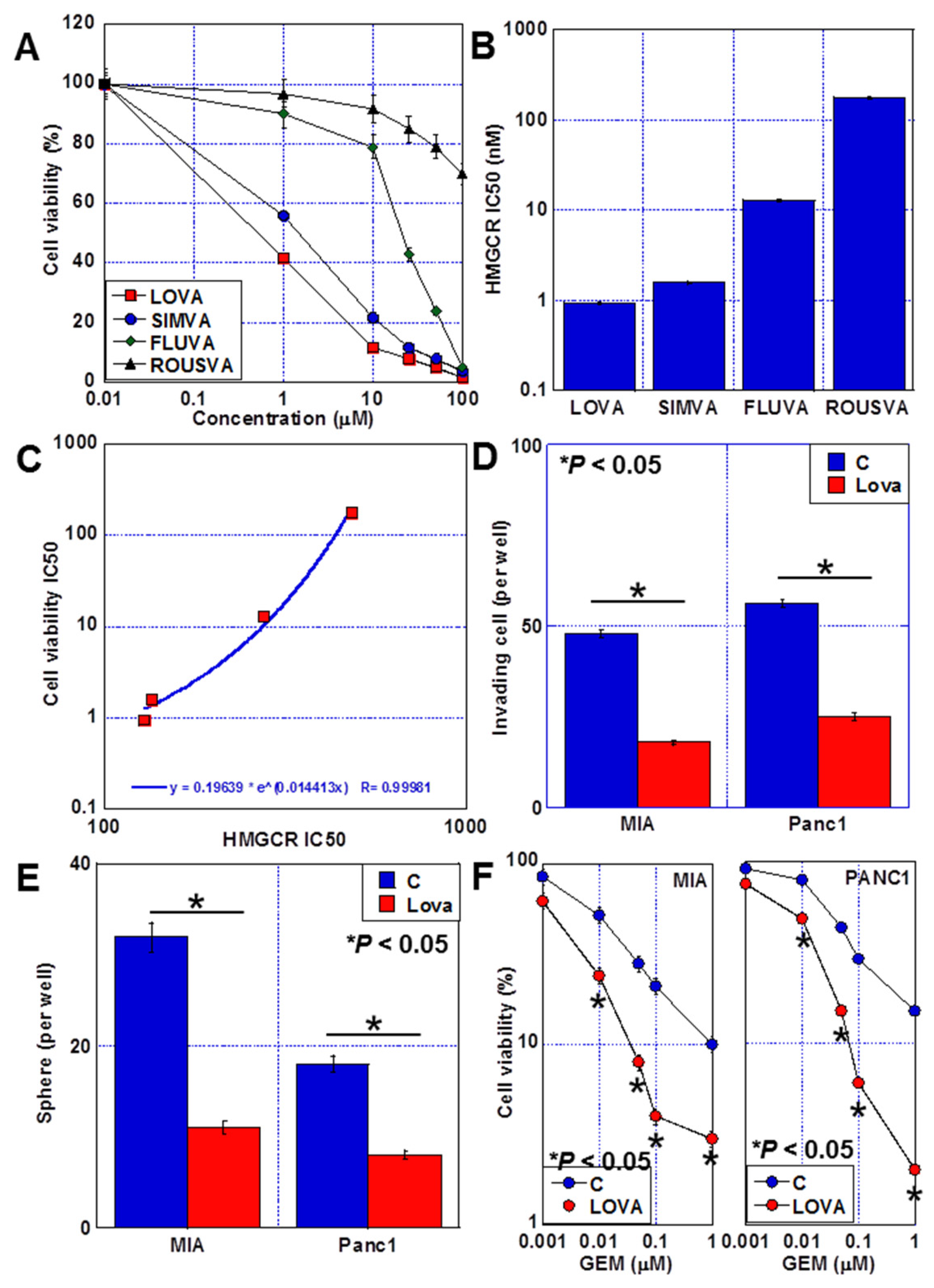
2.1. Antitumor Effects of Statins against PDAC Cells
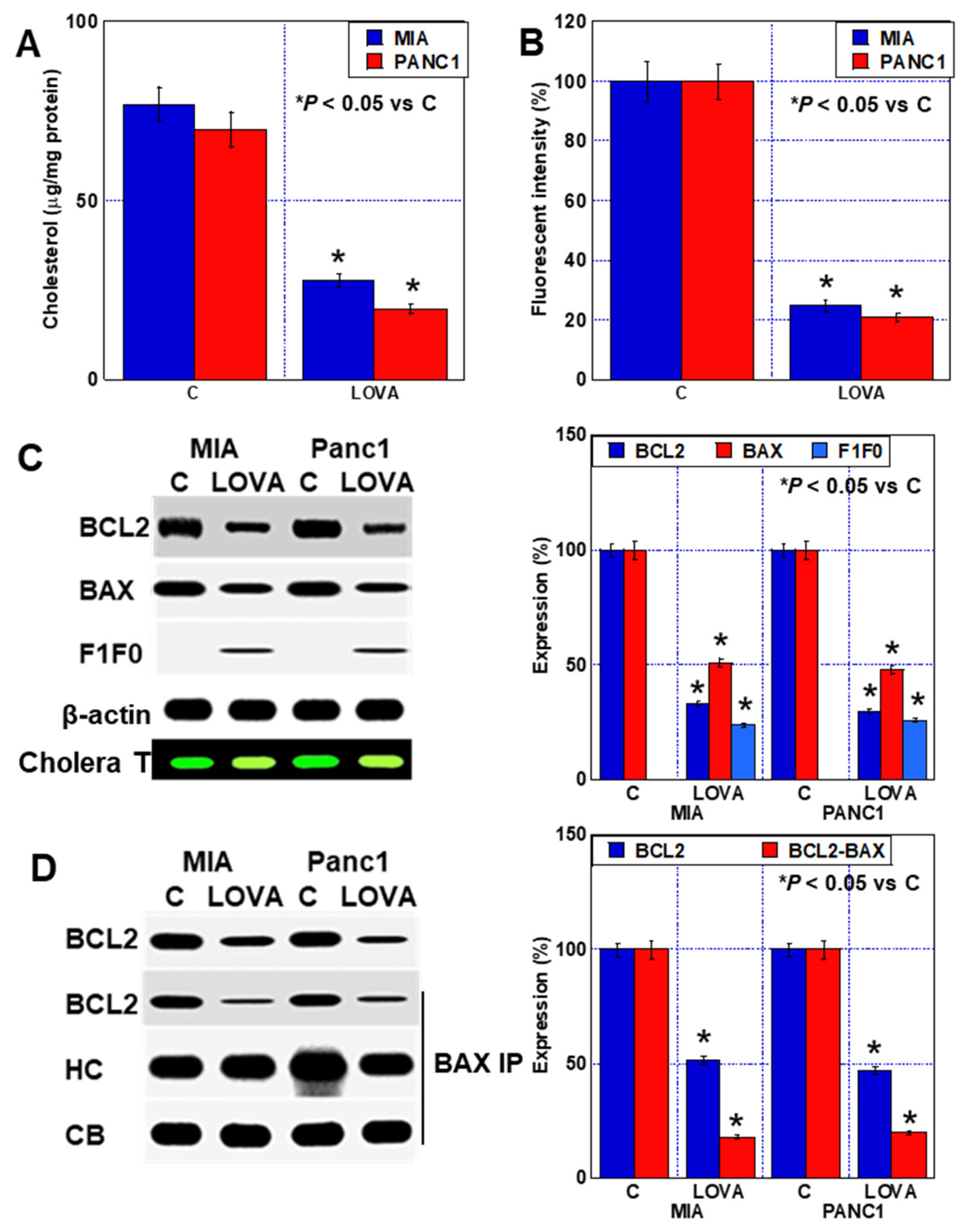
2.2. Cell Death Caused by LOVA
2.3. Inhibition of Plasma Membrane Rafts by LOVA Treatment
2.4. Inhibition of Mitochondrial Rafts by LOVA Treatment
