
Sex-Related Gene Network Revealed by Transcriptome Differentiation of Bisexual and Unisexual Flowers of Orchid Cymbidium tortisepalum

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Morphological Comparison of Sexual Phenotypes in C. tortisepalum
2.2. Transcriptome Sequencing, Assembly, and Annotation
2.3. Expression Levels among the Three Sexual Phenotypes
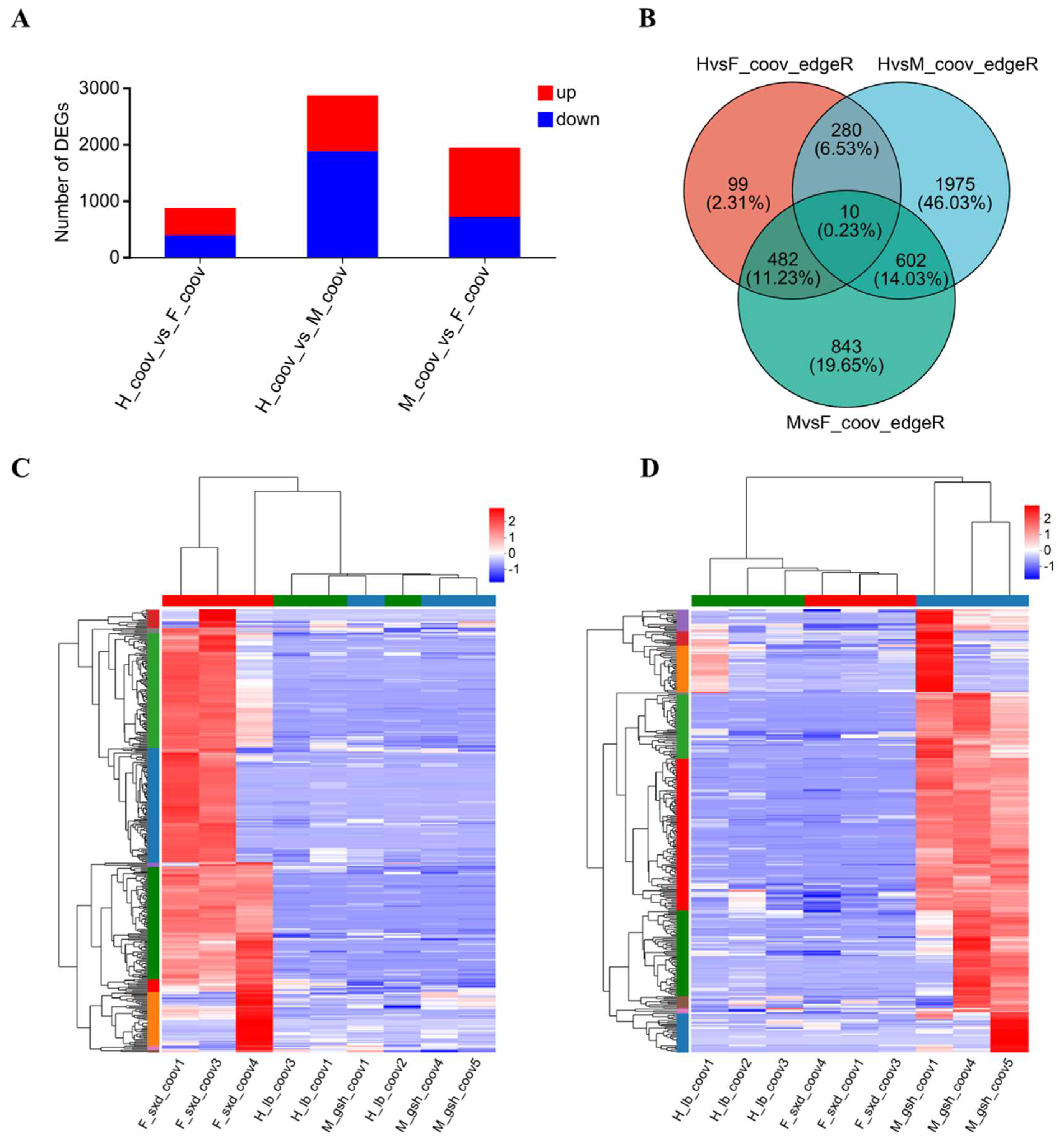
2.4. Sex-Biased DEGs among the Three Sexual Phenotypes
2.5. GO and KEGG Enrichments of Sex-Biased DEGs
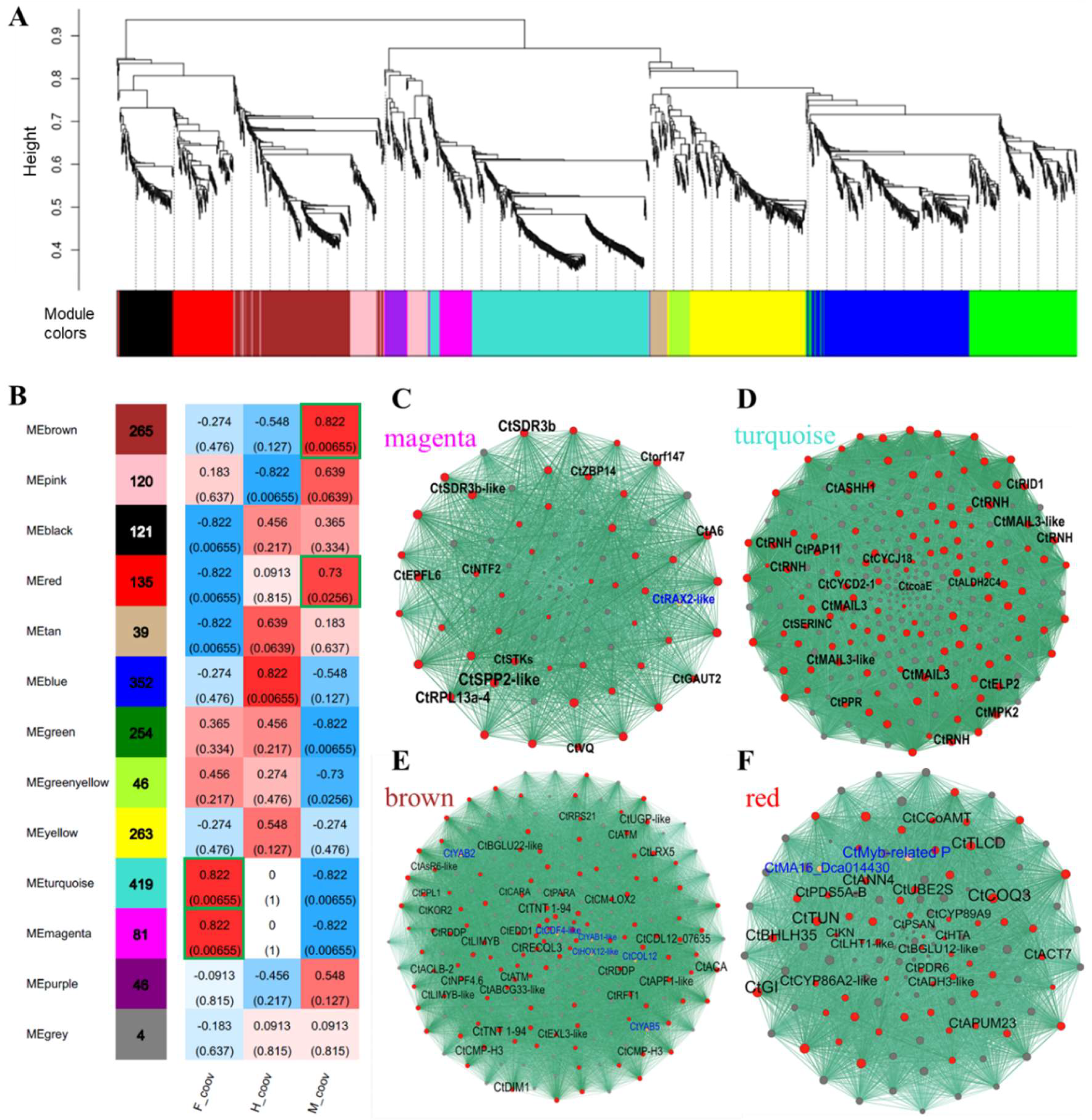
2.6. Co-Expression Networks of Sex-Biased DEGs
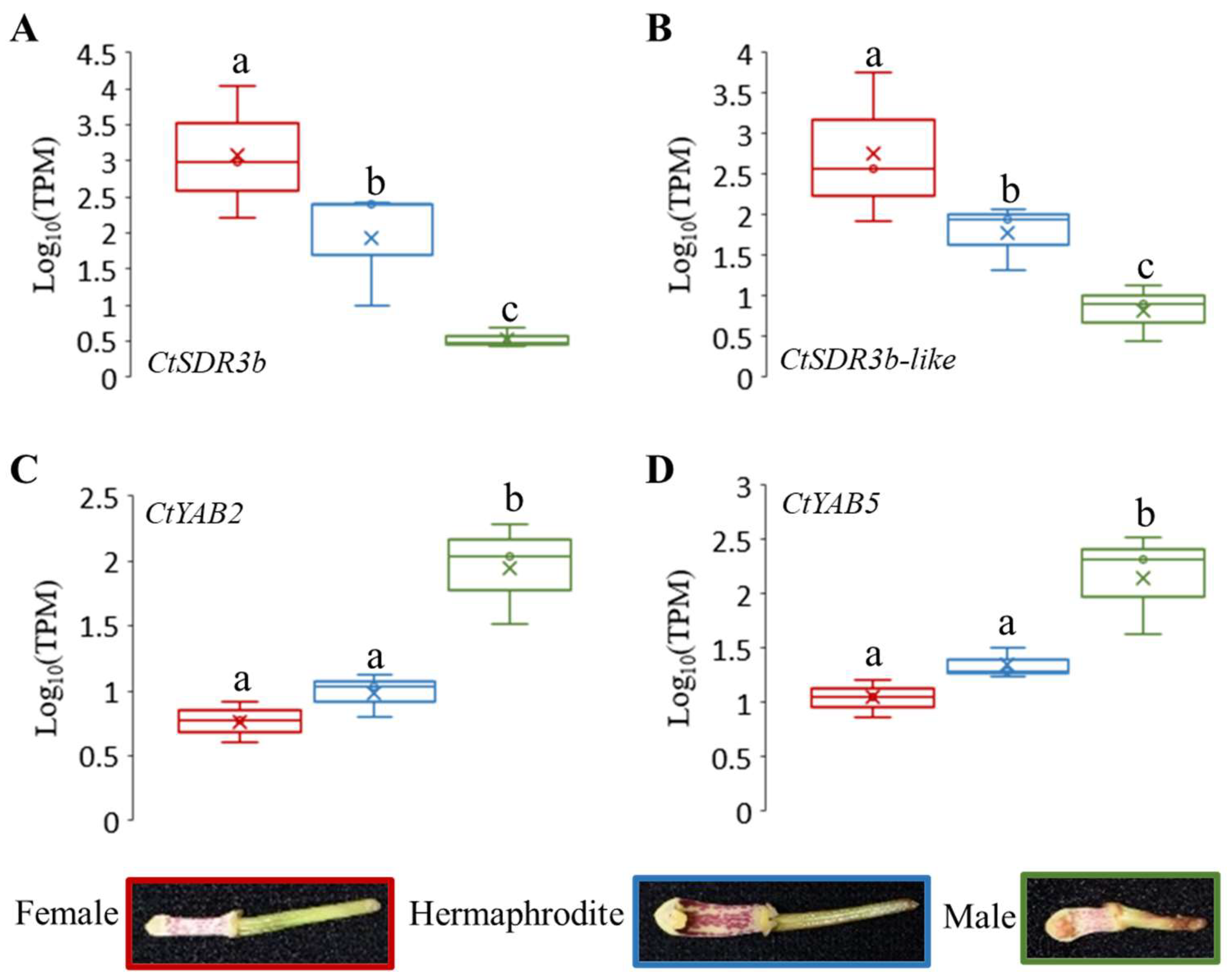
2.7. Candidate Genes of Sex Differentiation and Their Expression Patterns
2.8. qRT-PCR Validation of Sex Differentiation Candidate Genes
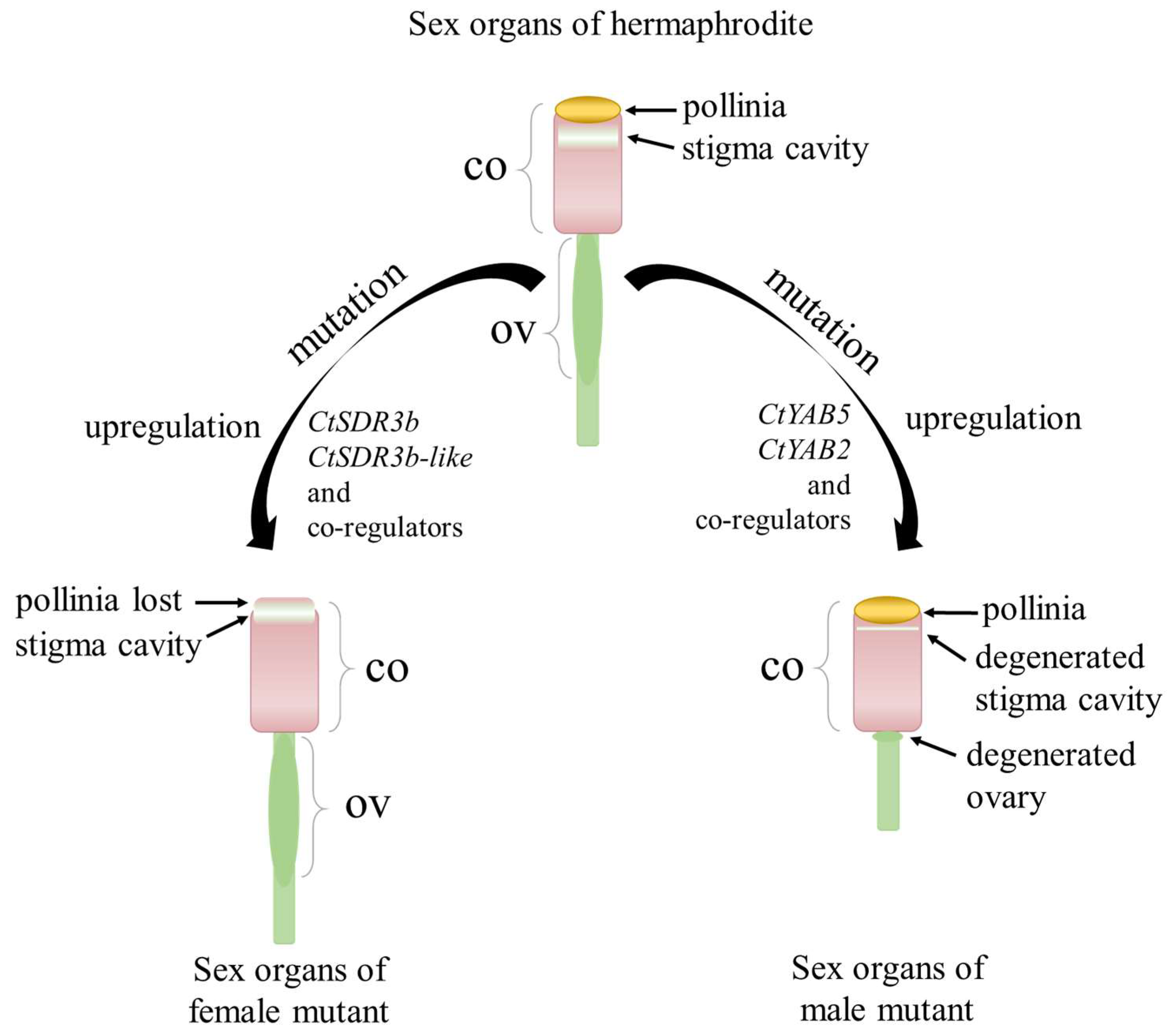
3. Discussion
4. Materials and Methods
4.1. Morphological Measurements of Three Sexual Phenotypes
4.2. RNA Sample Collection and Extraction
4.3. Construction of cDNA Library and Transcriptome Sequencing
4.4. De Novo Transcriptome Assembly
4.5. Functional Annotations and Transcription Factor Prediction
4.6. Identification of Differentially Expressed Genes (DEGs)
4.7. GO and KEGG Enrichment of DEGs
4.8. Construction of the Co-Expression Network
4.9. qRT-PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Renner, S.S.; Ricklefs, R.E. Dioecy and its correlates in the flowering plants. Am. J. Bot. 1995, 82, 596–606. [Google Scholar] [CrossRef]
- Diggle, P.K.; Di Stilio, V.S.; Gschwend, A.R.; Golenberg, E.M.; Moore, R.C.; Russell, J.R.; Sinclair, J.P. Multiple developmental processes underlie sex differentiation in angiosperms. Trends Genet. 2011, 27, 368–376. [Google Scholar] [CrossRef]
- Charlesworth, D. Plant sex determination and sex chromosomes. Heredity 2002, 88, 94–101. [Google Scholar] [CrossRef]
