
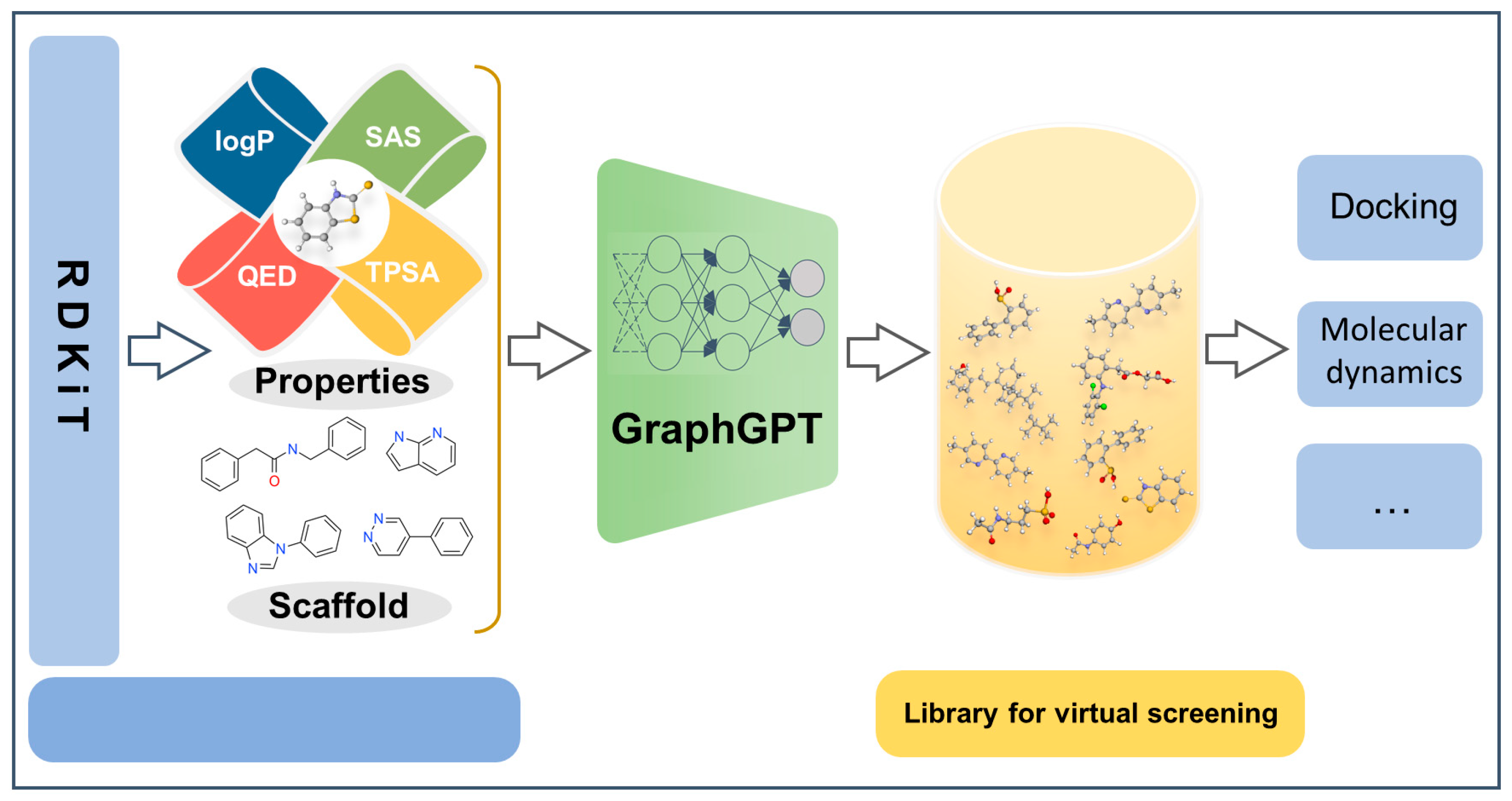
GraphGPT: A Graph Enhanced Generative Pretrained Transformer for Conditioned Molecular Generation


Abstract
:1. Introduction
2. Results
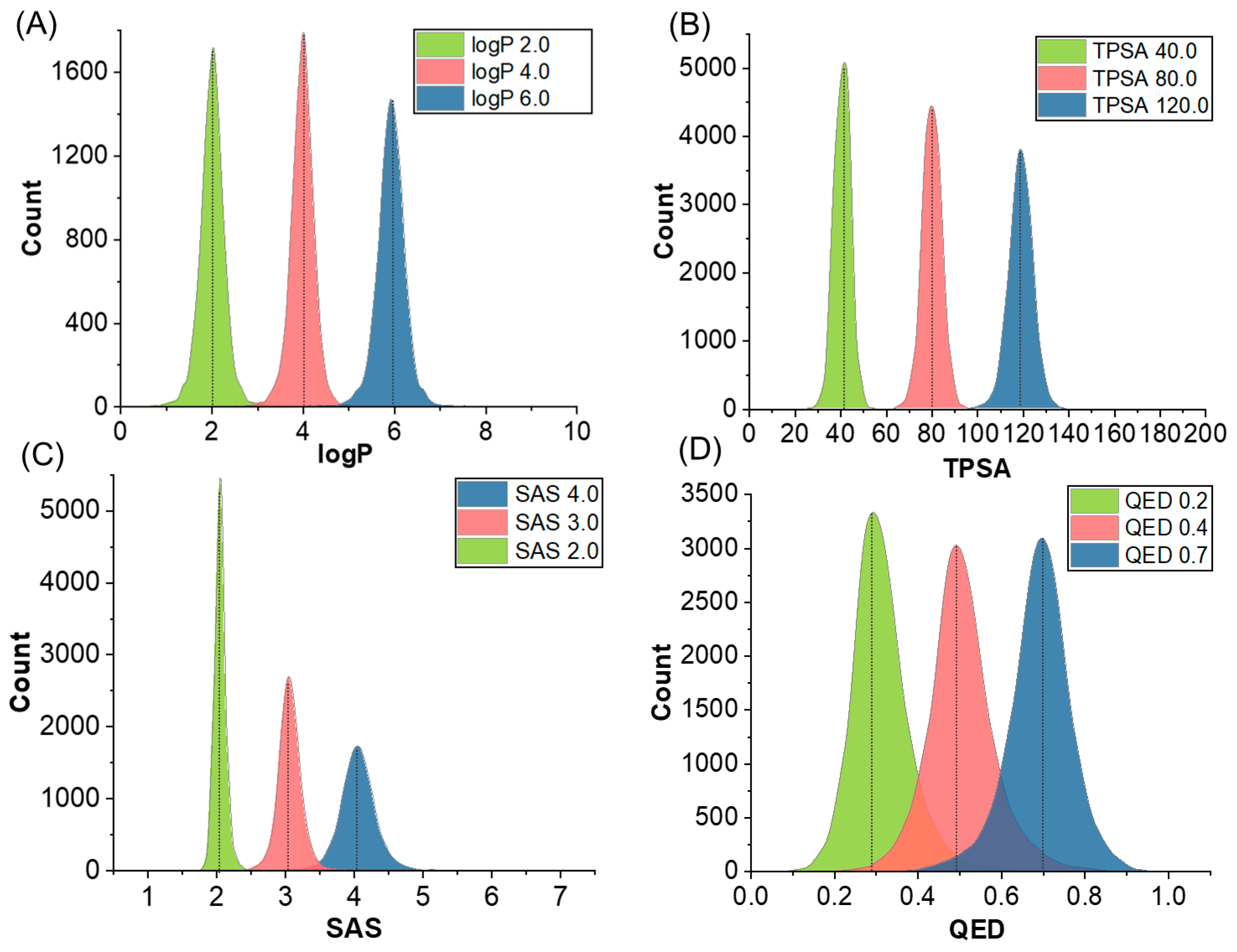
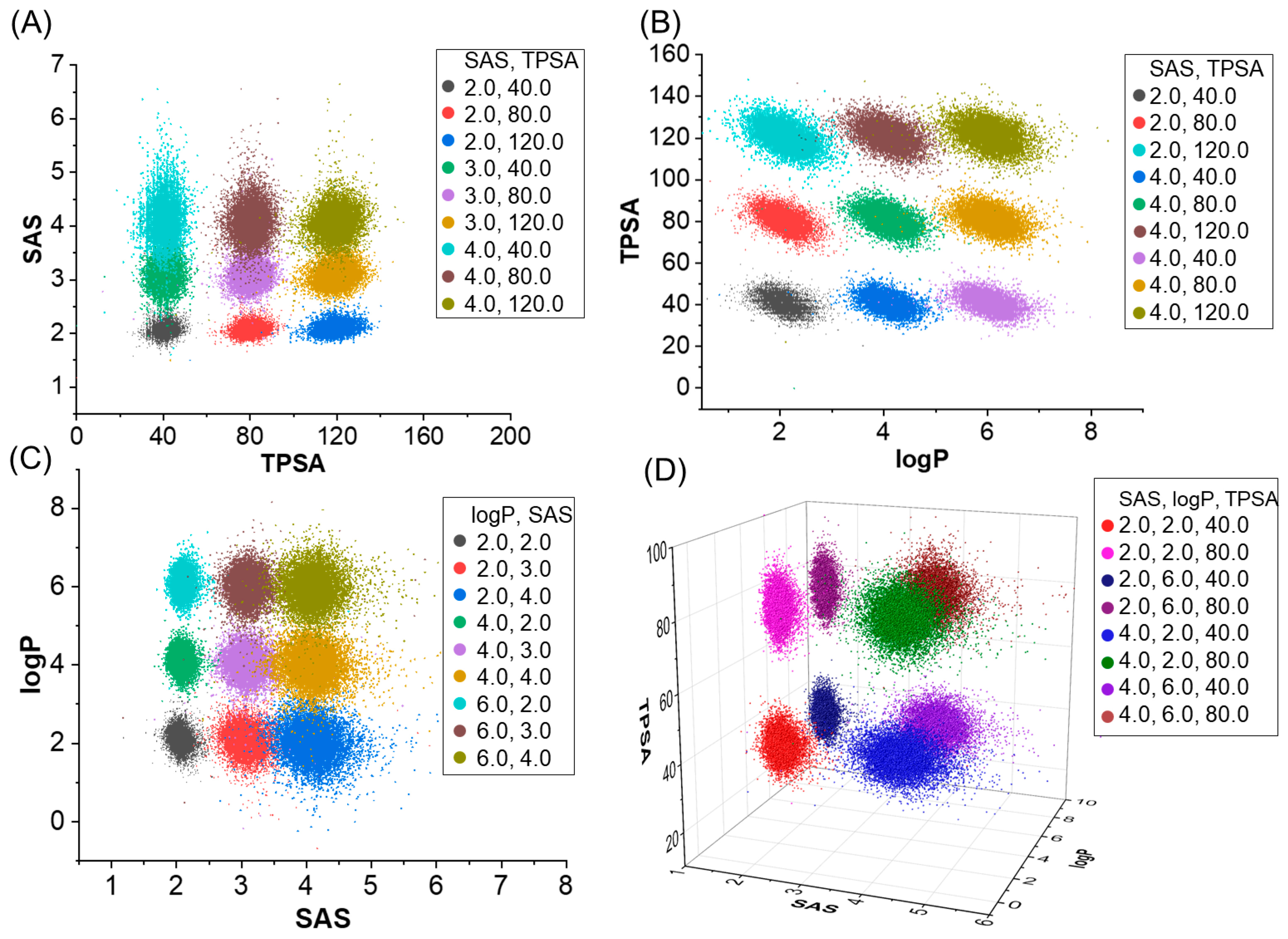
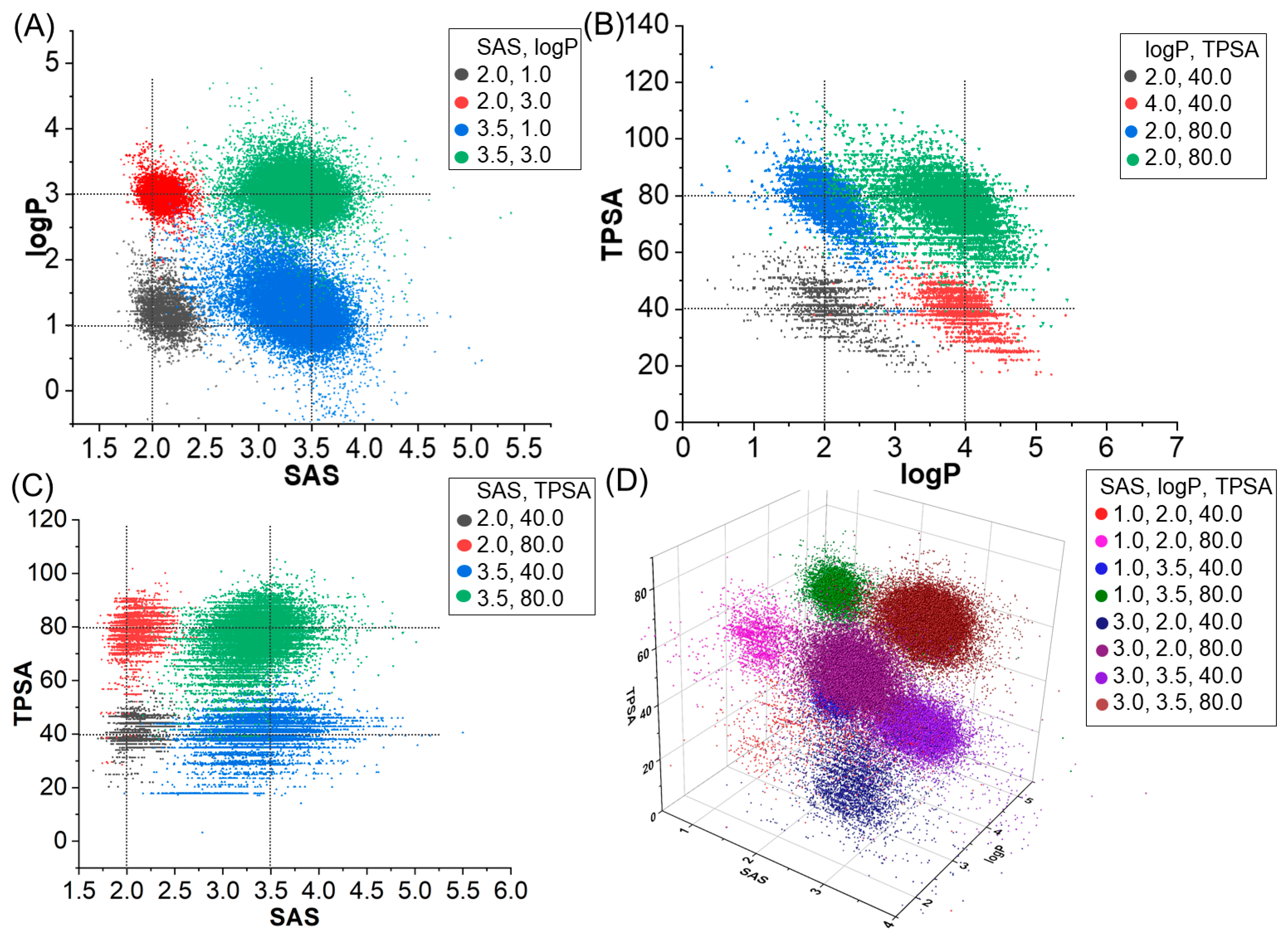
2.1. Molecular Generation Based on Properties
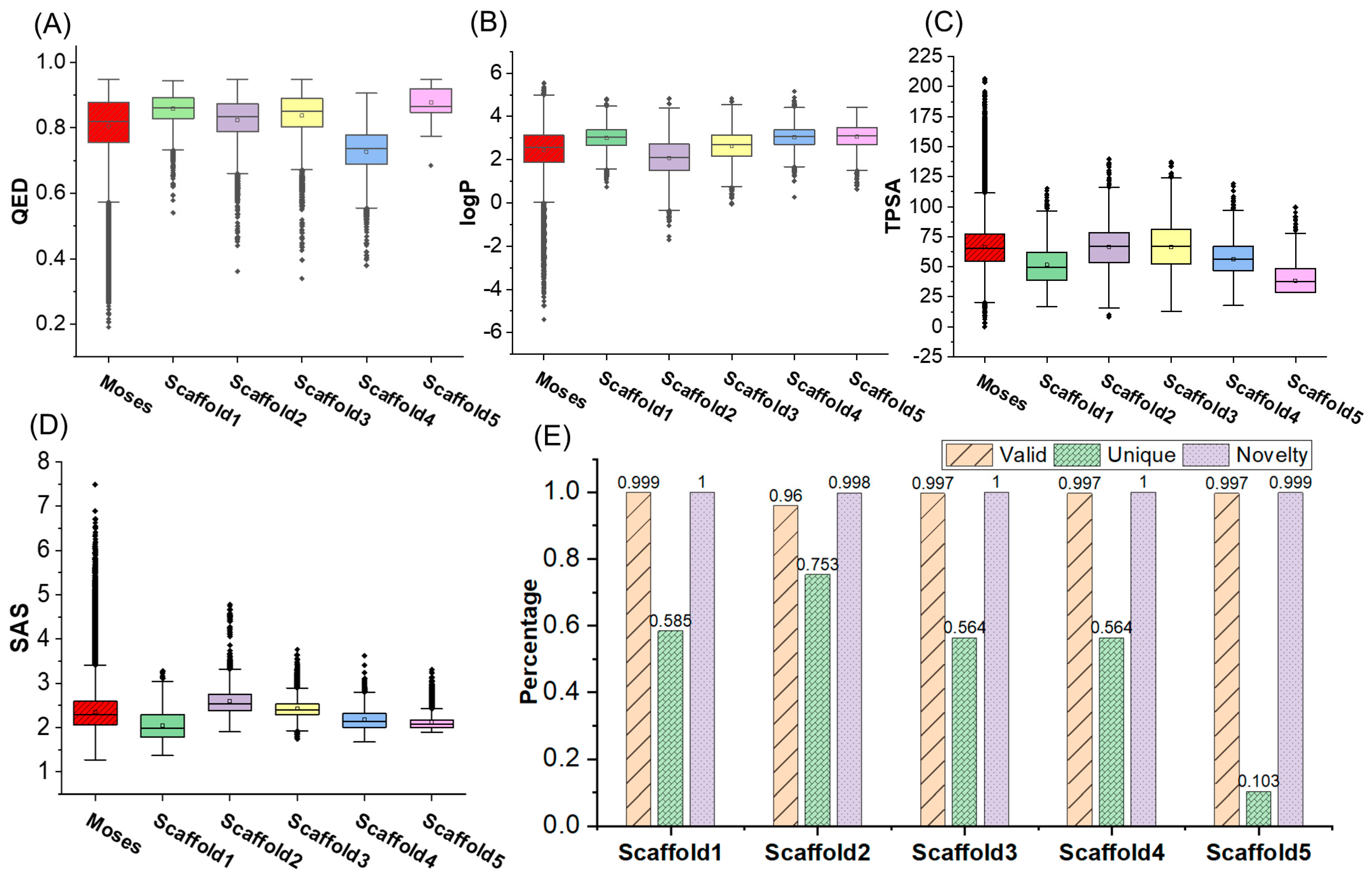
2.2. Molecular Generation Based on Properties and Scaffold
2.3. Unconditional Molecular Generation
