
Effects of the Long-Term Administration of Uridine on the Functioning of Rat Liver Mitochondria in Hyperthyroidism

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Effect of Uridine on Somatic and Biochemical Parameters of Rats with Hyperthyroidism
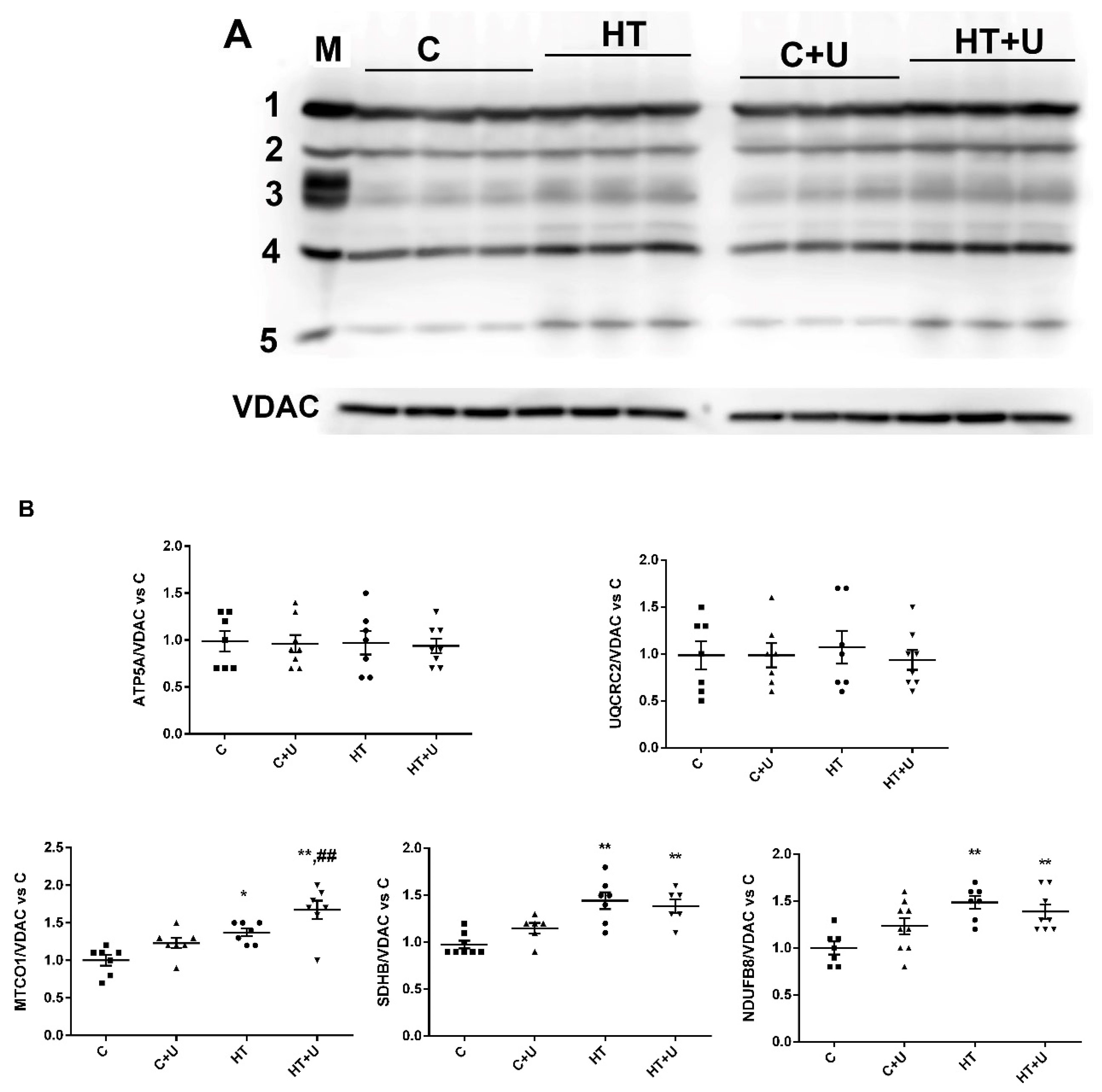
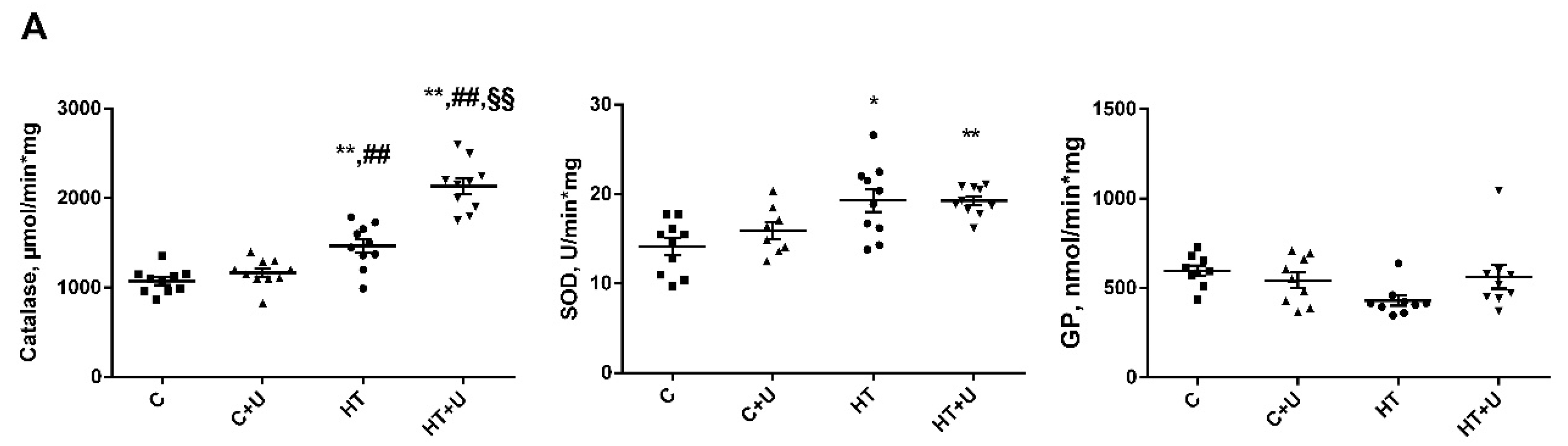
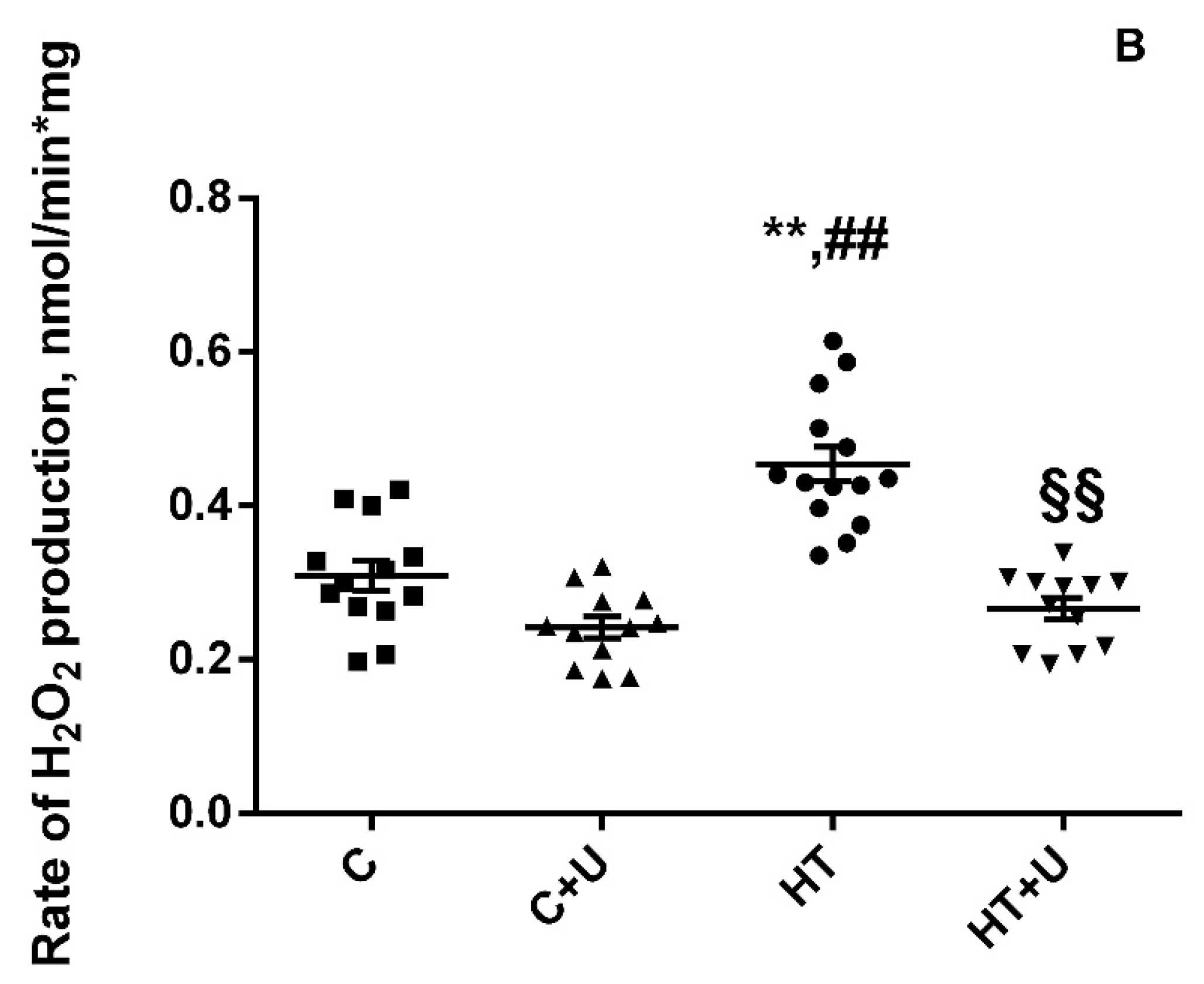
2.2. Effect of Uridine on the Functional State of Rat Liver Mitochondria with Hyperthyroidism
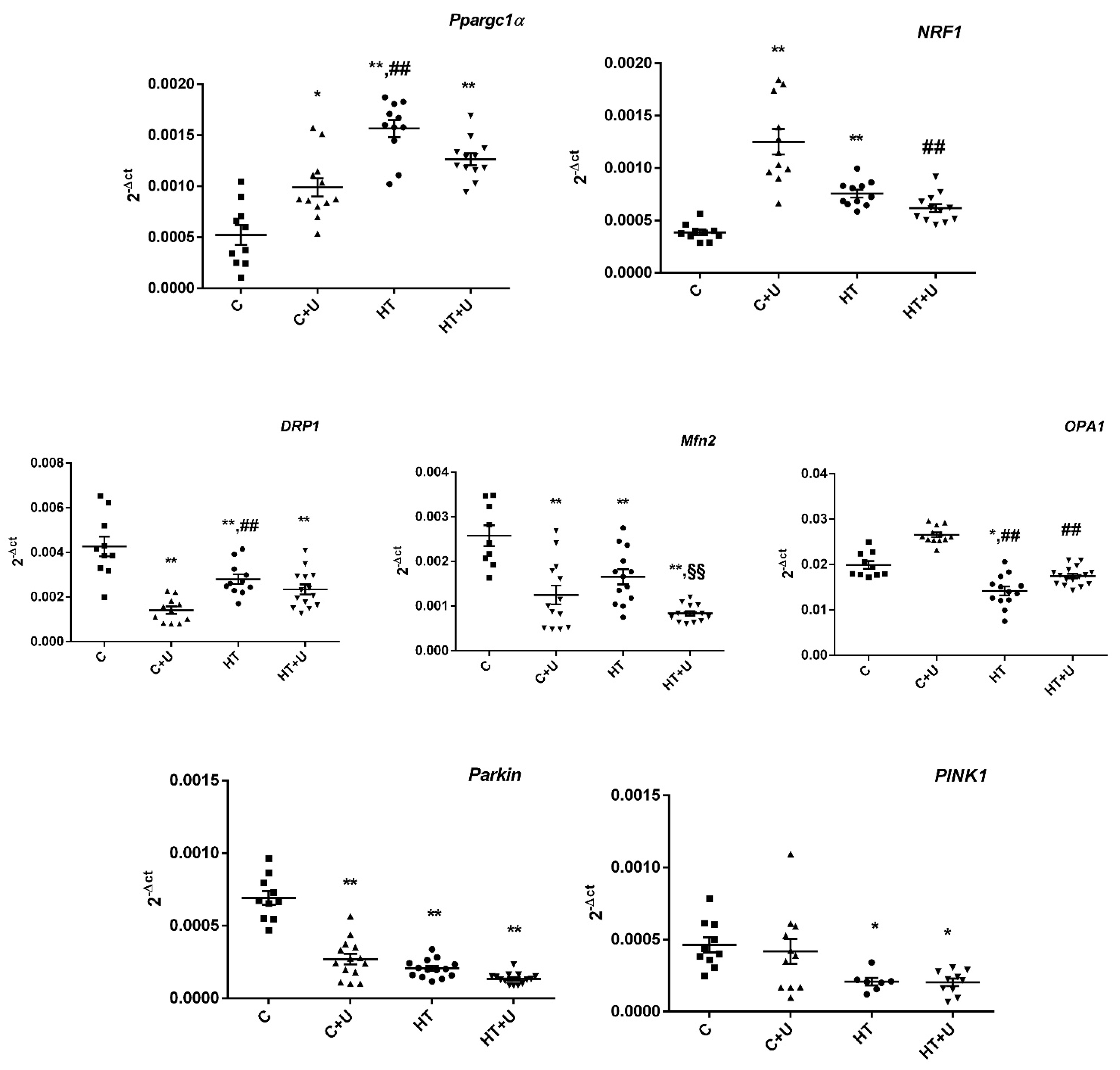
2.3. Effect of Uridine on HT-Induced Changes in the Expression of mRNA of the Proteins Responsible for Biogenesis and Mitochondrial Quality Control
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Isolation of the Liver Mitochondria and Assessment of Mitochondrial Functions in the Rats of the Experimental Groups
4.2.1. Enzyme Activity Assay
4.2.2. SOD Activity
4.2.3. CAT Activity
4.2.4. GPX Activity
4.3. Quantification of the Expression of the Genes of Mitochondrial Dynamics, Biogenesis, and Mitophagy Using Quantitative Real-Time PCR in the Rats of the Experimental Groups
4.4. Isolation of Rat Liver Mitochondria and Determination of Respiration and Oxidative Phosphorylation Electrophoresis and Immunoblotting of Mitochondrial OXPHOS Proteins
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bennett, C.F.; Latorre-Muro, P.; Puigserver, P. Mechanisms of mitochondrial respiratory adaptation. Nat. Rev. Mol. Cell Biol. 2022, 23, 817–835. [Google Scholar] [CrossRef]