- Charlesworth, D. Plant Sex Chromosomes. Annu. Rev. Plant Biol. 2016, 67, 397–420. [Google Scholar] [CrossRef]
- Ma, X.; Yu, L.; Fatima, M.; Wadlington, W.H.; Hulse-Kemp, A.M.; Zhang, X.; Zhang, S.; Xu, X.; Wang, J.; Huang, H.; et al. The spinach YY genome reveals sex chromosome evolution, domestication, and introgression history of the species. Genome Biol. 2022, 23, 75. [Google Scholar] [CrossRef]
- Jarne, P.; Charlesworth, D. The evolution of the selfing rate in functionally hermaphrodite plants and animals. Annu. Rev. Ecol. Syst. 1993, 24, 441–466. [Google Scholar] [CrossRef]
- Fatima, M.; Ma, X.; Zhou, P.; Zaynab, M.; Ming, R. Auxin regulated metabolic changes underlying sepal retention and development after pollination in spinach. BMC Plant Biol. 2021, 21, 166. [Google Scholar] [CrossRef]
- Ma, X.; Fatima, M.; Li, J.; Zhou, P.; Zaynab, M.; Ming, R. Post-pollination sepal longevity of female flower co-regulated by energy-associated multiple pathways in dioecious spinach. Front. Plant Sci. 2022, 13, 1010149. [Google Scholar] [CrossRef]
- Charlesworth, D. Plant sex chromosome evolution. J. Exp. Bot. 2013, 64, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Negrutiu, I.O.; Vyskot, B.; Barbacar, N.; Georgiev, S.; Moneger, F. Dioecious plants. A key to the early events of sex chromosome evolution. Plant Physiol. 2001, 127, 1418–1424. [Google Scholar] [CrossRef]
- Perl-Treves, R. Male to female conversion along the cucumber shoot: Approaches to studying sex genes and floral development in Cucumis sativus. In Sex Determination in Plants; Garland Science: New York, NY, USA, 2004; pp. 193–221. [Google Scholar]