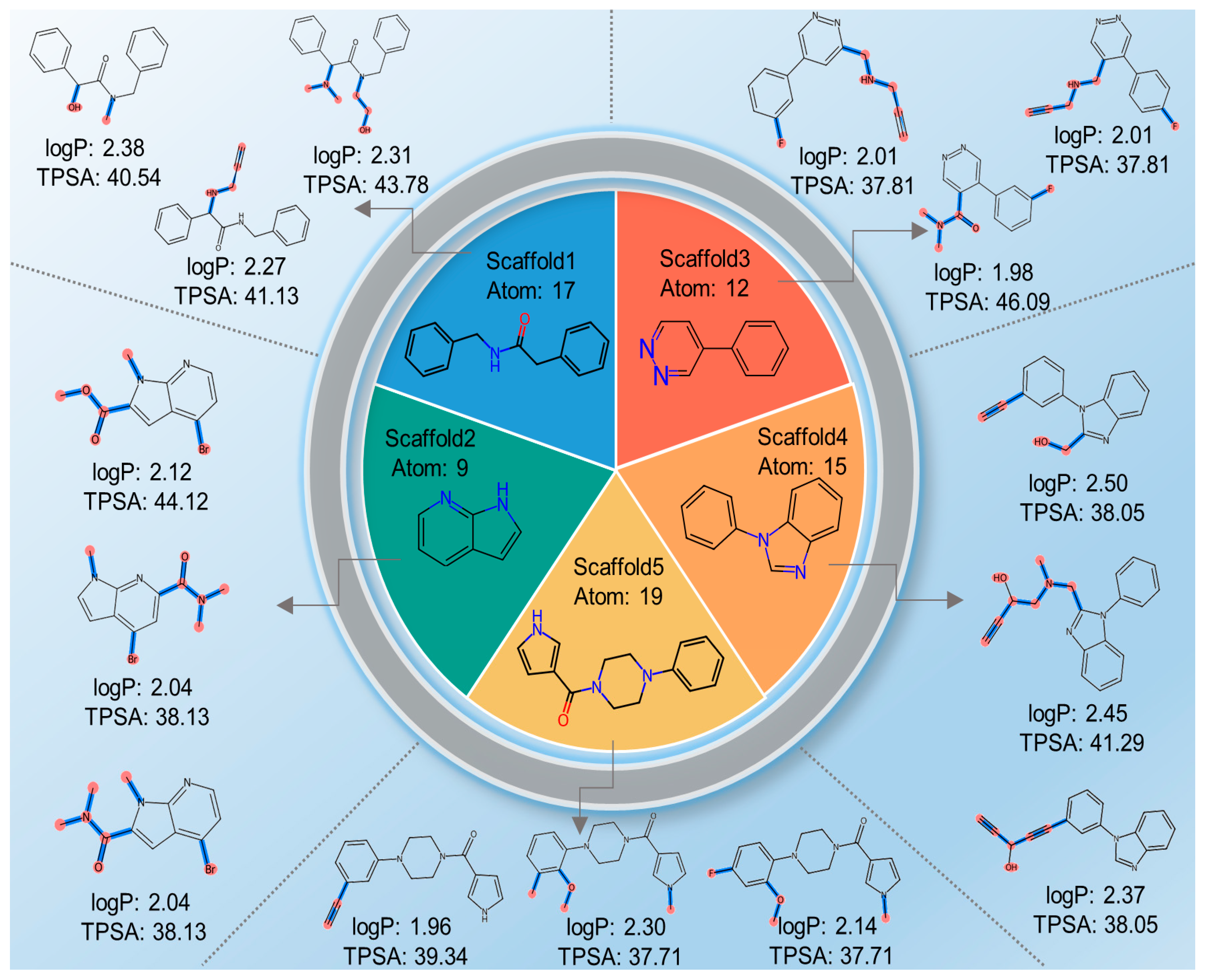
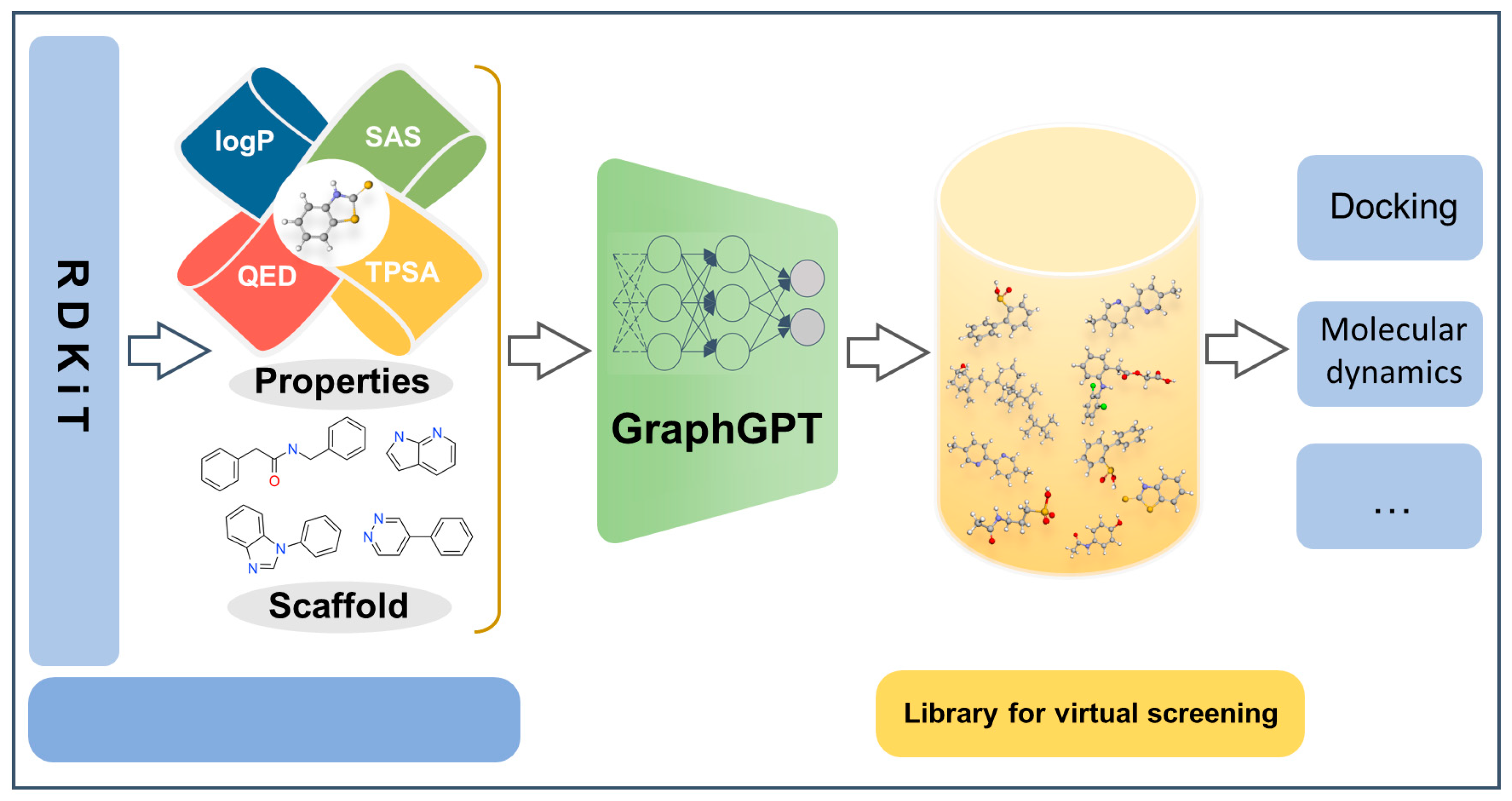
2.4. Case Study
2.5. Ablation Experiment
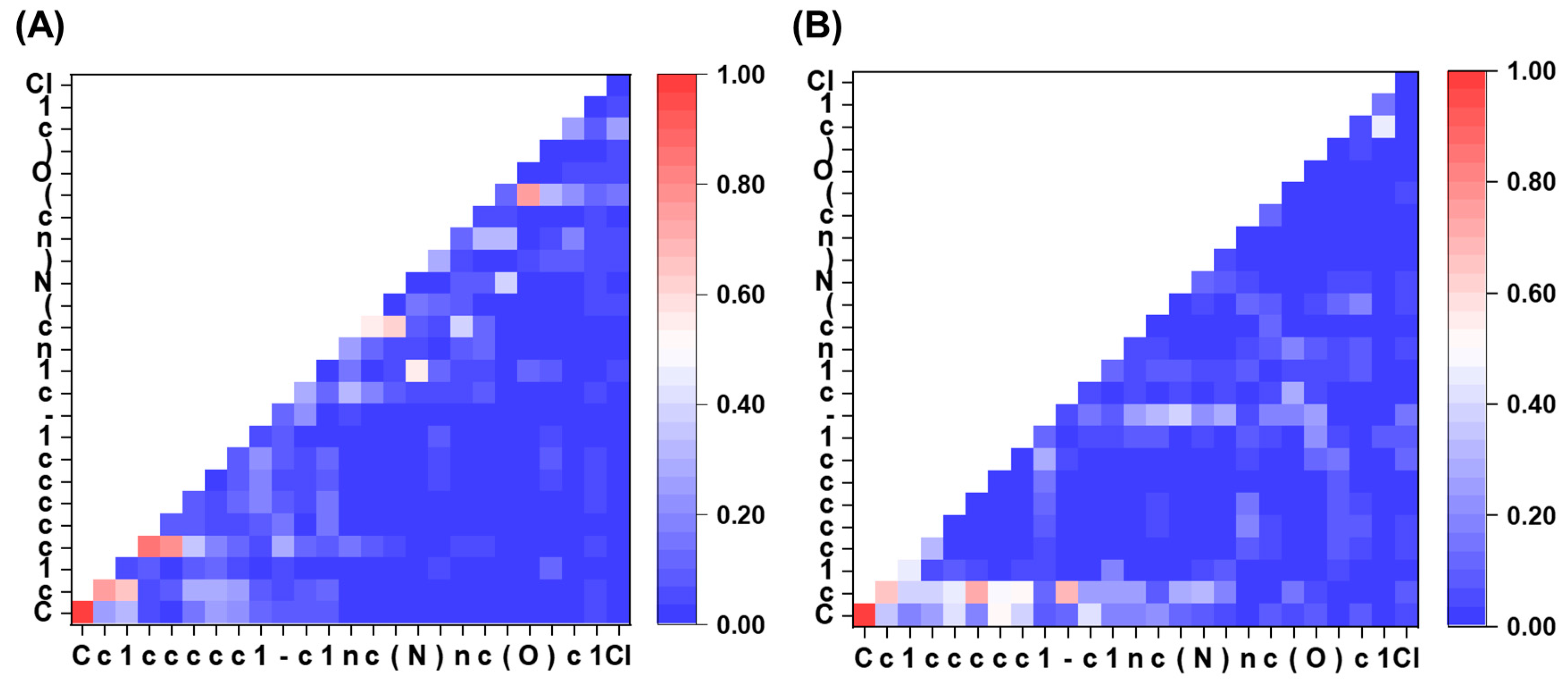
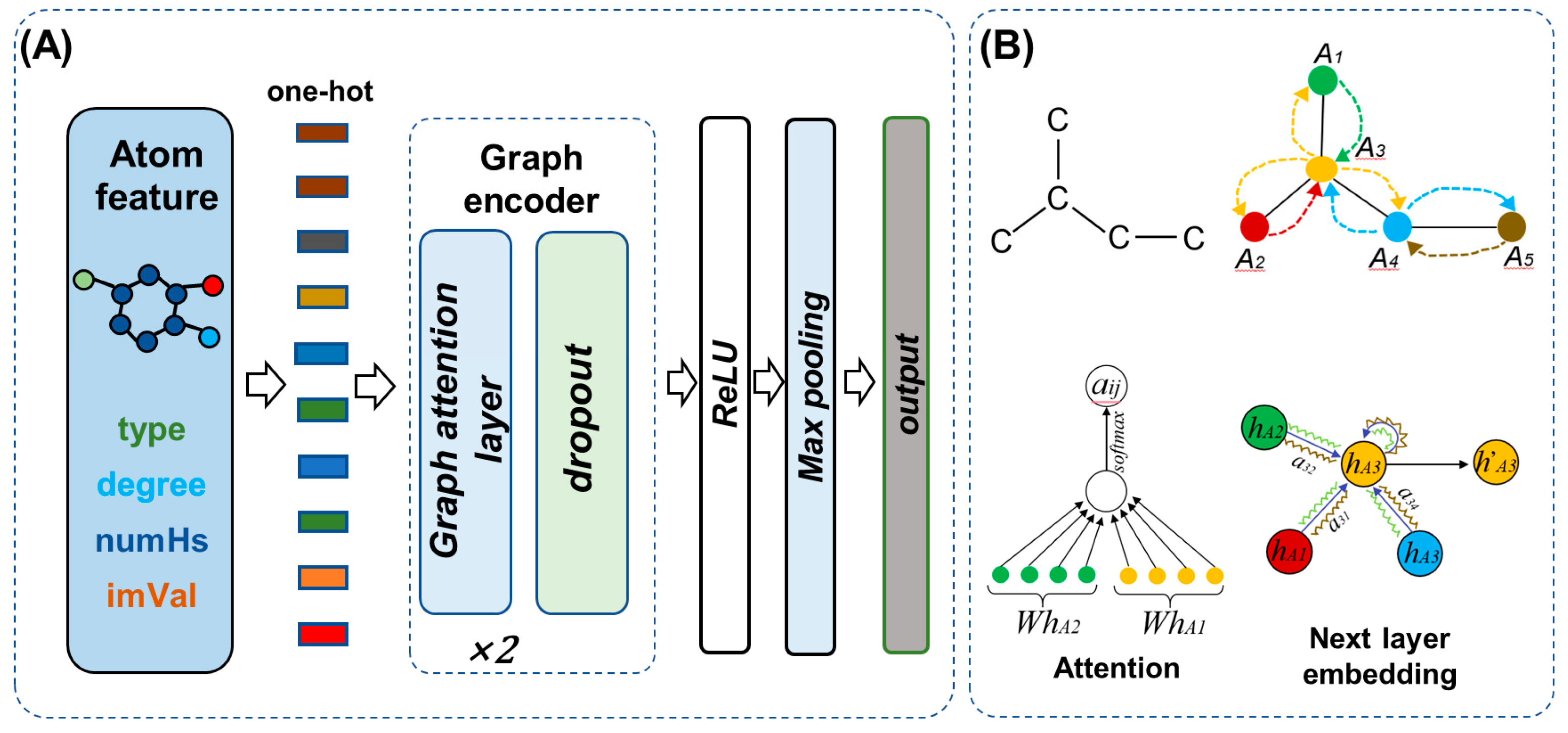
2.6. Attention Visualization
3. Discussion
4. Methods and Materials
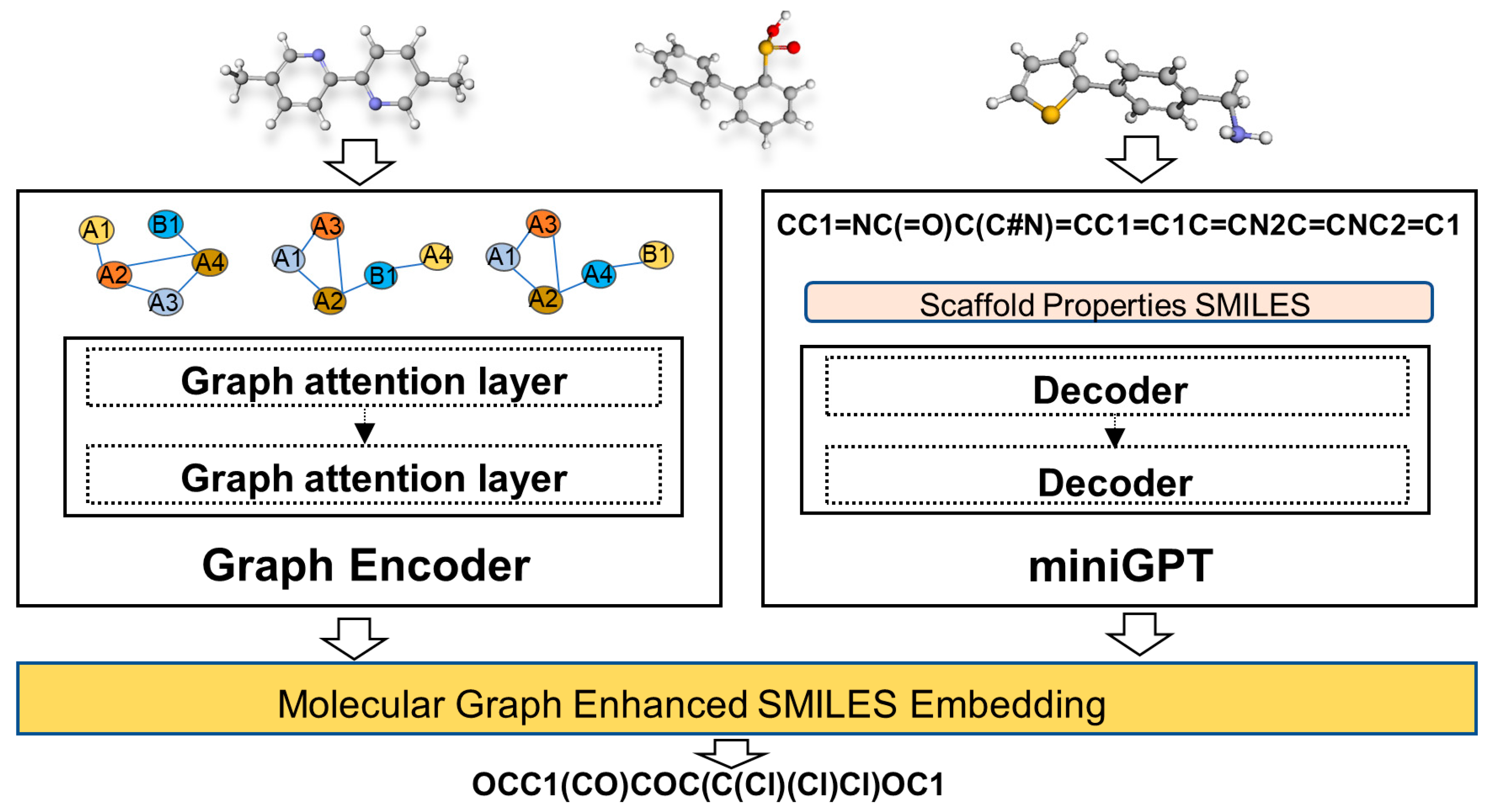
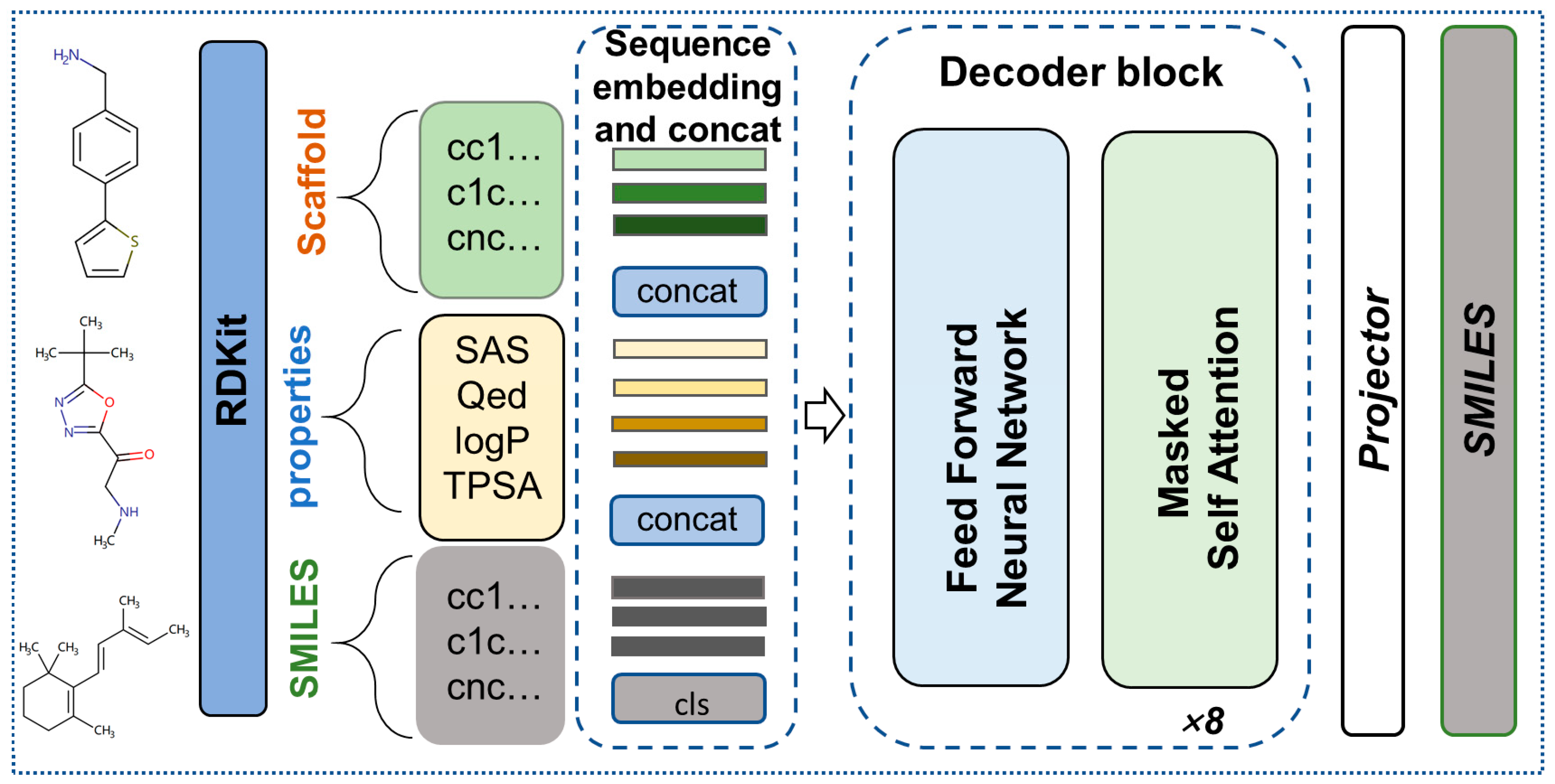
4.1. Overview of the Model
4.2. Graph Encoder
4.3. GPT encoding of SMILES and Properties
4.4. Optimum Objectives
4.5. Dataset