- Pietrobon, D.; Azzone, G.F.; Walz, D. Effect of funiculosin and antimycin A on the redox-driven H+-pumps in mitochondria: On the nature of “leaks”. Eur. J. Biochem. 1998, 117, 389–394. [Google Scholar] [CrossRef]
- Johnson, J.L.; Felicetta, J.V. Hyperthyroidism: A Comprehensive Review. J. Am. Acad. Nurse Pract. 1992, 4, 8–14. [Google Scholar] [CrossRef]
- Venediktova, N.; Solomadin, I.; Starinets, V.; Mironova, G. Structural and Dynamic Features of Liver Mitochondria and Mitophagy in Rats with Hyperthyroidism. Int. J. Mol. Sci. 2022, 23, 14327. [Google Scholar] [CrossRef]
- Venediktova, N.; Solomadin, I.; Nikiforova, A.; Starinets, V.; Mironova, G. Functional State of Rat Heart Mitochondria in Experimental Hyperthyroidism. Int. J. Mol. Sci. 2021, 22, 11744. [Google Scholar] [CrossRef]
- Venediktova, N.; Solomadin, I.; Starinets, V. Effect of Thyroxine on the Structural and Dynamic Features of Cardiac Mitochondria and Mitophagy in Rats. Cells 2023, 12, 396. [Google Scholar] [CrossRef]
- Harden, T.K.; Sesma, J.I.; Fricks, I.P.; Lazarowski, E.R.; Harden, T.K. Signalling and pharmacological properties of the P2Y receptor. Acta Physiol. 2010, 199, 149–160. [Google Scholar] [CrossRef]
- Iskra, D.A.; Butko, D.Y. Uridine and uridine-containing complexes for non-specific back pain. From pathogenesis to treatment. Nerv. Dis. 2020, 4, 20–24. [Google Scholar] [CrossRef]
- Lebrecht, D.; Vargas-Infante, Y.A.; Setzer, B.; Kirschner, J.; Walker, U.A. Uridine supplementation antagonizes zalcitabine-induced microvesicular steatohepatitis in mice. Hepatology 2007, 45, 72–79. [Google Scholar] [CrossRef]
- Le, T.T.; Urasaki, Y.; Pizzorno, G. Uridine prevents fenofibrate-induced fatty liver. PLoS ONE 2014, 9, e87179. [Google Scholar] [CrossRef]