- Martin, A.; Troadec, C.; Boualem, A.; Rajab, M.; Fernandez, R.; Morin, H.; Pitrat, M.; Dogimont, C.; Bendahmane, A. A transposon-induced epigenetic change leads to sex determination in melon. Nature 2009, 461, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Na, J.K.; Yu, Q.; Gschwend, A.R.; Han, J.; Zeng, F.; Aryal, R.; VanBuren, R.; Murray, J.E.; Zhang, W.; et al. Sequencing papaya X and Yh chromosomes reveals molecular basis of incipient sex chromosome evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 13710–13715. [Google Scholar] [CrossRef]
- Bowman, J.L.; Kohchi, T.; Yamato, K.T.; Jenkins, J.; Shu, S.; Ishizaki, K.; Yamaoka, S.; Nishihama, R.; Nakamura, Y.; Berger, F.; et al. Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cell 2017, 171, 287–304.e15. [Google Scholar] [CrossRef] [PubMed]
- Acosta, I.F.; Laparra, H.; Romero, S.P.; Schmelz, E.; Hamberg, M.; Mottinger, J.P.; Moreno, M.A.; Dellaporta, S.L. Tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize. Science 2009, 323, 262–265. [Google Scholar] [CrossRef]
- Bachtrog, D.; Mank, J.E.; Peichel, C.L.; Kirkpatrick, M.; Otto, S.P.; Ashman, T.L.; Hahn, M.W.; Kitano, J.; Mayrose, I.; Ming, R.; et al. Sex determination: Why so many ways of doing it? PLoS Biol. 2014, 12, e1001899. [Google Scholar] [CrossRef]
- Kafkas, S.; Ma, X.; Zhang, X.; Topcu, H.; Navajas-Perez, R.; Wai, C.M.; Tang, H.; Xu, X.; Khodaeiaminjan, M.; Guney, M.; et al. Pistachio genomes provide insights into nut tree domestication and ZW sex chromosome evolution. Plant Commun. 2022, 4, 100497. [Google Scholar] [CrossRef]
- Ross, M.D. The evolution of gynodioecy and subdioecy. Evolution 1978, 32, 174–188. [Google Scholar] [CrossRef]
- Xue, L.; Wu, H.; Chen, Y.; Li, X.; Hou, J.; Lu, J.; Wei, S.; Dai, X.; Olson, M.S.; Liu, J.; et al. Evidences for a role of two Y-specific genes in sex determination in Populus deltoides. Nat. Commun. 2020, 11, 5893. [Google Scholar] [CrossRef]