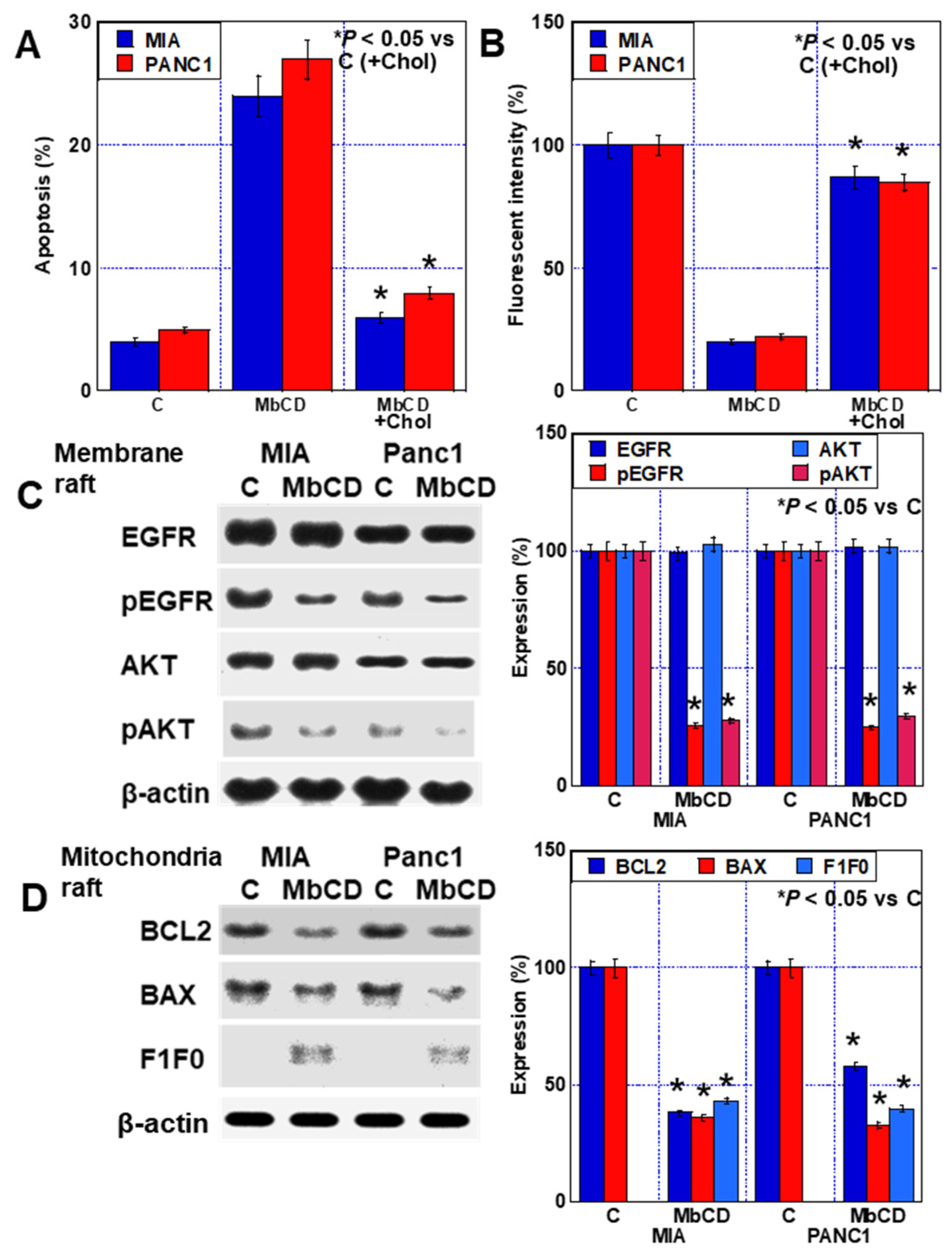
2.5. Effect of Cholesterol Adsorbent on PDA Cells
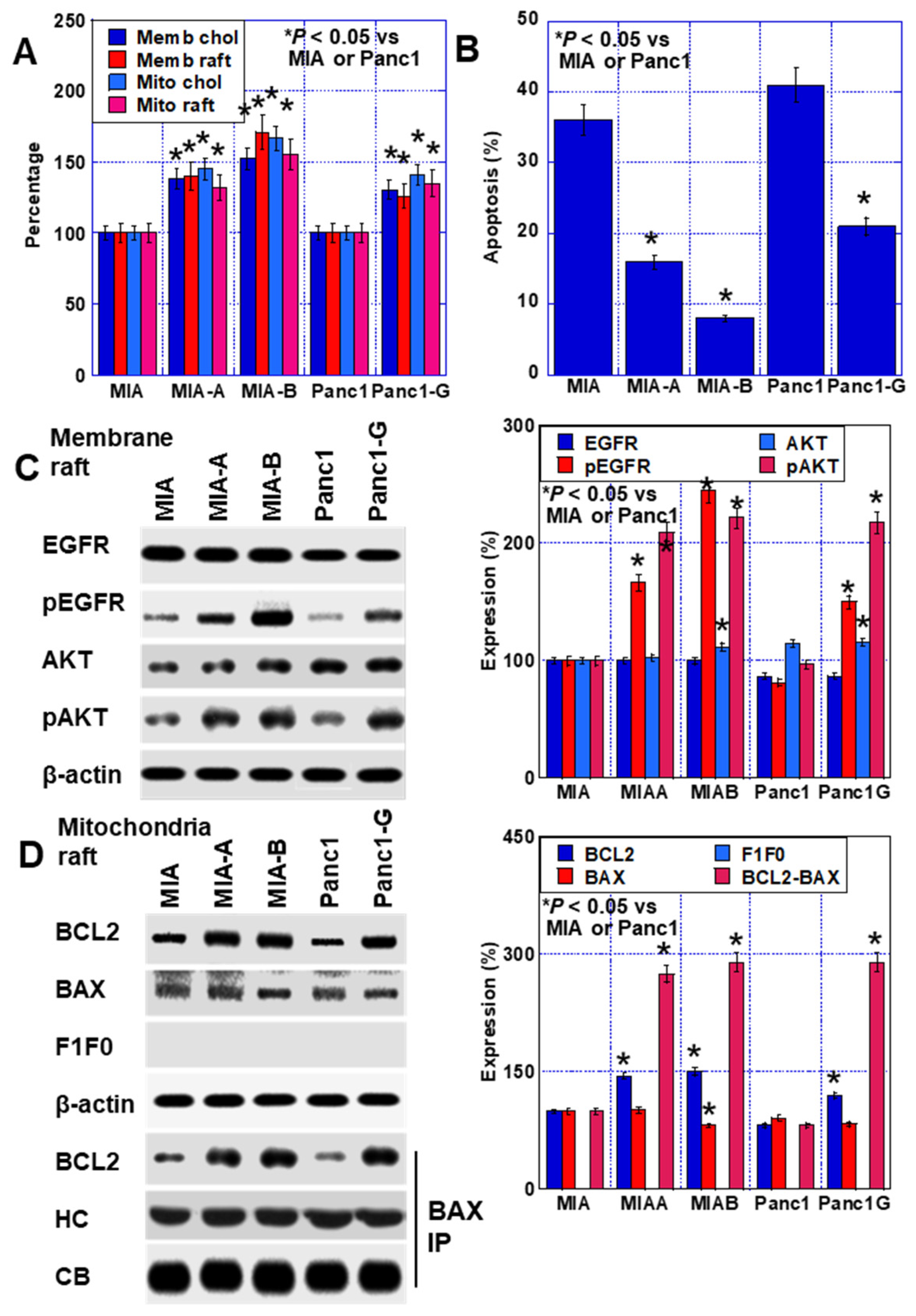
2.6. Effect of LOVA on GEM-Resistant PDAC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Line and Reagents
4.2. Reagents
4.3. Cell Growth, Cell Death, and Apoptosis
4.4. Fluorescent Imaging
4.5. Chamber Invasion Assay
4.6. Protein Extraction
4.7. Mitochondria Extraction
4.8. Extraction of Raft Fractions
4.9. Immunoblot Analysis
4.10. Immunoprecipitation
4.11. Enzyme-Linked Immunosorbent Assay (ELISA)
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Research Center Cancer Information Service. Available online: https://www.ncc.go.jp/en/icc/cancer-info/index.html (accessed on 15 September 2023).