- Le, T.T.; Urasaki, Y.; Pizzorno, G. Uridine prevents tamoxifen-induced liver lipid droplet accumulation. BMC Pharmacol. Toxicol. 2014, 23, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H. Modulation of ischemia by regulation of the ATP-sensitive potassium channel. Cardiovasc. Drugs Ther. 1993, 7, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Krylova, I.B.; Selina, E.N.; Bulion, V.V.; Rodionova, O.M.; Evdokimova, N.R.; Belosludtseva, N.V.; Shigaeva, M.I.; Mironova, G.D. Uridine treatment prevents myocardial injury in rat models of acute ischemia and ischemia/reperfusion by activating the mitochondrial ATP-dependent potassium channel. Sci. Rep. 2021, 11, 16999. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.L.; Zou, Z.; Yuan, H.B.; Wang, C.C.; Zhu, Q.F.; Xu, H.T.; Gao, X.; Shi, X.Y. Uridine 5’-triphosphate (UTP) protects against cerebral ischemia reperfusion injury in rats. Neurosci. Lett. 2009, 465, 55–60. [Google Scholar] [CrossRef]
- Venediktova, N.I.; Mashchenko, O.V.; Talanov, E.Y.; Belosludtseva, N.V.; Mironova, G.D. Energy metabolism and oxidative status of rat liver mitochondria in conditions of experimentally induced hyperthyroidism. Mitochondrion 2020, 52, 190–196. [Google Scholar] [CrossRef]
- Cioffi, F.; Giacco, A.; Goglia, F.; Silvestri, E. Bioenergetic Aspects of Mitochondrial Actions of Thyroid Hormones. Cells 2022, 11, 997. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, S.; Xie, C.; Fang, J. Uridine Metabolism and Its Role in Glucose, Lipid, and Amino Acid Homeostasis. Biomed. Res. Int. 2020, 2020, 7091718. [Google Scholar] [CrossRef]
