
Metformin Suppresses Stemness of Non-Small-Cell Lung Cancer Induced by Paclitaxel through FOXO3a

Abstract
:1. Introduction
2. Results
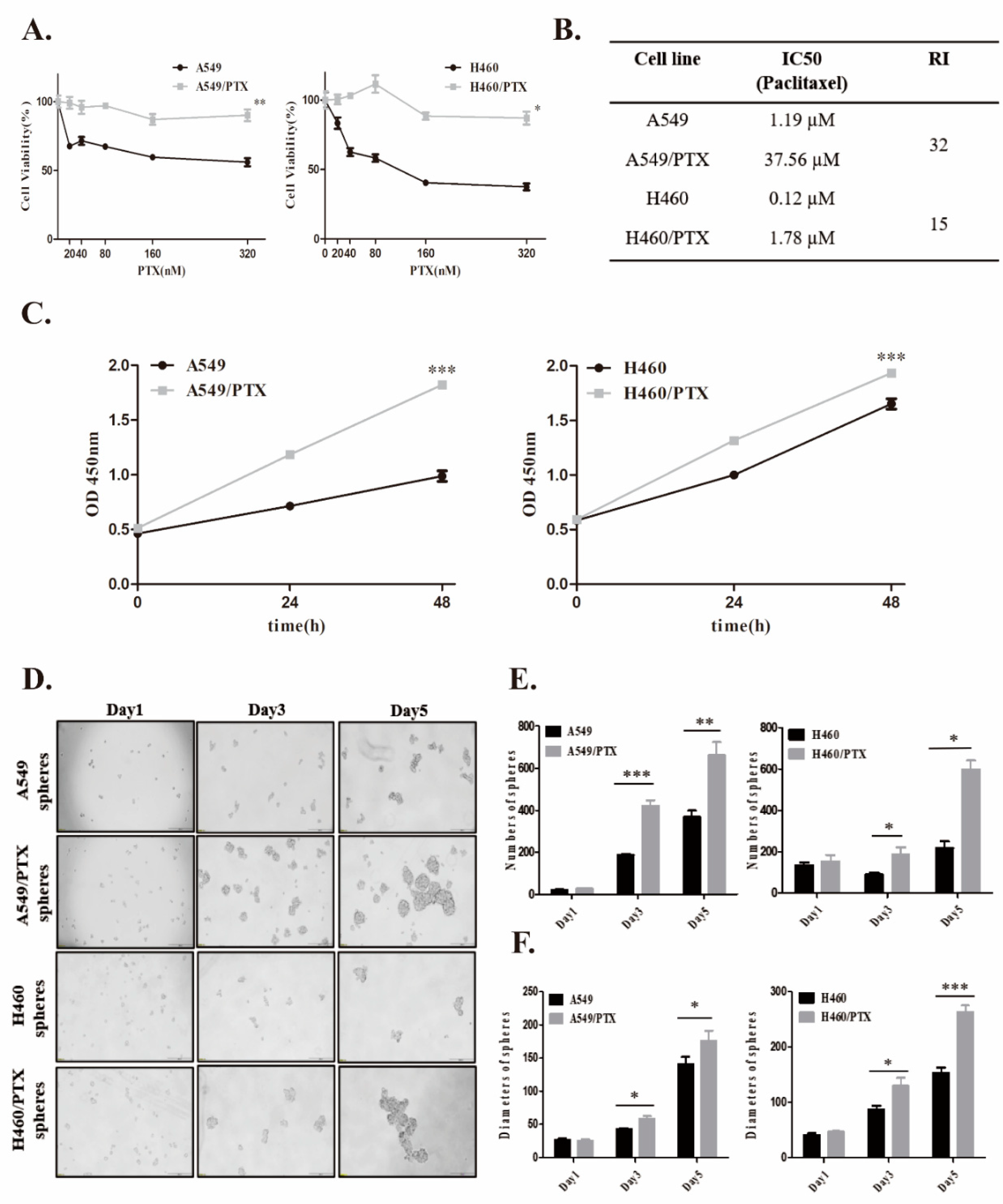
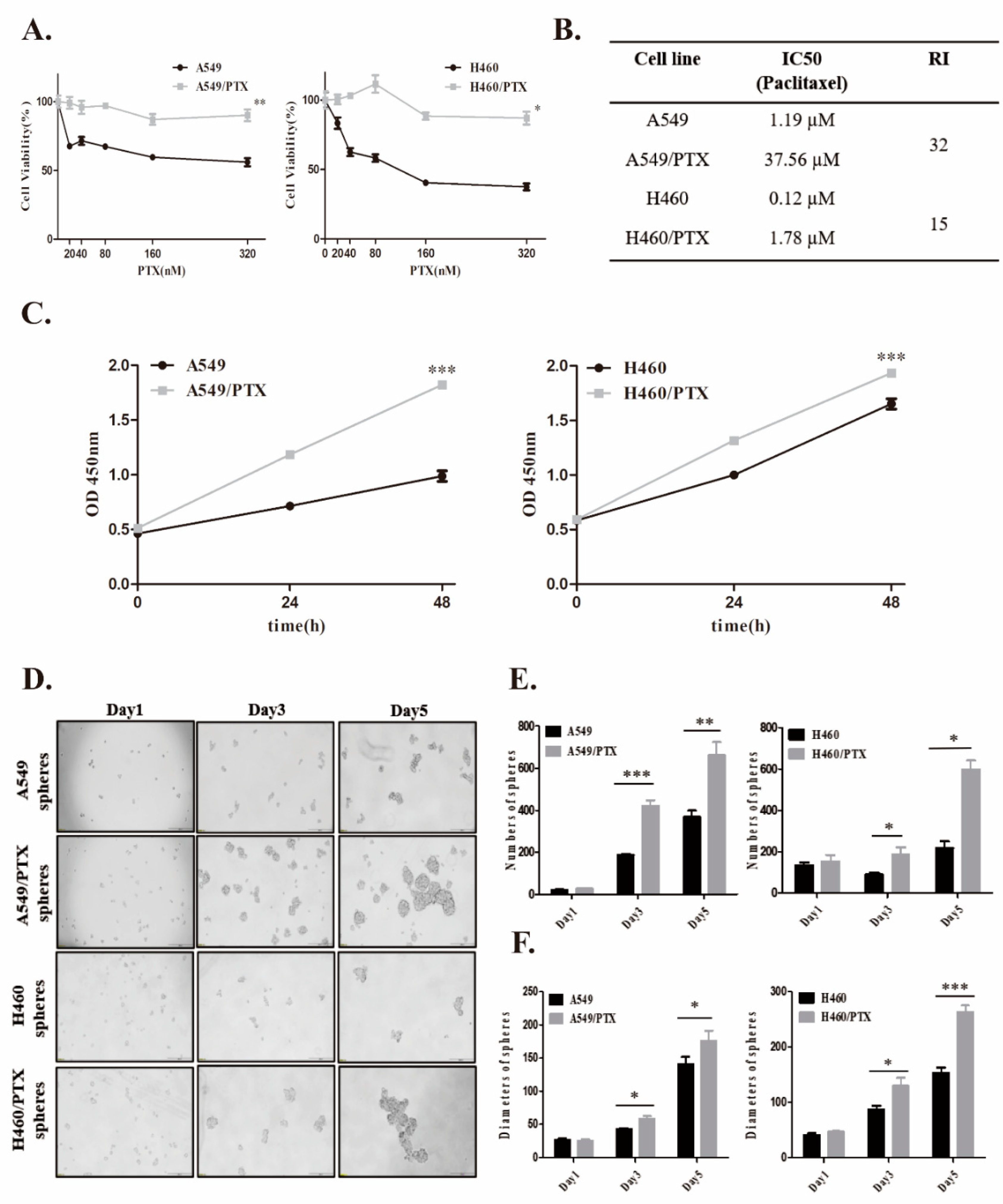
2.1. Establishment and Confirmation of PTX-Resistant Cell Lines
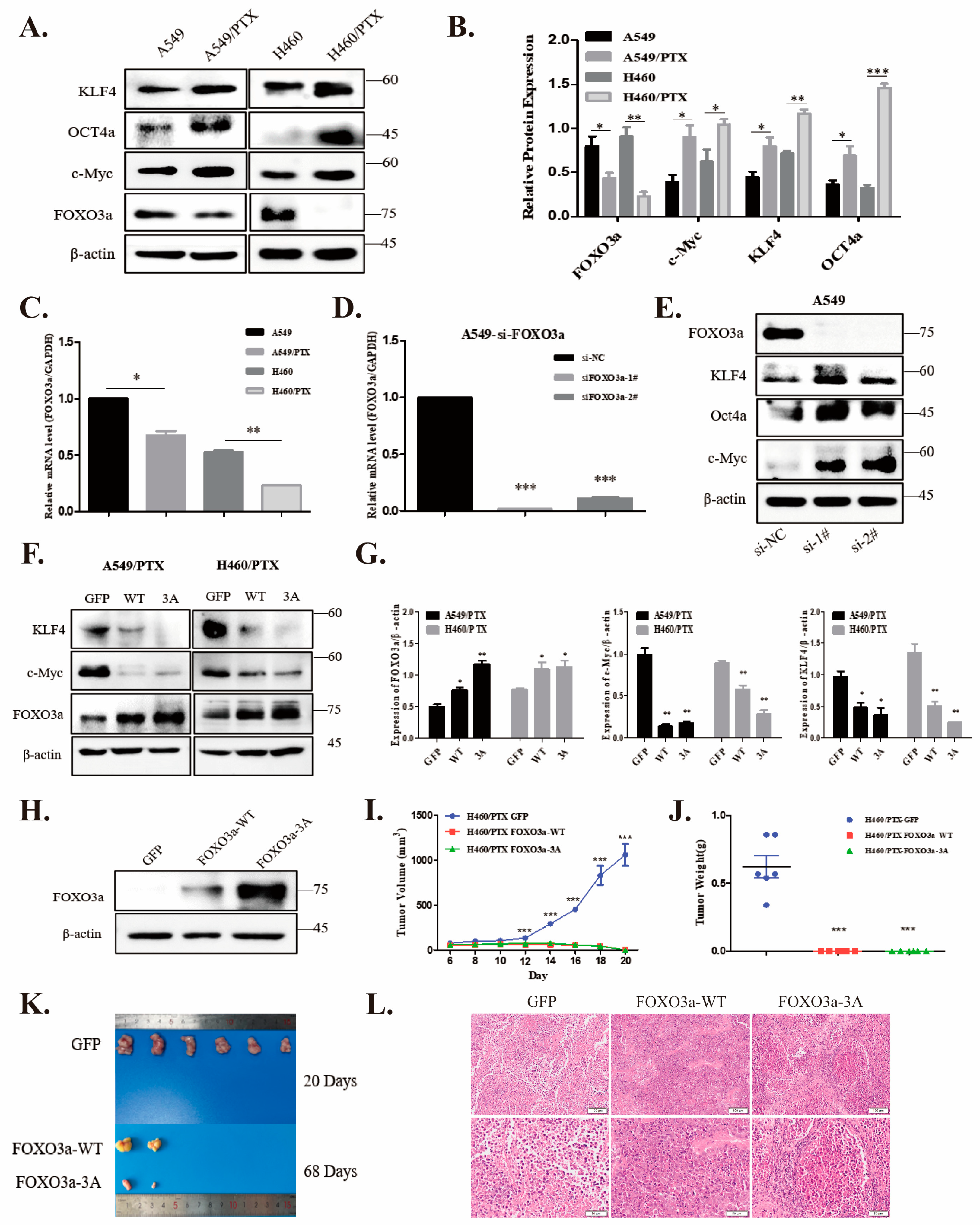
2.2. FOXO3a Regulates Stemness in NSCLC/PTX-Resistant Cells
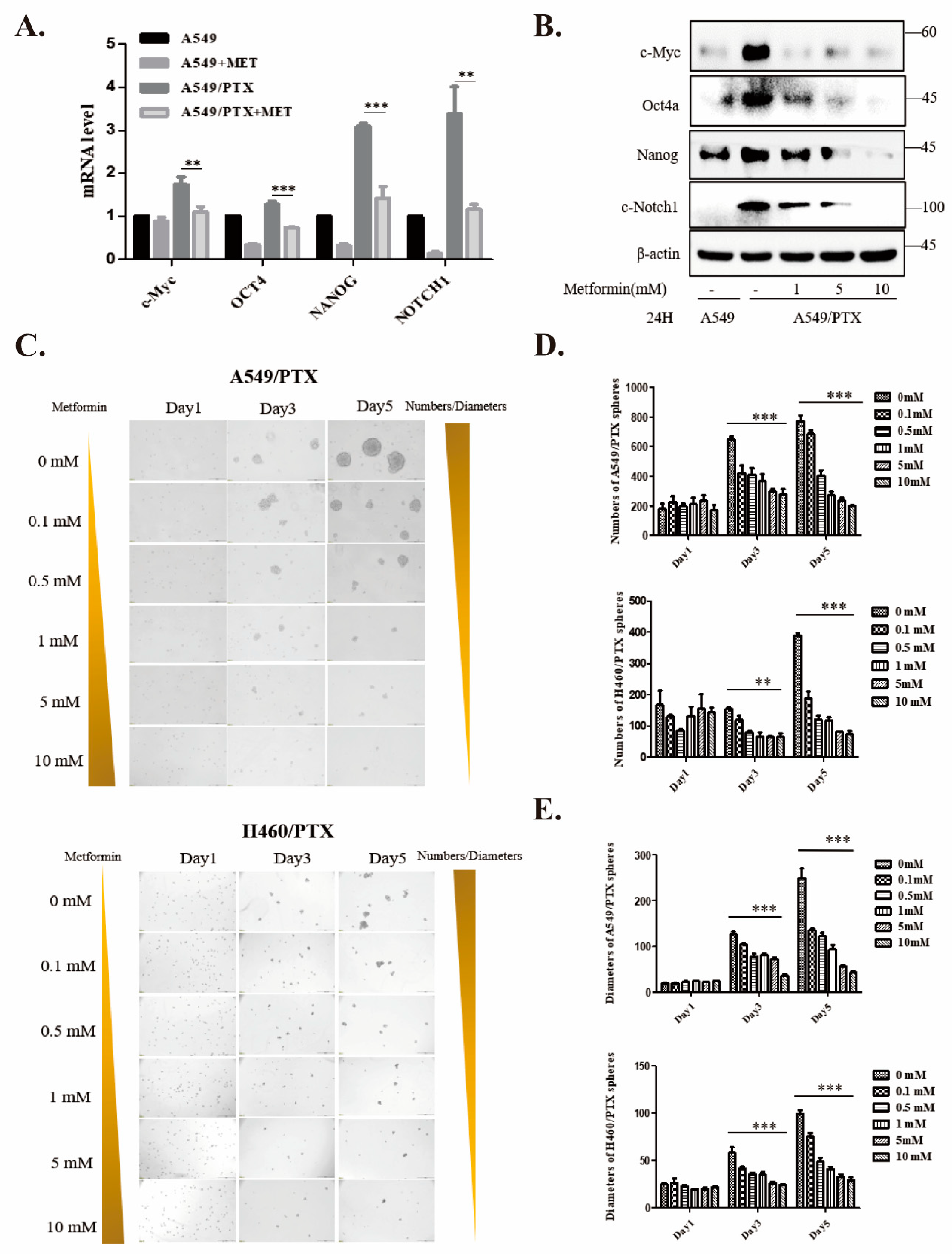
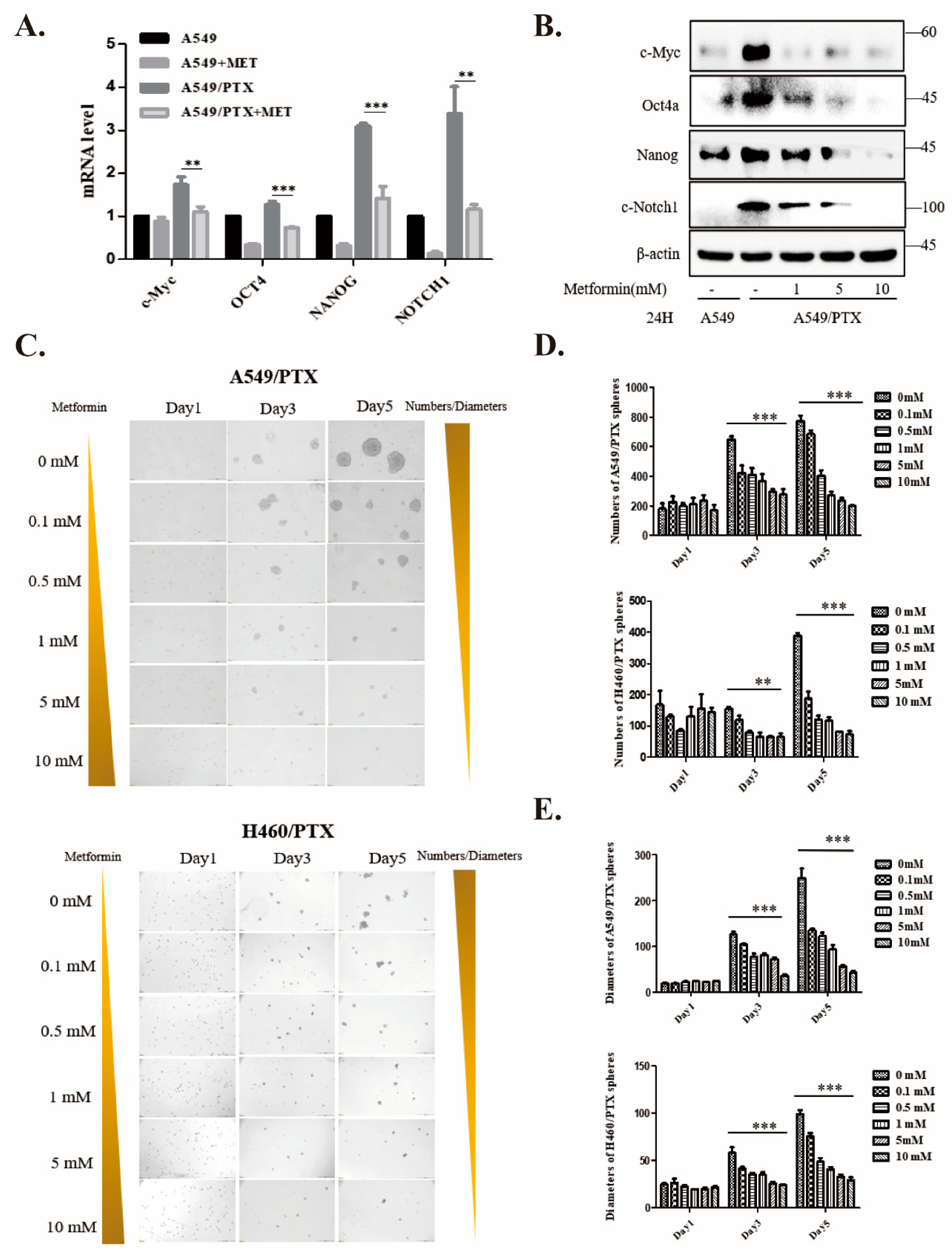
2.3. Metformin Attenuates Development of Cancer Stemness in PTX-Resistant Cells
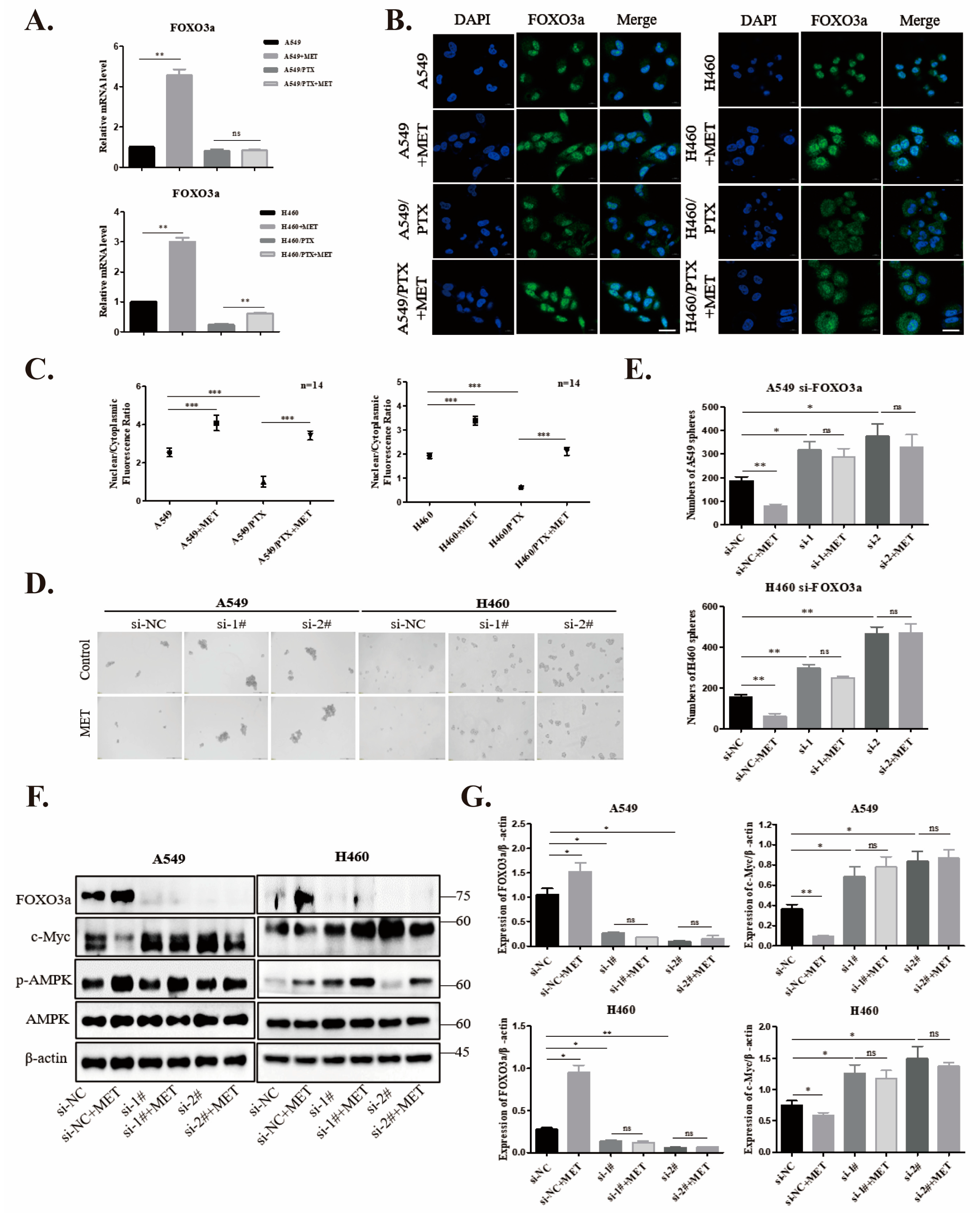
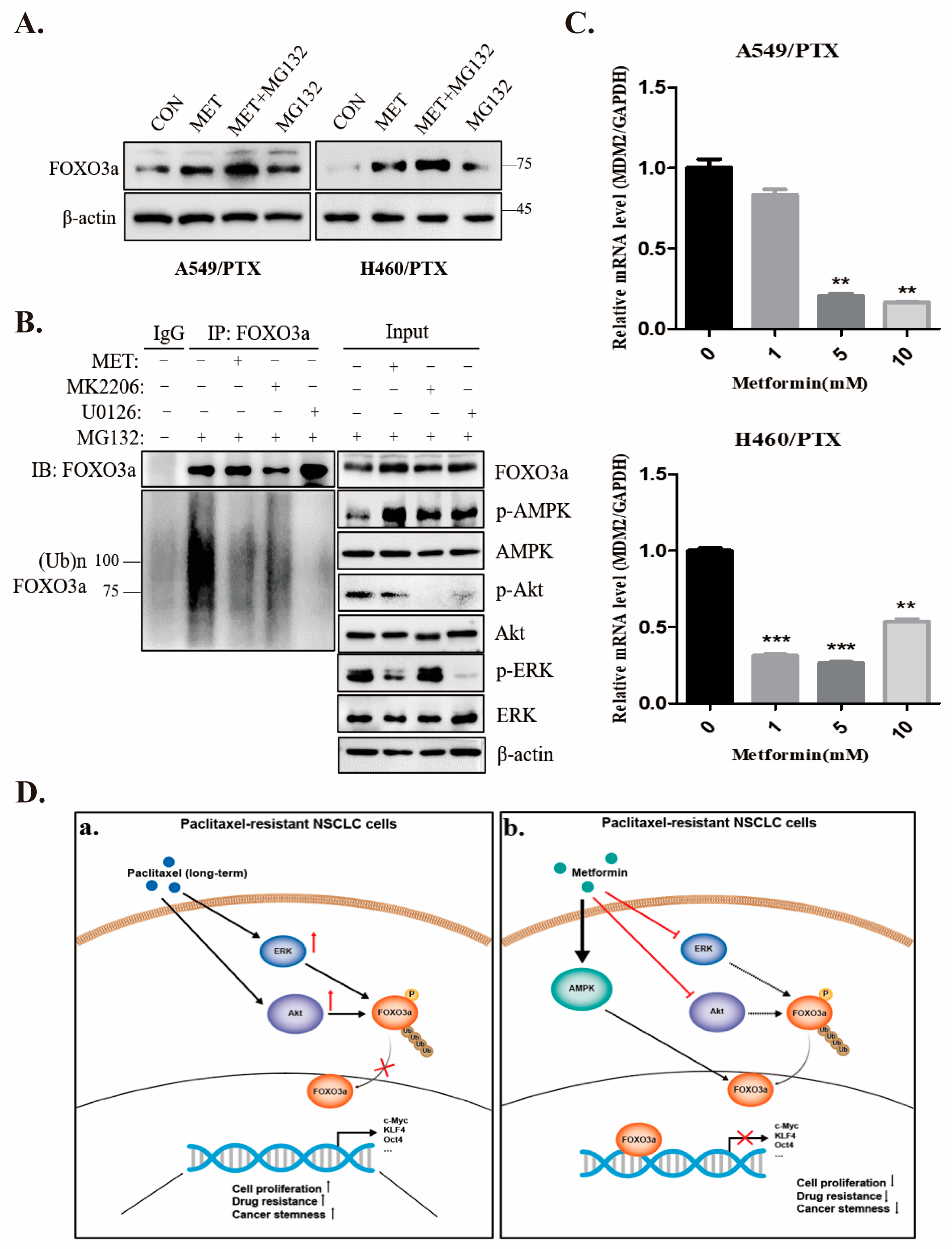
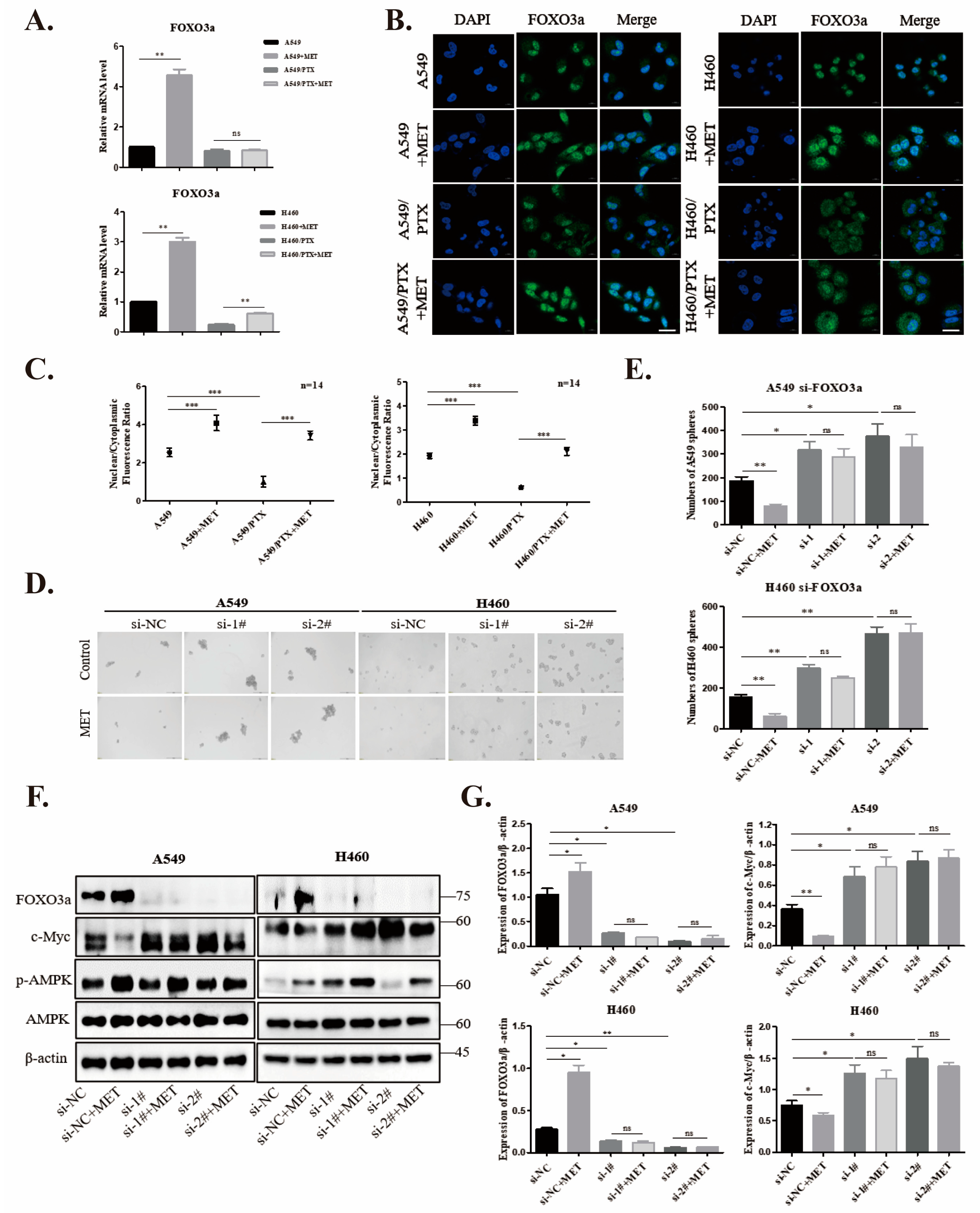
2.4. Metformin Upregulates FOXO3a and Induces FOXO3a Nuclear Localization
2.5. Knockdown of FOXO3a Reverses Metformin-Mediated Suppression of the Stemness in PTX-Sensitive Cells
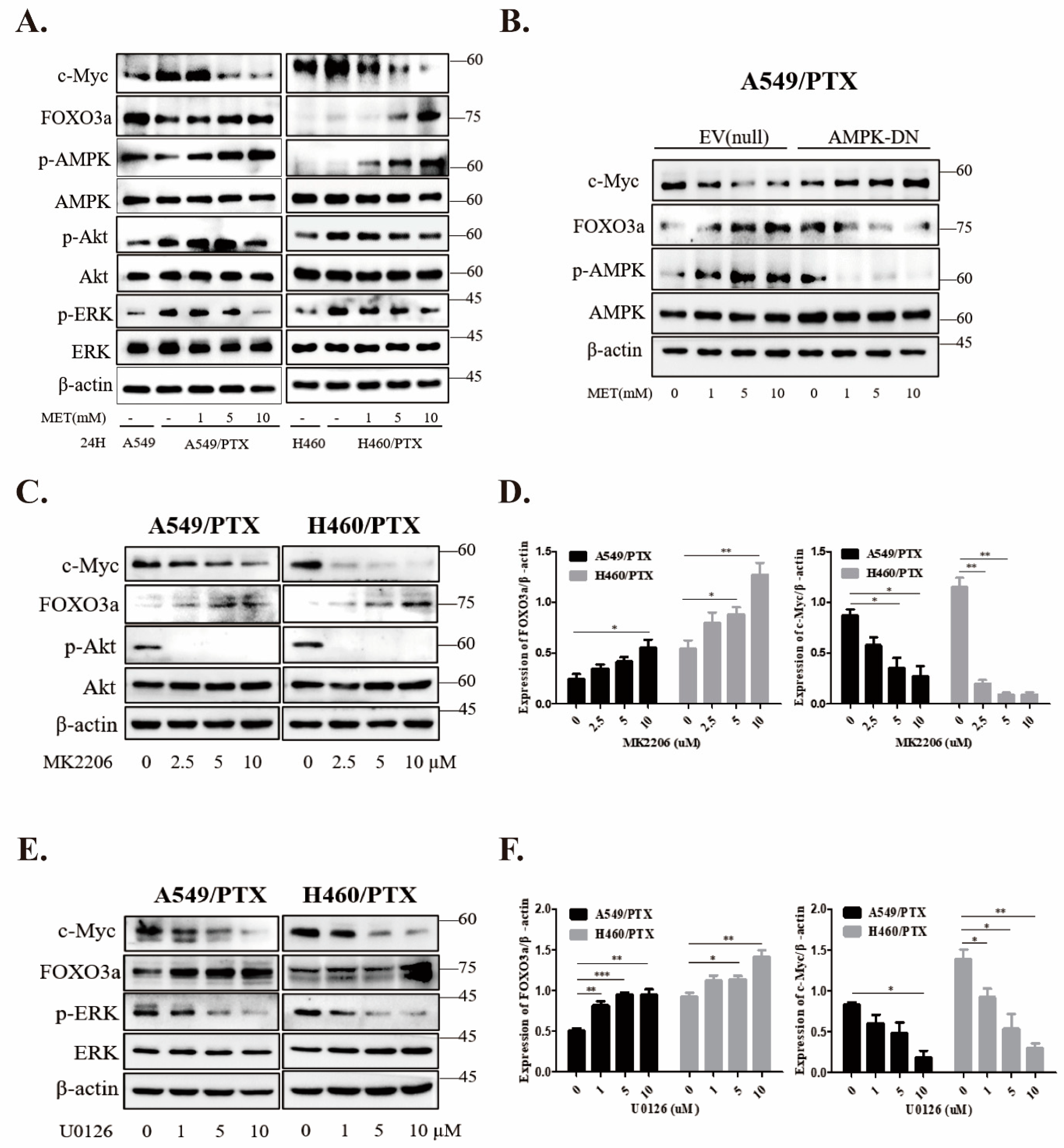
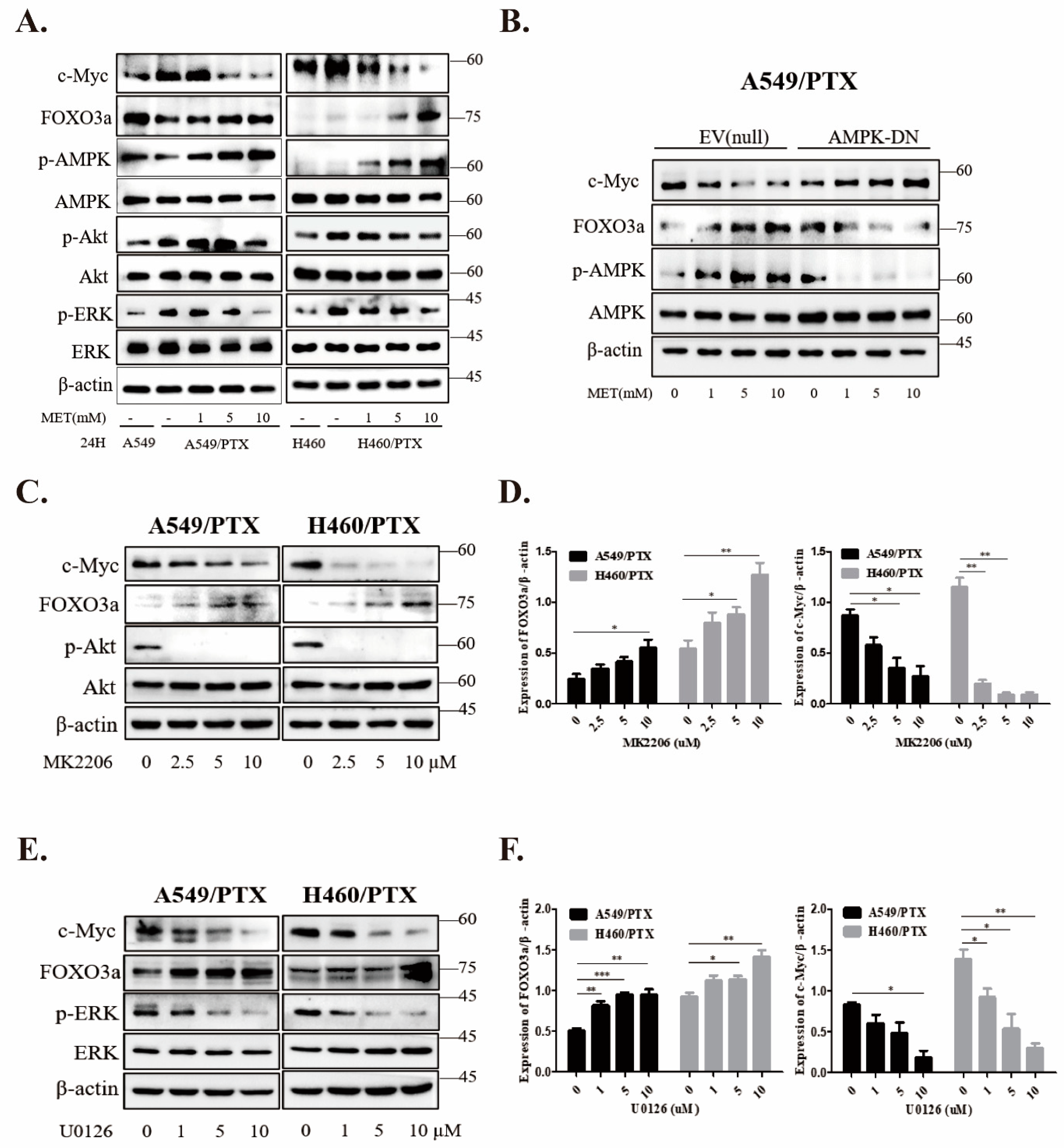
2.6. Metformin Inhibits Stemness through AMPK/Akt/ERK-FOXO3a Signaling
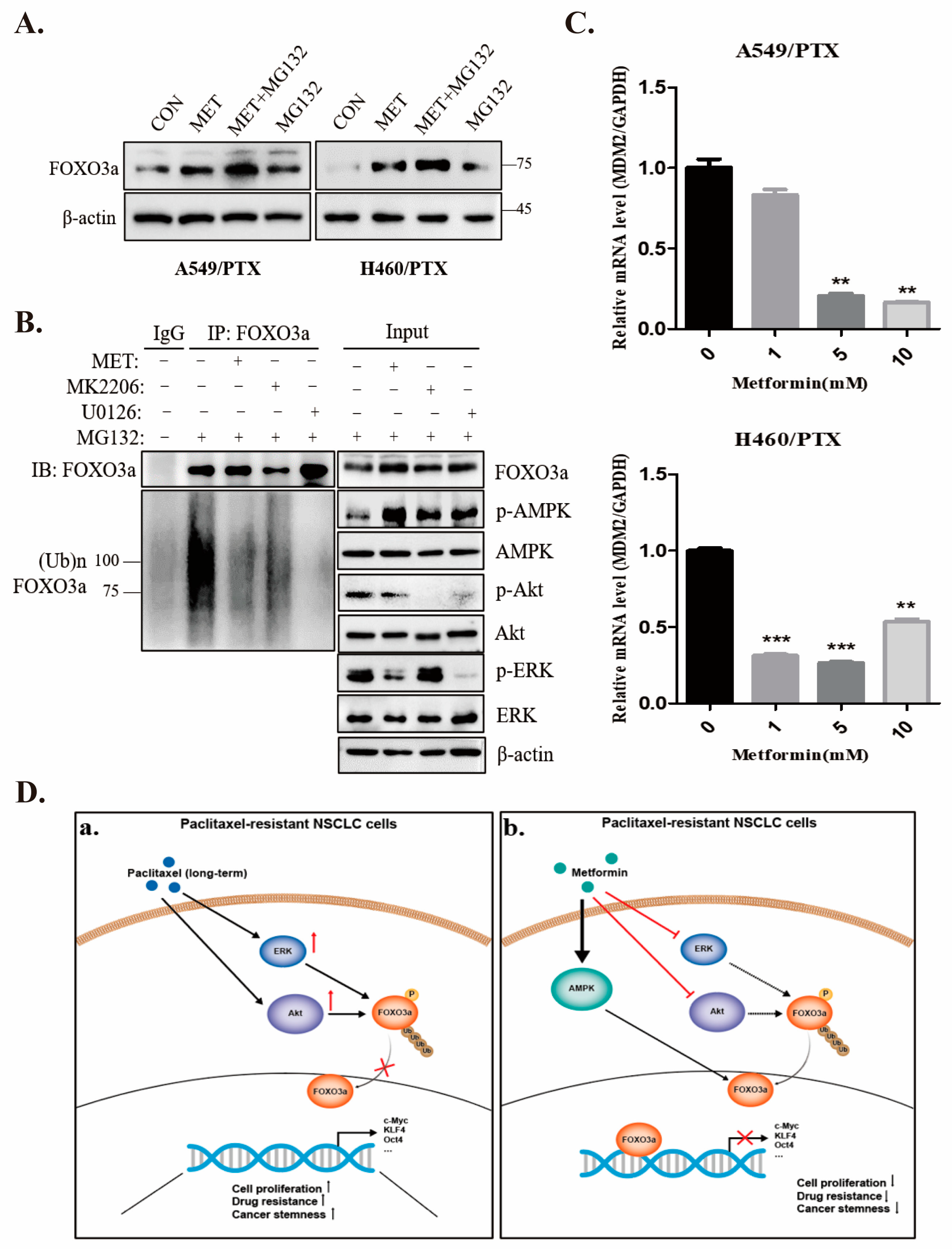
2.7. Metformin Stabilizes FOXO3a through Ubiquitin-Proteasome Pathway
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Viability Assays
4.4. Tests for Synergism and Interaction
4.5. Enrichment of Stem Cell-like Spheres
4.6. Flow Cytometric Analysis
4.7. Virus Production and Transfection
4.8. Real-Time PCR
4.9. Western Blotting
4.10. Xenograft Tumor Model
4.11. Hematoxylin and Eosin (H&E) Staining
4.12. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Crown, J.; O’Leary, M. The taxanes: An update. Lancet 2000, 355, 1176–1178. [Google Scholar] [CrossRef]
- Goldspiel, B.R. Clinical overview of the taxanes. Pharmacotherapy 1997, 17, 110S–125S. [Google Scholar] [CrossRef]
- Holmes, F.A.; Walters, R.S.; Theriault, R.L.; Forman, A.D.; Newton, L.K.; Raber, M.N.; Buzdar, A.U.; Frye, D.K.; Hortobagyi, G.N. Phase II trial of taxol, an active drug in the treatment of metastatic breast cancer. J. Natl. Cancer Inst. 1991, 83, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Torres, K.; Horwitz, S.B. Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res. 1998, 58, 3620–3626. [Google Scholar]