4.6. Metrics
- Valid: Valid pertains to the valid portions within the generated molecules based on SMILES syntax and atomic valency rules. We consider a molecule valid when the generated SMILES can be analyzed using an RDKit. A high valid score indicates that the model has learned the accurate representation of molecules and their chemical properties.
- Unique: Unique specifies that it is a case of duplicates in the generated molecule. If the newly generated molecule has not been generated before, then it is considered ideal. A lower uniqueness score suggests that the model is generating repetitive or redundant molecules.
- Novelty: Novelty refers to the segments present in the generated valid and unique molecules that are absent in the training dataset. This metric is employed to determine whether the model is overfitting, signifying that it has memorized the training data without generalizing to unseen molecules.
- Internal Diversity (IntDivp): Internal Diversity evaluates the similarity between generated molecules. As shown in Equation (8), s1 and s2 denote two molecules, and T represents Tanimoto similarity [48]. This entails similarity comparisons between all pairs of molecules within the generated set (S). The parameter p can be either 1 or 2.
- Frechet ChemNet Distance (FCD): This metric tests the similarity of the generated molecular data to the training molecular data. As shown in Equation (9), where μG is the mean and ΣG is the covariance of the distribution G. In the same way as Bagal et al. [36] for the Guacamol data set, the final value is −0.2 power of FCD.
- KL Divergence: KL Divergence was computed using a plethora of physicochemical descriptors for both the generated molecules and the training set. Lower values denote a proficient learning of the distribution of these properties by the model. The calculation is shown in Equations (10) and (11). Here, k represents the kth properties.