- Urasaki, Y.; Pizzorno, G.; Le, T.T. Uridine affects liver protein glycosylation, insulin signaling, and heme biosynthesis. PLoS ONE 2014, 9, e99728. [Google Scholar] [CrossRef]
- Zhu, D.; Wei, Y.; Yin, J.; Liu, D.; Ang, E.L.; Zhao, H.; Zhang, Y. A pathway for degradation of uracil to acetyl coenzyme A in Bacillus megaterium. Appl. Environ. Microbiol. 2020, 86, e02837-19. [Google Scholar] [CrossRef]
- Connolly, G.P.; Duley, J.A. Uridine and its nucleotides: Biological actions, therapeutic potentials. Trends Pharmacol. Sci. 1999, 20, 218–225. [Google Scholar] [CrossRef]
- Skinner, O.S.; Blanco-Fernández, J.; Goodman, R.P.; Kawakami, A.; Shen, H.; Kemény, L.V.; Joesch-Cohen, L.; Rees, M.G.; Roth, J.A.; Fisher, D.E.; et al. Salvage of ribose from uridine or RNA supports glycolysis in nutrient-limited conditions. Nat. Metab. 2023, 5, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Geng, Q.; Huang, H.; Yao, H.; Du, T.; Chen, L.; Wu, Z.; Miao, X.; Shi, P. Antioxidative and Cardioprotective Effects of Schisandra chinensis Bee Pollen Extract on Isoprenaline-Induced Myocardial Infarction in Rats. Molecules 2019, 24, 1090. [Google Scholar] [CrossRef]
- Jiang, N.; Zhao, Z. Intestinal aging is alleviated by uridine via regulating inflammation and oxidative stress in vivo and in vitro. Cell Cycle 2022, 21, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Battaglia, V.; Brunati, A.M.; La Rocca, N.; Tibaldi, E.; Pietrangeli, P.; Marcocci, L.; Mondovı, B.; Rossi, C.A.; Toninello, A. Catalase Takes Part in Rat Liver Mitochondria Oxidative Stress Defense. J. Biol. Chem. 2007, 282, 24407–24415. [Google Scholar] [CrossRef]