- van der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 440–450. [Google Scholar] [CrossRef]
- Gui, T.; Burgering, B.M.T. FOXOs: Masters of the equilibrium. FEBS J. 2022, 289, 7918–7939. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.D.; Johnston, D.A.; Kazianis, S.; Butler, A.P. Cloning and analysis of a FoxO transcription factor from Xiphophorus. Gene 2003, 302, 31–41. [Google Scholar] [CrossRef]
- Aoki, M.; Jiang, H.; Vogt, P.K. Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 13613–13617. [Google Scholar] [CrossRef]
- Essers, M.A.; Weijzen, S.; de Vries-Smits, A.M.; Saarloos, I.; de Ruiter, N.D.; Bos, J.L.; Burgering, B.M. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Seoane, J.; Le, H.V.; Shen, L.; Anderson, S.A.; Massague, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Matrone, A.; Ferrari, E.; Ingravallo, G.; Lo Sasso, G.; Murzilli, S.; Petruzzelli, M.; Salvatore, L.; Moschetta, A.; Simone, C. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ. 2009, 16, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Kim, S.; Salamone, R.J.; Walker, J.R.; Maira, S.M.; Garcia-Echeverria, C.; Schultz, P.G.; Reddy, V.A. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl. Acad. Sci. USA 2009, 106, 268–273. [Google Scholar] [CrossRef]
- Sunayama, J.; Sato, A.; Matsuda, K.; Tachibana, K.; Watanabe, E.; Seino, S.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; et al. FoxO3a functions as a key integrator of cellular signals that control glioblastoma stem-like cell differentiation and tumorigenicity. Stem Cells 2011, 29, 1327–1337. [Google Scholar] [CrossRef]
- Chiu, C.F.; Chang, Y.W.; Kuo, K.T.; Shen, Y.S.; Liu, C.Y.; Yu, Y.H.; Cheng, C.C.; Lee, K.Y.; Chen, F.C.; Hsu, M.K.; et al. NF-kappaB-driven suppression of FOXO3a contributes to EGFR mutation-independent gefitinib resistance. Proc. Natl. Acad. Sci. USA 2016, 113, E2526–E2535. [Google Scholar] [CrossRef]