- Li, Y.; Wang, D.; Wang, W.; Yang, W.; Gao, J.; Zhang, W.; Shan, L.; Kang, M.; Chen, Y.; Ma, T. A chromosome-level Populus qiongdaoensis genome assembly provides insights into tropical adaptation and a cryptic turnover of sex determination. Mol. Ecol. 2023, 32, 1366–1380. [Google Scholar] [CrossRef]
- Grumet, R.; Taft, J. Sex Expression in Cucurbits. In Genetics, Genomics and Breeding in Crop Plants; Science Publishers: Enfield, NH, USA, 2011; pp. 353–375. [Google Scholar]
- Bhowmick, B.K.; Jha, S. Dynamics of sex expression and chromosome diversity in Cucurbitaceae: A story in the making. J. Genet. 2015, 94, 793–808. [Google Scholar] [CrossRef]
- Wu, Z.; Raven, P.; Hong, D. Flora of China. Vol. 25 (Orchidaceae); Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2009. [Google Scholar]
- Cozzolino, S.; Widmer, A. Orchid diversity: An evolutionary consequence of deception? Trends Ecol. Evol. 2005, 20, 487–494. [Google Scholar] [CrossRef]
- Schluter, P.M.; Schiestl, F.P. Molecular mechanisms of floral mimicry in orchids. Trends Plant Sci. 2008, 13, 228–235. [Google Scholar] [CrossRef]
- Cai, J.; Liu, X.; Vanneste, K.; Proost, S.; Tsai, W.C.; Liu, K.W.; Chen, L.J.; He, Y.; Xu, Q.; Bian, C.; et al. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 2015, 47, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Q.; Liu, K.W.; Li, Z.; Lohaus, R.; Hsiao, Y.Y.; Niu, S.C.; Wang, J.Y.; Lin, Y.C.; Xu, Q.; Chen, L.J.; et al. The Apostasia genome and the evolution of orchids. Nature 2017, 549, 379–383. [Google Scholar] [CrossRef]
- Huang, S.Q.; Lu, Y.; Chen, Y.Z.; Luo, Y.B.; Delph, L.F. Parthenogenesis maintains male sterility in a gynodioecious orchid. Am. Nat. 2009, 174, 578–584. [Google Scholar] [CrossRef]