- Lino, C.A.; Demasi, M.; Barreto-Chaves, M.L. Modulation of the ubiquitin proteasome system (UPS) in thyroid hormone-induced cardiac hypertrophy. FASEB J. 2012, 26, 1139. [Google Scholar] [CrossRef]
- Chi, H.C.; Chen, S.L.; Lin, S.L.; Tsai, C.Y.; Chuang, W.Y.; Lin, Y.H.; Huang, Y.H.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnover. Oncogene 2017, 36, 5274–5284. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation: III. The steady state. J. Biol. Chem. 1955, 217, 409–427. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Ermani, M.; Salviati, L.; Angelini, C. Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion 2011, 11, 893–904. [Google Scholar] [CrossRef]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Aebi, H.E. Catalase. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Elsevier: Amsterdam, The Netherlands, 1984; Volume 3, pp. 273–286. [Google Scholar]
- Kosenko, E.; Kaminsky, Y.; Kaminsky, A.; Valencia, M.; Lee, L.; Hermenegildo, C.; Felipo, V. Superoxide production and antioxidant enzymes in ammonia intoxication in rats. Free Radic. Res. 1997, 27, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | C + U | HT | HT + U | |
|---|---|---|---|---|
| T3 free, pmol/L | 5.2 ± 0.3 | 5.8 ± 0.4 | 9.3 ± 1.2 ** | 10 ± 1 **, ## |
| T4 free, pmol/L | 19.2 ± 1.0 | 19.1 ± 2.9 | 66.2 ± 4.4 ** | 70 ± 8 **, ## |
| Body weight, g | 272 ± 3.8 | 292 ± 8.3 | 238 ± 5.7 **, ## | 271 ± 6.8 § |