- Liu, H.; Song, Y.; Qiu, H.; Liu, Y.; Luo, K.; Yi, Y.; Jiang, G.; Lu, M.; Zhang, Z.; Yin, J.; et al. Downregulation of FOXO3a by DNMT1 promotes breast cancer stem cell properties and tumorigenesis. Cell Death Differ. 2020, 27, 966–983. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Allen, J.E.; Dicker, D.T.; El-Deiry, W.S. Small-Molecule ONC201/TIC10 Targets Chemotherapy-Resistant Colorectal Cancer Stem-like Cells in an Akt/Foxo3a/TRAIL-Dependent Manner. Cancer Res. 2015, 75, 1423–1432. [Google Scholar] [CrossRef]
- Zhou, M.; Xia, L.; Wang, J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab. Dispos. 2007, 35, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Liu, M.; Huang, Y.; Wu, K.; Huang, X.; Zhao, Y.; He, W.; Zhang, R. Metformin Use and Lung Cancer Risk in Diabetic Patients: A Systematic Review and Meta-Analysis. Dis. Markers 2019, 2019, 6230162. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Lei, Y.; Yi, Y.; Liu, Y.; Liu, X.; Keller, E.T.; Qian, C.N.; Zhang, J.; Lu, Y. Metformin targets multiple signaling pathways in cancer. Chin. J. Cancer 2017, 36, 17. [Google Scholar] [CrossRef] [PubMed]
- Martin-Castillo, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Metformin and cancer: Doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 2010, 9, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Emami Riedmaier, A.; Fisel, P.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Metformin and cancer: From the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol. Sci. 2013, 34, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Soo, J.S.; Ng, C.H.; Tan, S.H.; Malik, R.A.; Teh, Y.C.; Tan, B.S.; Ho, G.F.; See, M.H.; Taib, N.A.; Yip, C.H.; et al. Metformin synergizes 5-fluorouracil, epirubicin, and cyclophosphamide (FEC) combination therapy through impairing intracellular ATP production and DNA repair in breast cancer stem cells. Apoptosis 2015, 20, 1373–1387. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Guimaraes, I.; Ladislau-Magescky, T.; Tessarollo, N.G.; Dos Santos, D.Z.; Gimba, E.R.P.; Sternberg, C.; Silva, I.V.; Rangel, L.B.A. Chemosensitizing effects of metformin on cisplatin- and paclitaxel-resistant ovarian cancer cell lines. Pharmacol. Rep. 2018, 70, 409–417. [Google Scholar] [CrossRef]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Suwei, D.; Liang, Z.; Zhimin, L.; Ruilei, L.; Yingying, Z.; Zhen, L.; Chunlei, G.; Zhangchao, L.; Yuanbo, X.; Jinyan, Y.; et al. NLK functions to maintain proliferation and stemness of NSCLC and is a target of metformin. J. Hematol. Oncol. 2015, 8, 120. [Google Scholar] [CrossRef]
- Xiao, Z.; Sperl, B.; Ullrich, A.; Knyazev, P. Metformin and salinomycin as the best combination for the eradication of NSCLC monolayer cells and their alveospheres (cancer stem cells) irrespective of EGFR, KRAS, EML4/ALK and LKB1 status. Oncotarget 2014, 5, 12877–12890. [Google Scholar] [CrossRef]
- Hu, T.; Chung, Y.M.; Guan, M.; Ma, M.; Ma, J.; Berek, J.S.; Hu, M.C. Reprogramming ovarian and breast cancer cells into non-cancerous cells by low-dose metformin or SN-38 through FOXO3 activation. Sci. Rep. 2014, 4, 5810. [Google Scholar] [CrossRef]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef]
- Paterson, C.; Kilmister, E.J.; Brasch, H.D.; Bockett, N.; Patel, J.; Paterson, E.; Purdie, G.; Galvin, S.; Davis, P.F.; Itinteang, T.; et al. Cell Populations Expressing Stemness-Associated Markers in Lung Adenocarcinoma. Life 2021, 11, 1106. [Google Scholar] [CrossRef]
- Brownawell, A.M.; Kops, G.J.; Macara, I.G.; Burgering, B.M. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol. Cell Biol. 2001, 21, 3534–3546. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef]
- Fu, W.; Ma, Q.; Chen, L.; Li, P.; Zhang, M.; Ramamoorthy, S.; Nawaz, Z.; Shimojima, T.; Wang, H.; Yang, Y.; et al. MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J. Biol. Chem. 2009, 284, 13987–14000. [Google Scholar] [CrossRef]
- Cui, F.C.; Chen, Y.; Wu, X.Y.; Hu, M.; Qin, W.S. MicroRNA-493-5p suppresses colorectal cancer progression via the PI3K-Akt-FoxO3a signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4212–4223. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Lee, K.H.; Lai, I.L.; Wang, D.; Mo, X.; Kulp, S.K.; Shapiro, C.L.; Chen, C.S. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014, 74, 4783–4795, Erratum in Cancer Res. 2018, 78, 3401. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Wu, W.; Hu, Y.; Li, J.; Xu, J.; Chen, X.; Xu, Z.; Yang, X.; Yang, B.; He, Q.; et al. Regorafenib inhibits EphA2 phosphorylation and leads to liver damage via the ERK/MDM2/p53 axis. Nat. Commun. 2023, 14, 2756. [Google Scholar] [CrossRef]
- Zechner, D.; Burtin, F.; Albert, A.C.; Zhang, X.; Kumstel, S.; Schonrogge, M.; Graffunder, J.; Shih, H.Y.; Muller, S.; Radecke, T.; et al. Intratumoral heterogeneity of the therapeutical response to gemcitabine and metformin. Oncotarget 2016, 7, 56395–56407. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.W.; Jin, X.; Zhao, Y.; Pan, Y.; Yang, J.; Karnes, R.J.; Zhang, J.; Wang, L.; Huang, H. AKT-phosphorylated FOXO1 suppresses ERK activation and chemoresistance by disrupting IQGAP1-MAPK interaction. EMBO J. 2017, 36, 995–1010. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Luo, C.; Ren, K.; Quan, M.; Cao, J. FOXO3a-mediated suppression of the self-renewal capacity of sphere-forming cells derived from the ovarian cancer SKOV3 cell line by 7-difluoromethoxyl-5,4′-di-n-octyl genistein. Mol. Med. Rep. 2014, 9, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Capristo, M.; Manara, M.C.; Mancarella, C.; Landuzzi, L.; Belfiore, A.; Lollini, P.L.; Picci, P.; Scotlandi, K. Metformin as an adjuvant drug against pediatric sarcomas: Hypoxia limits therapeutic effects of the drug. PLoS ONE 2013, 8, e83832. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ogo, A.; Yamura, M.; Yamaguchi, Y.; Nakashima, H. Metformin suppresses sonic hedgehog expression in pancreatic cancer cells. Anticancer Res. 2014, 34, 1765–1769. [Google Scholar]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.; Mak, C.S.; Yung, M.M.; Leung, T.H.; Wong, O.G.; Cheung, A.N.; Ngan, H.Y. AMPK activators suppress cervical cancer cell growth through inhibition of DVL3 mediated Wnt/beta-catenin signaling activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef]