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updat. 2015, 23, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Okusaka, T.; Nakamura, M.; Yoshida, M.; Kitano, M.; Uesaka, K.; Ito, Y.; Furuse, J.; Hanada, K.; Okazaki, K. Clinical Practice Guidelines for Pancreatic Cancer 2019 From the Japan Pancreas Society: A Synopsis. Pancreas 2020, 49, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Beatty, G.L. Tumor Microenvironment in Pancreatic Cancer Pathogenesis and Therapeutic Resistance. Annu. Rev. Pathol. 2023, 18, 123–148. [Google Scholar] [CrossRef]
- Fujiwara-Tani, R.; Sasaki, T.; Takagi, T.; Mori, S.; Kishi, S.; Nishiguchi, Y.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Gemcitabine Resistance in Pancreatic Ductal Carcinoma Cell Lines Stems from Reprogramming of Energy Metabolism. Int. J. Mol. Sci. 2022, 23, 7824. [Google Scholar] [CrossRef]
- Takagi, T.; Fujiwara-Tani, R.; Mori, S.; Kishi, S.; Nishiguchi, Y.; Sasaki, T.; Ogata, R.; Ikemoto, A.; Sasaki, R.; Ohmori, H.; et al. Lauric Acid Overcomes Hypoxia-Induced Gemcitabine Chemoresistance in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7506. [Google Scholar] [CrossRef]
- Tummala, R.; Gupta, M.; Devanabanda, A.R.; Bandyopadhyay, D.; Aronow, W.S.; Ray, K.K.; Mamas, M.; Ghosh, R.K. Bempedoic acid and its role in contemporary management of hyperlipidemia in atherosclerosis. Ann. Med. 2022, 54, 1287–1296. [Google Scholar] [CrossRef]
- Nelson, R.H. Hyperlipidemia as a risk factor for cardiovascular disease. Prim. Care 2013, 40, 195–211. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Kostantinou, A.; Kougias, M.; Kazazis, C. Statins and cancer. Anticancer Agents Med. Chem. 2014, 14, 706–712. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Lipid Raft Isolation by Sucrose Gradient Centrifugation and Visualization of Raft-Located Proteins by Fluorescence Microscopy: The Use of Combined Techniques to Assess Fas/CD95 Location in Rafts During Apoptosis Triggering. Methods Mol. Biol. 2021, 2187, 147–186. [Google Scholar]
- George, K.S.; Wu, S. Lipid raft: A floating island of death or survival. Toxicol. Appl. Pharmacol. 2012, 259, 311–319. [Google Scholar] [CrossRef]
- Mannino, G.; Iovino, P.; Lauria, A.; Genova, T.; Asteggiano, A.; Notarbartolo, M.; Porcu, A.; Serio, G.; Chinigò, G.; Occhipinti, A.; et al. Bioactive Triterpenes of Protium heptaphyllum Gum Resin Extract Display Cholesterol-Lowering Potential. Int. J. Mol. Sci. 2021, 22, 2664. [Google Scholar] [CrossRef]
- Mantell, G. Extended worldwide experience. HMG-CoA reductase inhibitors: Lovastatin and simvastatin. Therapie 1992, 47, 161–164. [Google Scholar]
- Kishi, S.; Fujiwara-Tani, R.; Luo, Y.; Kawahara, I.; Goto, K.; Fujii, K.; Ohmori, H.; Nakashima, C.; Sasaki, T.; Kuniyasu, H. Pro-metastatic signaling of the trans fatty acid elaidic acid is associated with lipid rafts. Oncol. Lett. 2018, 15, 4423–4426. [Google Scholar] [CrossRef]
- Saito, K.; Sato, Y.; Nakatani, E.; Kaneda, H.; Yamamoto, S.; Miyachi, Y.; Itoh, H. Statin Exposure and Pancreatic Cancer Incidence: A Japanese Regional Population-Based Cohort Study, the Shizuoka Study. Cancer Prev. Res. 2021, 14, 863–872. [Google Scholar] [CrossRef]