| Liver weight, g | 12.4 ± 0.4 | 12.1 ± 0.6 | 8.7 ± 0.4 **, ## | 9.8 ± 0.6 *, # |
| Body weight gain, g | 42.6 ± 5.1 | 56 ± 4.5 | 14.3 ± 1.4 **, ## | 30 ± 4.2 §, ## |
| ALT, µmol/min·mg | 42.7 ± 2.1 | 35 ± 1.1 | 43.9 ± 3.6 | 47 ± 2.4 |
| AST, µmol/min·mg | 65.5 ± 1.7 | 75.4 ± 1.5 | 106.7 ± 14 **, ## | 105.4 ± 6 **, # |
| LDH, µmol/min·mg | 258 ± 6.4 | 292 ± 16 | 393 ± 30 **, ## | 316 ± 11 § |
| State 2 | State 3 | State 4 | State DNP | RCR | ADP/O | |
|---|---|---|---|---|---|---|
| C | 3.7 ± 0.3 | 41.3 ± 0.8 | 4.5 ± 0.2 | 41 ± 0.8 | 9.2 ± 0.2 | 2.9 ± 0.04 |
| C + U | 3.4 ± 0.2 | 41.5 ± 0.8 | 4.6 ± 0.2 | 43 ± 1.3 | 9.1 ± 0.2 | 2.9 ± 0.02 |
| HT | 5.9 ± 0.4 **, ## | 52 ± 1.3 **, ## | 7.9 ± 0.4 **, ## | 47 ± 1.8 | 6.6 ± 0.3 **, ## | 2.6 ± 0.04 **, ## |
| HT + U | 5.6 ± 0.3 **, ## | 51.4 ± 1.3 **, ## | 7.2 ± 0.3 **, ## | 47 ± 1.6 | 7.2 ± 0.2 **, ## | 2.9 ± 0.03 §§ |
| C | C + U | HT | HT + U | |
|---|---|---|---|---|
| CS, nmol/min·mg | 222 ± 11 | 250 ± 6 | 300 ± 9 **, ## | 299 ± 9 **, ## |
| CI, nmol/min·mg | 101 ± 8 | 125 ± 8 | 135 ± 7 * | 145 ± 11 ** |
| CII, nmol/min·mg | 24 ± 0.6 | 29 ± 2.7 | 32 ± 1 ** | 36 ± 3.4 ** |
| CII+III, nmol/min·mg | 240 ± 18 | 200 ± 20 | 185 ± 13 | 233 ± 27 |
| CIII, nmol/min·mg | 400 ± 19 | 470 ± 19 | 320 ± 20 *, ## | 506 ± 54 *, §§ |
| CIV, nmol/min·mg | 1453 ± 91 | 1265 ± 85 | 1028 ± 117 ** | 1173 ± 70 |
| CV, nmol/min·mg | 820 ± 25 | 899 ± 53 | 701 ± 29 *, # | 922 ± 96 §§ |
| Gene | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| Drp1 | GATCCAGATGGGCGCAGAAC | ATGTCCAGTTGGCTCCTGTT |
| Mfn2 | AGCGTCCTCTCCCTCTGACA | TTCCACACCACTCCTCCGAC |