- Subramaniam, N.; Sherman, M.H.; Rao, R.; Wilson, C.; Coulter, S.; Atkins, A.R.; Evans, R.M.; Liddle, C.; Downes, M. Metformin-mediated Bambi expression in hepatic stellate cells induces prosurvival Wnt/beta-catenin signaling. Cancer Prev. Res. 2012, 5, 553–561. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Cufi, S.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010, 9, 3807–3814. [Google Scholar] [CrossRef]
- Koeck, S.; Amann, A.; Huber, J.M.; Gamerith, G.; Hilbe, W.; Zwierzina, H. The impact of metformin and salinomycin on transforming growth factor beta-induced epithelial-to-mesenchymal transition in non-small cell lung cancer cell lines. Oncol. Lett. 2016, 11, 2946–2952. [Google Scholar] [CrossRef]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef]
- Li, X.N.; Song, J.; Zhang, L.; LeMaire, S.A.; Hou, X.; Zhang, C.; Coselli, J.S.; Chen, L.; Wang, X.L.; Zhang, Y.; et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 2009, 58, 2246–2257. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Colombo, S.L.; Moncada, S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem. J. 2009, 421, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Potocnjak, I.; Simic, L.; Gobin, I.; Vukelic, I.; Domitrovic, R. Antitumor activity of luteolin in human colon cancer SW620 cells is mediated by the ERK/FOXO3a signaling pathway. Toxicol. Vitr. 2020, 66, 104852. [Google Scholar] [CrossRef]
- Lin, L.; Ding, D.; Jiang, Y.; Li, Y.; Li, S. MEK inhibitors induce apoptosis via FoxO3a-dependent PUMA induction in colorectal cancer cells. Oncogenesis 2018, 7, 67. [Google Scholar] [CrossRef]
- Liu, H.; Jia, X.; Luo, K.; Chen, X.; Zhang, Z.; Chen, D.; Gu, Y.; He, Z.; Zheng, G. TCRP1 activated by mutant p53 promotes NSCLC proliferation via inhibiting FOXO3a. Oncogenesis 2022, 11, 19. [Google Scholar] [CrossRef]
- Adamowicz, M.; Vermezovic, J.; d’Adda di Fagagna, F. NOTCH1 Inhibits Activation of ATM by Impairing the Formation of an ATM-FOXO3a-KAT5/Tip60 Complex. Cell Rep. 2016, 16, 2068–2076. [Google Scholar] [CrossRef]