- Jian-Yu, E.; Graber, J.M.; Lu, S.E.; Lin, Y.; Lu-Yao, G.; Tan, X.L. Effect of Metformin and Statin Use on Survival in Pancreatic Cancer Patients: A Systematic Literature Review and Meta-analysis. Curr. Med. Chem. 2018, 25, 2595–2607. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, S.H.; Lee, H.J.; Chung, M.J.; Park, J.Y.; Park, S.W.; Song, S.Y.; Bang, S. Statin Use and Its Impact on Survival in Pancreatic Cancer Patients. Medicine 2016, 95, e3607. [Google Scholar] [CrossRef]
- Müller, C.; Bockhorn, A.G.; Klusmeier, S.; Kiehl, M.; Roeder, C.; Kalthoff, H.; Koch, O.M. Lovastatin inhibits proliferation of pancreatic cancer cell lines with mutant as well as with wild-type K-ras oncogene but has different effects on protein phosphorylation and induction of apoptosis. Int. J. Oncol. 1998, 12, 717–723. [Google Scholar] [CrossRef]
- Lee, E.J.; Yun, U.J.; Koo, K.H.; Sung, J.Y.; Shim, J.; Ye, S.K.; Hong, K.M.; Kim, Y.N. Down-regulation of lipid raft-associated onco-proteins via cholesterol-dependent lipid raft internalization in docosahexaenoic acid-induced apoptosis. Biochim. Biophys. Acta 2014, 1841, 190–203. [Google Scholar] [CrossRef]
- Yu, S.; Wang, L.; Che, D.; Zhang, M.; Li, M.; Naito, M.; Xin, W.; Zhou, L. Targeting CRABP-II overcomes pancreatic cancer drug resistance by reversing lipid raft cholesterol accumulation and AKT survival signaling. J. Exp. Clin. Cancer Res. 2022, 41, 88. [Google Scholar] [CrossRef]
- Kumar, N.; Mandal, C.C. Cholesterol-Lowering Drugs on Akt Signaling for Prevention of Tumorigenesis. Front. Genet. 2021, 12, 724149. [Google Scholar] [CrossRef]
- Mahammad, S.; Parmryd, I. Cholesterol depletion using methyl-β-cyclodextrin. Methods Mol. Biol. 2015, 1232, 91–102. [Google Scholar]
- Feo, F.; Canuto, R.A.; Garcea, R.; Gabriel, L. Effect of cholesterol content on some physical and functional properties of mitochondria isolated from adult rat liver, fetal liver, cholesterol-enriched liver and hepatomas AH-130, 3924A and 5123. Biochim. Biophys. Acta 1975, 413, 116–134. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine. Pharmaceutics 2021, 13, 763. [Google Scholar] [CrossRef]
- Cheng, B.; Hsu, D.K.; Kimura, T. Utilization of intramitochondrial membrane cholesterol by cytochrome P-450-dependent cholesterol side-chain cleavage reaction in bovine adrenocortical mitochondria: Steroidogenic and non-steroidogenic pools of cholesterol in the mitochondrial inner membranes. Mol. Cell. Endocrinol. 1985, 40, 233–243. [Google Scholar]
- Selvaraj, V.; Stocco, D.M.; Tu, L.N. Minireview: Translocator protein (TSPO) and steroidogenesis: A reappraisal. Mol. Endocrinol. 2015, 29, 490–501. [Google Scholar] [CrossRef]
- Garofalo, T.; Manganelli, V.; Grasso, M.; Mattei, V.; Ferri, A.; Misasi, R.; Sorice, M. Role of mitochondrial raft-like microdomains in the regulation of cell apoptosis. Apoptosis 2015, 20, 621–634. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond - mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Szabadkai, G. What makes you can also break you: Mitochondrial permeability transition pore formation by the c subunit of the F(1)F(0) ATP-synthase? Front. Oncol. 2013, 3, 25. [Google Scholar] [CrossRef]