- Perez-Escobar, O.A.; Gottschling, M.; Whitten, W.M.; Salazar, G.; Gerlach, G. Sex and the Catasetinae (Darwin’s favourite orchids). Mol. Phylogenet. Evol. 2016, 97, 1–10. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, Y.; Lin, H.; Ma, X. The complete chloroplast genome of a Cymbidium tortisepalum (Orchidaceae) male mutant. Mitochondrial DNA B Resour. 2019, 4, 4087–4088. [Google Scholar] [CrossRef]
- Lin, H.; Sun, Y.; Lan, S.; Ma, X. The complete plastome of Cymbidium tortisepalum (Orchidaceae) hermaphrodite. Mitochondrial DNA Part B 2020, 5, 1265–1266. [Google Scholar] [CrossRef]
- DeLong, A.; Calderon-Urrea, A.; Dellaporta, S.L. Sex determination gene TASSELSEED2 of maize encodes a short-chain alcohol dehydrogenase required for stage-specific floral organ abortion. Cell 1993, 74, 757–768. [Google Scholar] [CrossRef]
- Massonnet, M.; Cochetel, N.; Minio, A.; Vondras, A.M.; Lin, J.; Muyle, A.; Garcia, J.F.; Zhou, Y.; Delledonne, M.; Riaz, S.; et al. The genetic basis of sex determination in grapes. Nat. Commun. 2020, 11, 2902. [Google Scholar] [CrossRef]
- Malcomber, S.T.; Kellogg, E.A. Evolution of unisexual flowers in grasses (Poaceae) and the putative sex-determination gene, TASSELSEED2 (TS2). New Phytol. 2006, 170, 885–899. [Google Scholar] [CrossRef]
- Li, D.; Blakey, C.A.; Dewald, C.; Dellaporta, S.L. Evidence for a common sex determination mechanism for pistil abortion in maize and in its wild relative Tripsacum. Proc. Natl. Acad. Sci. USA 1997, 94, 4217–4222. [Google Scholar] [CrossRef]
- Kinney, M.S.; Columbus, J.T.; Friar, E.A. Molecular evolution of the maize sex-determining gene TASSELSEED2 in Bouteloua (Poaceae). Mol. Phylogenet. Evol. 2003, 29, 519–528. [Google Scholar] [CrossRef]