- Chung, Y.M.; Khan, P.P.; Wang, H.; Tsai, W.B.; Qiao, Y.; Yu, B.; Larrick, J.W.; Hu, M.C. Sensitizing tumors to anti-PD-1 therapy by promoting NK and CD8+ T cells via pharmacological activation of FOXO3. J. Immunother. Cancer 2021, 9, e002772. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef]
- Xu, S.P.; Sun, G.P.; Shen, Y.X.; Wei, W.; Peng, W.R.; Wang, H. Antiproliferation and apoptosis induction of paeonol in HepG2 cells. World J. Gastroenterol. 2007, 13, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.L.; Gong, J.; Yin, T.J.; Lu, Y.J.; Xia, J.J.; Xie, Y.Y.; Di, Y.; He, L.; Guo, J.L.; Sun, J.; et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur. J. Pharmacol. 2011, 654, 17–25. [Google Scholar] [CrossRef]
- Zhao, W.; Sachsenmeier, K.; Zhang, L.; Sult, E.; Hollingsworth, R.E.; Yang, H. A New Bliss Independence Model to Analyze Drug Combination Data. J. Biomol. Screen. 2014, 19, 817–821. [Google Scholar] [CrossRef]
- Feng, Y.; Guo, X.; Huang, X.; Wu, M.; Li, X.; Wu, S.; Luo, X. Metformin reverses stem cell-like HepG2 sphere formation and resistance to sorafenib by attenuating epithelial-mesenchymal transformation. Mol. Med. Rep. 2018, 18, 3866–3872. [Google Scholar] [CrossRef]
- Yi, Y.; Chen, D.; Ao, J.; Zhang, W.; Yi, J.; Ren, X.; Fei, J.; Li, F.; Niu, M.; Chen, H.; et al. Transcriptional suppression of AMPKalpha1 promotes breast cancer metastasis upon oncogene activation. Proc. Natl. Acad. Sci. USA 2020, 117, 8013–8021. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jin, J.; Guo, W.; Tang, Z.; Luo, Y.; Ying, Y.; Lin, H.; Luo, Z. PEBP4 Directs the Malignant Behavior of Hepatocellular Carcinoma Cells via Regulating mTORC1 and mTORC2. Int. J. Mol. Sci. 2022, 23, 8798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, A.; Xu, R.; Tao, X.; Dong, Y.; Lv, X.; Wei, D. Cell-penetrating and endoplasmic reticulum-locating TAT-IL-24-KDEL fusion protein induces tumor apoptosis. J. Cell Physiol. 2016, 231, 84–93. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Source | Catalog No. |
|---|---|---|
| p-Akt (Ser473) | Cell Signaling Tech. (Danvers, MA, USA) | 4060S |
| Akt | Cell Signaling Tech. | 13038 |