- Gerle, C. Mitochondrial F-ATP synthase as the permeability transition pore. Pharmacol. Res. 2020, 160, 105081. [Google Scholar] [CrossRef]
- Babu, S.; Manoharan, S.; Ottappilakkil, H.; Perumal, E. Role of oxidative stress-mediated cell death and signaling pathways in experimental fluorosis. Chem. Biol. Interact. 2022, 365, 110106. [Google Scholar] [CrossRef]
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Oxidative stress, redox signaling, and autophagy: Cell death versus survival. Antioxid. Redox Signal 2014, 21, 66–85. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef]
- Fujiwara-Tani, R.; Fujii, K.; Mori, S.; Kishi, S.; Sasaki, T.; Ohmori, H.; Nakashima, C.; Kawahara, I.; Nishiguchi, Y.; Mori, T.; et al. Role of Clostridium perfringens Enterotoxin on YAP Activation in Colonic Sessile Serrated Adenoma/ Polyps with Dysplasia. Int. J. Mol. Sci. 2020, 21, 3840. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Clone | Dilution | Code | Manufacturer | Location |
|---|---|---|---|---|---|
| PARP | - | 1:400 | GTX100573 | GeneTex | Irvine, CA, USA |
| Caveoin-1 | A-6 | 1:300 | sc-393013 | Santa-Cruz | Santa Cruz, CA, USA |
| EGFR | CDX2-88 | 1:500 | ab157524 | Abcam | Waltham, MA, USA |
| pEGFR | PAb240 | 1:300 | ab26 | Abcam | Waltham, MA, USA |
| AKT | - | 1:400 | ab70346 | Abcam | Waltham, MA, USA |
| pAKT | - | 1:400 | #9271 | Cell Signaling ser473 | Danvers, MA, USA |
| PI3K | OTI4H9 | 1:500 | ab139307 | Abcam | Waltham, MA, USA |
| BCL2 | C-2 | 1:300 | sc-7382 | Santa-Cruz | Santa Cruz, CA, USA |
| BAX | E63 | 1:500 | ab205822 | Abcam | Waltham, MA, USA |
| F1F0 | - | 1:200 | ab96655 | Abcam | Waltham, MA, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyoten, M.; Luo, Y.; Fujiwara-Tani, R.; Mori, S.; Ogata, R.; Kishi, S.; Kuniyasu, H. Lovastatin Treatment Inducing Apoptosis in Human Pancreatic Cancer Cells by Inhibiting Cholesterol Rafts in Plasma Membrane and Mitochondria. Int. J. Mol. Sci. 2023, 24, 16814. https://doi.org/10.3390/ijms242316814
Gyoten M, Luo Y, Fujiwara-Tani R, Mori S, Ogata R, Kishi S, Kuniyasu H. Lovastatin Treatment Inducing Apoptosis in Human Pancreatic Cancer Cells by Inhibiting Cholesterol Rafts in Plasma Membrane and Mitochondria. International Journal of Molecular Sciences. 2023; 24(23):16814. https://doi.org/10.3390/ijms242316814
Chicago/Turabian StyleGyoten, Momoko, Yi Luo, Rina Fujiwara-Tani, Shiori Mori, Ruiko Ogata, Shingo Kishi, and Hiroki Kuniyasu. 2023. "Lovastatin Treatment Inducing Apoptosis in Human Pancreatic Cancer Cells by Inhibiting Cholesterol Rafts in Plasma Membrane and Mitochondria" International Journal of Molecular Sciences 24, no. 23: 16814. https://doi.org/10.3390/ijms242316814
APA StyleGyoten, M., Luo, Y., Fujiwara-Tani, R., Mori, S., Ogata, R., Kishi, S., & Kuniyasu, H. (2023). Lovastatin Treatment Inducing Apoptosis in Human Pancreatic Cancer Cells by Inhibiting Cholesterol Rafts in Plasma Membrane and Mitochondria. International Journal of Molecular Sciences, 24(23), 16814. https://doi.org/10.3390/ijms242316814