- Dellaporta, S.L.; Calderon-Urrea, A. The sex determination process in maize. Science 1994, 266, 1501–1505. [Google Scholar] [CrossRef]
- Wang, F.; Yuan, Z.; Zhao, Z.; Li, C.; Zhang, X.; Liang, H.; Liu, Y.; Xu, Q.; Liu, H. Tasselseed5 encodes a cytochrome C oxidase that functions in sex determination by affecting jasmonate catabolism in maize. J. Integr. Plant Biol. 2020, 62, 247–255. [Google Scholar] [CrossRef]
- Sun, Q.; Qu, J.; Yu, Y.; Yang, Z.; Wei, S.; Wu, Y.; Yang, J.; Peng, Z. TaEPFL1, an EPIDERMAL PATTERNING FACTOR-LIKE (EPFL) secreted peptide gene, is required for stamen development in wheat. Genetica 2019, 147, 121–130. [Google Scholar] [CrossRef]
- Cai, H.; Huang, Y.; Liu, L.; Zhang, M.; Chai, M.; Xi, X.; Aslam, M.; Wang, L.; Ma, S.; Su, H.; et al. Signaling by the EPFL-ERECTA family coordinates female germline specification through the BZR1 family in Arabidopsis. Plant Cell 2023, 35, 1455–1473. [Google Scholar] [CrossRef]
- Han, Z.; Qin, Y.; Deng, Y.; Kong, F.; Wang, Z.; Shen, G.; Wang, J.; Duan, B.; Li, R. Expression profiles of a cytoplasmic male sterile line of Gossypium harknessii and its fertility restorer and maintainer lines revealed by RNA-Seq. Plant Physiol. Biochem. 2017, 116, 106–115. [Google Scholar] [CrossRef]
- Bowman, J.L. The YABBY gene family and abaxial cell fate. Curr. Opin. Plant Biol. 2000, 3, 17–22. [Google Scholar] [CrossRef]
- Finet, C.; Floyd, S.K.; Conway, S.J.; Zhong, B.; Scutt, C.P.; Bowman, J.L. Evolution of the YABBY gene family in seed plants. Evol. Dev. 2016, 18, 116–126. [Google Scholar] [CrossRef]
- Sawa, S.; Watanabe, K.; Goto, K.; Liu, Y.G.; Shibata, D.; Kanaya, E.; Morita, E.H.; Okada, K. Filamentous flower, a meristem and organ identity gene of Arabidopsis, encodes a protein with a zinc finger and HMG-related domains. Genes. Dev. 1999, 13, 1079–1088. [Google Scholar] [CrossRef]