| OPA1 | GCAGAAGACAGCTTGAGGGT | TGCGTCCCACTGTTGCTTAT |
| PINK1 | GATGTGGAATATCTCGGCAGGA | TGTTTGCTGAACCCAAGGCT |
| Parkin | GGCCAGAGGAAAGTCACCTG | CACCCGGTATGCCTGAGAAG |
| Ppargc1α | TGACATAGAGTGTGCTGCCC | GCTGTCTGTGTCCAGGTCAT |
| Nrf1 | TACAAGGCGGGGGACAGATA | TGCATGAACTCCATCTGGGC |
| Actb | GACCCAGATCATGTTTGAGACCT | CCAGAGGCATACAGGGACAAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venediktova, N.; Solomadin, I.; Nikiforova, A.; Belosludtsev, K.N.; Mironova, G. Effects of the Long-Term Administration of Uridine on the Functioning of Rat Liver Mitochondria in Hyperthyroidism. Int. J. Mol. Sci. 2023, 24, 16730. https://doi.org/10.3390/ijms242316730
Venediktova N, Solomadin I, Nikiforova A, Belosludtsev KN, Mironova G. Effects of the Long-Term Administration of Uridine on the Functioning of Rat Liver Mitochondria in Hyperthyroidism. International Journal of Molecular Sciences. 2023; 24(23):16730. https://doi.org/10.3390/ijms242316730
Chicago/Turabian StyleVenediktova, Natalya, Ilya Solomadin, Anna Nikiforova, Konstantin N. Belosludtsev, and Galina Mironova. 2023. "Effects of the Long-Term Administration of Uridine on the Functioning of Rat Liver Mitochondria in Hyperthyroidism" International Journal of Molecular Sciences 24, no. 23: 16730. https://doi.org/10.3390/ijms242316730
APA StyleVenediktova, N., Solomadin, I., Nikiforova, A., Belosludtsev, K. N., & Mironova, G. (2023). Effects of the Long-Term Administration of Uridine on the Functioning of Rat Liver Mitochondria in Hyperthyroidism. International Journal of Molecular Sciences, 24(23), 16730. https://doi.org/10.3390/ijms242316730