| p-AMPK (Thr172) | Cell Signaling Tech. | 8208 |
| AMPK | Cell Signaling Tech. | 5832 |
| p-ERK (Thr202/Tyr204) | Cell Signaling Tech. | 4370 |
| ERK | Cell Signaling Tech. | 4695S |
| Nanog | Cell Signaling Tech. | 4903P |
| Cleaved-Notch1 | Cell Signaling Tech. | 2421S |
| Oct4a | Cell Signaling Tech. | 2840P |
| KLF4 | Cell Signaling Tech. | 4038P |
| c-Myc | Cell Signaling Tech. | 5605P |
| FOXO3a | Proteintech (Wuhan, China) | 10849-1-AP |
| Ubiquitin | Proteintech (Wuhan, China) | 10201-2-AP |
| β-actin | OriGene (Rockville, MD, USA) | TA811000 |
| Gene Names | Primers (5′–3′) |
|---|---|
| GAPDH | F: ACGGGAAGCTCACTGGCATGG |
| R: GGTCCACCACCCTGTTGCTGTA | |
| Oct-4 | F: CTTCTCGCCCCCTCCAGGT |
| R: AAATAGAACCCCCAGGGTGAGC | |
| Nanog | F: TGAACCTCAGCTACAAACAG |
| R: TGGTGGTAGGAAGAGTAAAG | |
| Notch1 | F: TCAGCGGGATCCACTGTGAG |
| R: ACACAGGCAGGTGAACGAGTTG | |
| c-MYC | F: GGACGACGAGACCTTCATCAA |
| R: CCAGCTTCTCTGAGACGAGCTT | |
| FOXO3a | F: CGGACAAACGGCTCACTCT |
| R: GGACCCGCATGAATCGACTAT | |
| MDM2 | F: GAATCATCGGACTCAGGTACATC |
| R: TCTGTCTCACTAATTGCTCTCCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Z.; Zhang, Y.; Yu, Z.; Luo, Z. Metformin Suppresses Stemness of Non-Small-Cell Lung Cancer Induced by Paclitaxel through FOXO3a. Int. J. Mol. Sci. 2023, 24, 16611. https://doi.org/10.3390/ijms242316611
Tang Z, Zhang Y, Yu Z, Luo Z. Metformin Suppresses Stemness of Non-Small-Cell Lung Cancer Induced by Paclitaxel through FOXO3a. International Journal of Molecular Sciences. 2023; 24(23):16611. https://doi.org/10.3390/ijms242316611
Chicago/Turabian StyleTang, Zhimin, Yilan Zhang, Zhengyi Yu, and Zhijun Luo. 2023. "Metformin Suppresses Stemness of Non-Small-Cell Lung Cancer Induced by Paclitaxel through FOXO3a" International Journal of Molecular Sciences 24, no. 23: 16611. https://doi.org/10.3390/ijms242316611
APA StyleTang, Z., Zhang, Y., Yu, Z., & Luo, Z. (2023). Metformin Suppresses Stemness of Non-Small-Cell Lung Cancer Induced by Paclitaxel through FOXO3a. International Journal of Molecular Sciences, 24(23), 16611. https://doi.org/10.3390/ijms242316611