4.7. Baselines
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, Y.; Gong, Y.; Liu, Y.; Song, B.; Zou, Q. Molecular design in drug discovery: A comprehensive review of deep generative models. Brief. Bioinform. 2021, 22, bbab344. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, M.; Wang, S.; Zhang, S. Deep learning methods for molecular representation and property prediction. Drug Discov. Today 2023, 27, 103373. [Google Scholar] [CrossRef] [PubMed]
- Urbina, F.; Lentzos, F.; Invernizzi, C.; Ekins, S. Dual use of artificial-intelligence-powered drug discovery. Nat. Mach. Intell. 2022, 4, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Nagra, N.S.; Lieven, V.D.V.; Stanzl, E.; Champagne, D.; Devereson, A.; Macak, M. The company landscape for artificial intelligence in large-molecule drug discovery. Nat. Rev. Drug Discov. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Chen, G.; Seukep, A.J.; Guo, M. Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs. Mar. Drugs 2020, 18, 545. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, H.; Zheng, H.; Wang, L.; Zhang, G.; Yang, J.; Lu, X.; Bai, Y.; Zhang, H.; Li, J.; et al. Evaluation of drug efficacy based on the spatial position comparison of drug–target interaction centers. Brief. Bioinform. 2020, 21, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, O.; Shen, C.; Qu, W.; Chen, S.; Cao, H.; Kang, Y.; Wang, Z.; Wang, E.; Zhang, J.; et al. Efficient and accurate large library ligand docking with KarmaDock. Nat. Comput. Sci. 2023, 3, 789–804. [Google Scholar] [CrossRef]
- Kuan, J.; Radaeva, M.; Avenido, A.; Cherkasov, A.; Gentile, F. Keeping pace with the explosive growth of chemical libraries with structure-based virtual screening. Wires Comput. Mol. 2023, 13, e1678. [Google Scholar] [CrossRef]
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput.-Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef]
- Sarkar, C.; Das, B.; Rawat, V.S.; Wahlang, J.B.; Nongpiur, A.; Tiewsoh, I.; Lyngdoh, N.M.; Das, D.; Bidarolli, M.; Sony, H.T. Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development. Int. J. Mol. Sci. 2023, 24, 2026. [Google Scholar] [CrossRef]
- Westermayr, J.; Gilkes, J.; Barrett, R.; Maurer, R. High-throughput property-driven generative design of functional organic molecules. Nat. Comput. Sci. 2023, 3, 139–148. [Google Scholar] [CrossRef]
- Liu, Q.; Allamanis, M.; Brockschmidt, M.; Gaunt, A.L. Constrained Graph Variational Autoencoders for Molecule Design. In Proceedings of the 2020 IEEE Symposium Series on Computational Intelligence SSCI, Canberra, Australia, 1–4 December 2020. [Google Scholar]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. In Proceedings of the International Conference on Learning Representations ICLR, Banff, AB, Canada, 14–16 April 2014. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Networks. In Proceedings of the 27th International Conference on Neural Information Processing Systems NIPS, Montreal, QC, Canada, 8–13 December 2014; Volume 2, pp. 2672–2680. [Google Scholar]
- Zhou, Z.; Kearnes, S.; Li, L.; Zare, R.N.; Riley, P. Optimization of Molecules via Deep Reinforcement Learning. Sci. Rep. 2019, 9, 10752. [Google Scholar] [CrossRef]
- Bilodeau, C.; Jin, W.; Jaakkola, T.; Barzilay, R.; Jensen, K.F. Generative models for molecular discovery: Recent advances and challenges. Wires Comput. Mol. 2022, 12, e1608. [Google Scholar] [CrossRef]
- Li, S.; Hu, C.; Ke, S.; Yang, C.; Chen, J.; Xiong, Y.; Liu, H.; Hong, L. LS-MolGen: Ligand-and-Structure Dual-Driven Deep Reinforcement Learning for 411 Target-Specific Molecular Generation Improves Binding Affinity and Novelty. J. Chem. Inf. Model. 2023, 63, 4207–4215. [Google Scholar] [CrossRef]
- Liu, X.; Ye, K.; van Vlijmen, H.W.T.; IJzerman, A.P.; van Westen, G.J.P. DrugEx v3: Scaffold-constrained drug design with graph transformer-based reinforcement learning. J. Cheminform. 2023, 37, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Z.; Sun, H.; Wang, H.; Wang, J.; Shen, C.; Weng, G.; Chai, X.; Li, H.; Cao, D.; et al. Deep learning approaches for de novo drug design: An overview. Curr. Opin. Struct. Biol. 2022, 72, 135–144. [Google Scholar] [CrossRef]
- Cai, C.; Wang, S.; Xu, Y.; Zhang, W.; Tang, K.; Ouyang, Q.; Lai, L.; Pei, J. Transfer Learning for Drug Discovery. J. Med. Chem. 2020, 63, 8683–8694. [Google Scholar] [CrossRef]
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language Models are Few-Shot Learners. Adv. Neural Inf. Process. Syst. 2020, 33, 1877–1901. [Google Scholar]
- OPENAI. GPT-4 Technical Report. arXiv 2023, arXiv:2303.08774. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Tysinger, E.P.; Rai, B.K.; Sinitskiy, A.V. Can We Quickly Learn to “Translate” Bioactive Molecules with Transformer Models. J. Chem. Inf. Model. 2023, 63, 1734–1744. [Google Scholar] [CrossRef]
- Mokaya, M.; Imrie, F.; Van Hoorn, W.P.; Kalisz, A.; Bradley, A.R.; Deane, C.M. Testing the limits of SMILES-based de novo molecular generation with curriculum and deep reinforcement learning. Nat. Mach. Intell. 2023, 5, 386–394. [Google Scholar] [CrossRef]
- Dwivedi, V.P.; Bresson, X. A Generalization of Transformer Networks to Graphs. In Proceedings of the AAAI 2021 Workshop on Deep Learning on Graphs: Methods and Applications, DGL-AAAI, Virtual, 8–9 February 2021. [Google Scholar]
- Vaswani, A.; Shazeer, N.M.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. In Proceedings of the 31st International Conference on 441 Neural Information Processing Systems, NIPS, Long Beach, CA, USA, 4–9 December 2017; pp. 6000–6010. [Google Scholar]
- Ying, C.; Cai, T.; Luo, S.; Luo, S.; Zheng, S.; Ke, G.; He, D.; Shen, Y.; Liu, T.-Y. Do Transformers Really Perform Bad for Graph Representation? Arxiv 2021, arXiv:2106.05234. [Google Scholar]
- Chen, D.; O’Bray, L.; Borgwardt, K.M. Structure-Aware Transformer for Graph Representation Learning. In Proceedings of the International Conference on Machine Learning, ICML, Baltimore, MD, USA, 17–23 July 2022; Volume 162, pp. 3469–3489. [Google Scholar]
- Luo, S.; Chen, T.; Xu, Y.; Zheng, S.; Liu, T.Y.; He, D.; Wang, L. One Transformer Can Understand Both 2D & 3D Molecular Data. In Proceedings of the International Conference on Learning Representations, ICLR, Kigali, Rwanda, 1–5 May 2023. [Google Scholar]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Abraham, M.H.; Chadha, H.S.; Leitao, R.E.; Mitchell, R.C.; Lambert, W.J.; Kaliszan, R.; Nasal, A.; Haber, P. Determination of solute lipophilicity, as log P(octanol) and log P(alkane) using 481 poly(styrene–divinylbenzene) and immobilised artificial membrane stationary phases in reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1997, 766, 35–47. [Google Scholar] [CrossRef]
- Zhong, H.; Mashinson, V.; Woolman, T.A.; Zha, M. Understanding the Molecular Properties and Metabolism of Top Prescribed Drugs. Curr. Top. Med. Chem. 2013, 13, 1290–1307. [Google Scholar] [CrossRef]
- Bagal, V.; Aggarwal, R.; Vinod, P.K.; Priyakumar, U.D. MolGPT: Molecular Generation Using a Transformer-Decoder Model. J. Chem. Inf. Model. 2022, 62, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; Fiscato, M.; Segler, M.H.S.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Preuer, K.; Renz, P.; Unterthiner, T.; Hochreiter, S.; Klambauer, G. Frechet ChemNet Distance: A Metric for Generative Models for Molecules in Drug Discovery. J. Chem. Inf. Model. 2018, 58, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovabov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 2020, 11, 565644. [Google Scholar] [CrossRef]
- Krenn, M.; Häse, F.; Nigam, A.; Friederich, P.; Apuru-Guzik, A. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 2020, 1, 045024. [Google Scholar] [CrossRef]
- Zhong, Z.; Song, J.; Feng, Z.; Liu, T.; Jia, L.; Yao, S.; Wu, M.; Hou, T.; Song, M. Root-aligned SMILES: A tight representation for chemical reaction prediction. Chem. Sci. 2022, 13, 9023–9034. [Google Scholar] [CrossRef]
- Velickovic, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph Attention Networks. International Conference on Learning Representations. arXiv 2018, arXiv:1710.10903. [Google Scholar]
- RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org (accessed on 22 November 2023).