- Lugassi, N.; Nakayama, N.; Bochnik, R.; Zik, M. A novel allele of filamentous flower reveals new insights on the link between inflorescence and floral meristem organization and flower morphogenesis. BMC Plant Biol. 2010, 10, 131. [Google Scholar] [CrossRef]
- Tanaka, W.; Toriba, T.; Ohmori, Y.; Yoshida, A.; Kawai, A.; Mayama-Tsuchida, T.; Ichikawa, H.; Mitsuda, N.; Ohme-Takagi, M.; Hirano, H.Y. The YABBY gene TONGARI-BOUSHI1 is involved in lateral organ development and maintenance of meristem organization in the rice spikelet. Plant Cell 2012, 24, 80–95. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Hsiao, Y.Y.; Li, C.I.; Yeh, C.M.; Mitsuda, N.; Yang, H.X.; Chiu, C.C.; Chang, S.B.; Liu, Z.J.; Tsai, W.C. The ancestral duplicated DL/CRC orthologs, PeDL1 and PeDL2, function in orchid reproductive organ innovation. J. Exp. Bot. 2021, 72, 5442–5461. [Google Scholar] [CrossRef]
- Ma, X.; He, B.; Lan, S.; Lin, H.; Han, X. Establishment of in vitro Regeneration System and Rapid Propagation Method for Unisexual Varieties of Cymbidium tortisepalum. China Patent ZL201910310848.7, 17 June 2022. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Hu, Z.; Mellor, J.; DeLisi, C. Analyzing networks with VisANT. Curr. Protoc. Bioinform. 2004, 8, 8.8.1–8.8.24. [Google Scholar] [CrossRef]
- Csárdi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Zhang, J.; Wu, K.; Zeng, S.; Teixeira da Silva, J.A.; Zhao, X.; Tian, C.E.; Xia, H.; Duan, J. Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genom. 2013, 14, 279. [Google Scholar] [CrossRef]
- Lalitha, S. Primer Premier 5. Biotech Softw. Internet Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ (–delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71. [Google Scholar]
- Yang, J.; Lee, H.J.; Shin, D.H.; Oh, S.K.; Seon, J.H.; Paek, K.Y.; Han, K.H. Genetic transformation of Cymbidium orchid by particle bombardment. Plant Cell Rep. 1999, 18, 978–984. [Google Scholar] [CrossRef]
- Chin, D.P.; Mishiba, K.; Mii, M. Agrobacterium-mediated transformation of protocorm-like bodies in Cymbidium. Plant Cell Rep. 2007, 26, 735–743. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Ju, S.; Lin, H.; Huang, H.; Huang, J.; Peng, D.; Ming, R.; Lan, S.; Liu, Z.-J. Sex-Related Gene Network Revealed by Transcriptome Differentiation of Bisexual and Unisexual Flowers of Orchid Cymbidium tortisepalum. Int. J. Mol. Sci. 2023, 24, 16627. https://doi.org/10.3390/ijms242316627
Ma X, Ju S, Lin H, Huang H, Huang J, Peng D, Ming R, Lan S, Liu Z-J. Sex-Related Gene Network Revealed by Transcriptome Differentiation of Bisexual and Unisexual Flowers of Orchid Cymbidium tortisepalum. International Journal of Molecular Sciences. 2023; 24(23):16627. https://doi.org/10.3390/ijms242316627
Chicago/Turabian StyleMa, Xiaokai, Song Ju, Han Lin, Huaxing Huang, Jie Huang, Donghui Peng, Ray Ming, Siren Lan, and Zhong-Jian Liu. 2023. "Sex-Related Gene Network Revealed by Transcriptome Differentiation of Bisexual and Unisexual Flowers of Orchid Cymbidium tortisepalum" International Journal of Molecular Sciences 24, no. 23: 16627. https://doi.org/10.3390/ijms242316627
APA StyleMa, X., Ju, S., Lin, H., Huang, H., Huang, J., Peng, D., Ming, R., Lan, S., & Liu, Z.-J. (2023). Sex-Related Gene Network Revealed by Transcriptome Differentiation of Bisexual and Unisexual Flowers of Orchid Cymbidium tortisepalum. International Journal of Molecular Sciences, 24(23), 16627. https://doi.org/10.3390/ijms242316627