- Zbontar, J.; Jing, L.; Misra, I.; LeCun, Y.; Deny, S. Barlow Twins: Self-Supervised Learning via Redundancy Reduction. arXiv 2021, arXiv:2103.03230. [Google Scholar]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Vogt, M.; Bajorath, J. Modeling Tanimoto Similarity Value Distributions and Predicting Search Results. Mol. Inform. 2017, 67, 1600131. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- Gomez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernandez-Lobato, J.M.; Sanchez-Lengeling, S.; Sheberla, D.; Aguilera-Lparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zhebrak, A.; Vetrov, D.; Ivanenkov, Y.; Aladinskiy, V.; Mamoshina, P.; Bozdaganyan, M.; Aliper, A.; Zhavoronkov, A.; Kadurin, A. Entangled Conditional Adversarial Autoencoder for de Novo Drug Discovery. Mol. Pharm. 2018, 15, 4398–4405. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction Tree Variational Autoencoder for Molecular Graph Generation. arXiv 2018, arXiv:1802.04364. [Google Scholar]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.C.; Arus-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef]
- Guimaraes, G.; Sanchez-Lengeling, B.; Outeiral, C.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models. arXiv 2018, arXiv:1705.10843. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Valid ↑ | Unique ↑ | Novelty ↑ | SD ↓ | MAD ↓ | |
|---|---|---|---|---|---|---|
| logP | MolGPT | 0.971 | 0.998 | 1 | 0.31 | 0.23 |
| ours | 0.969 | 0.998 | 1 | 0.29 | 0.22 | |
| TPSA | MolGPT | 0.972 | 0.996 | 1 | 4.66 | 3.52 |
| ours | 0.971 | 0.997 | 1 | 4.21 | 3.31 | |
| QED | MolGPT | 0.977 | 0.995 | 1 | 0.20 | 0.13 |
| ours | 0.968 | 0.999 | 1 | 0.07 | 0.05 | |
| SAS | MolGPT | 0.975 | 0.998 | 1 | 0.20 | 0.13 |
| ours | 0.977 | 0.996 | 1 | 0.19 | 0.14 |
| Model | Valid ↑ | Unique ↑ | Novelty ↑ | SD/MAD ↓ | |||
|---|---|---|---|---|---|---|---|
| logP | SAS | TPSA | |||||
| logP + SAS | MolGPT | 0.972 | 0.992 | 1 | 0.340/0.250 | 0.210/0.140 | |
| ours | 0.971 | 0.991 | 1 | 0.331/0.252 | 0.208/0.151 | ||
| SAS + TPSA | MolGPT | 0.971 | 0.988 | 1 | 0.220/0.150 | 4.940/3.760 | |
| ours | 0.970 | 0.988 | 1 | 0.217/0.158 | 4.705/3.647 | ||
| logP + TPSA | MolGPT | 0.965 | 0.994 | 1 | 0.320/0.240 | 4.770/3.710 | |
| ours | 0.961 | 0.995 | 1 | 0.314/0.240 | 4.575/3.607 | ||
| logP + TPSA + SAS | MolGPT | 0.973 | 0.969 | 1 | 0.350/0.270 | 0.260/0.180 | 4.800/3.790 |
| ours | 0.966 | 0.964 | 1 | 0.335/0.259 | 0.247/0.183 | 4.461/3.532 | |
| Valid ↑ | Unique ↑ | Novelty ↑ | FCD ↑ | KL Divergence ↑ | |
|---|---|---|---|---|---|
| SMILES LSTM | 0.959 | 1 | 0.912 | 0.913 | 0.991 |
| AAE | 0.822 | 1 | 0.998 | 0.529 | 0.886 |
| Organ | 0.379 | 0.841 | 0.687 | 0 | 0.267 |
| VAE | 0.87 | 0.999 | 0.974 | 0.863 | 0.982 |
| MolGPT | 0.981 | 0.998 | 1 | 0.907 | 0.992 |
| ours | 0.975 | 0.999 | 1 | 1.009 | 0.992 |
| Valid ↑ | Unique ↑ | Novelty ↑ | IntDiv1 ↓ | IntDiv2 ↓ | |
|---|---|---|---|---|---|
| charRNN | 0.975 | 0.999 | 0.842 | 0.856 | 0.850 |
| VAE | 0.977 | 0.998 | 0.695 | 0.856 | 0.850 |
| AAE | 0.937 | 0.997 | 0.793 | 0.856 | 0.850 |
| LatentGAN | 0.897 | 0.997 | 0.949 | 0.857 | 0.850 |
| JT-VAE | 1 | 0.999 | 0.914 | 0.855 | 0.849 |
| MolGPT | 0.994 | 1 | 0.797 | 0.857 | 0.851 |
| ours | 0.995 | 0.999 | 0.787 | 0.851 | 0.845 |
| Graph Encoder | Decoder Layer | Valid ↑ | Unique ↑ | Novelty ↑ | SD ↓ | MAD ↓ | |
|---|---|---|---|---|---|---|---|
| miniGPT_a | × | 4 | 0.946 | 0.999 | 1 | 5.017 | 3.806 |
| miniGPT_b | × | 8 | 0.972 | 0.997 | 1 | 4.474 | 3.485 |
| miniGPT_c | × | 12 | 0.977 | 0.996 | 1 | 4.240 | 3.238 |
| GraphGPT | √ | 8 | 0.971 | 0.997 | 1 | 4.208 | 3.307 |
| GPT-1 | GraphGPT | |
|---|---|---|
| Decoder layer | 12 | 8 |
| Attention header | 12 | 8 |
| Dimensions of vocab | 768 | 256 |
| Sequence length | 512 | 100 |
| Parameter | 117 million | 7.07 million |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Wei, Z.; Wang, X.; Zhang, K.; Liu, H. GraphGPT: A Graph Enhanced Generative Pretrained Transformer for Conditioned Molecular Generation. Int. J. Mol. Sci. 2023, 24, 16761. https://doi.org/10.3390/ijms242316761
Lu H, Wei Z, Wang X, Zhang K, Liu H. GraphGPT: A Graph Enhanced Generative Pretrained Transformer for Conditioned Molecular Generation. International Journal of Molecular Sciences. 2023; 24(23):16761. https://doi.org/10.3390/ijms242316761
Chicago/Turabian StyleLu, Hao, Zhiqiang Wei, Xuze Wang, Kun Zhang, and Hao Liu. 2023. "GraphGPT: A Graph Enhanced Generative Pretrained Transformer for Conditioned Molecular Generation" International Journal of Molecular Sciences 24, no. 23: 16761. https://doi.org/10.3390/ijms242316761
APA StyleLu, H., Wei, Z., Wang, X., Zhang, K., & Liu, H. (2023). GraphGPT: A Graph Enhanced Generative Pretrained Transformer for Conditioned Molecular Generation. International Journal of Molecular Sciences, 24(23), 16761. https://doi.org/10.3390/ijms242316761