
The Subtype Selectivity in Search of Potent Hypotensive Agents among 5,5-Dimethylhydantoin Derived α1-Adrenoceptors Antagonists

 , , , ,
, , , ,
Abstract
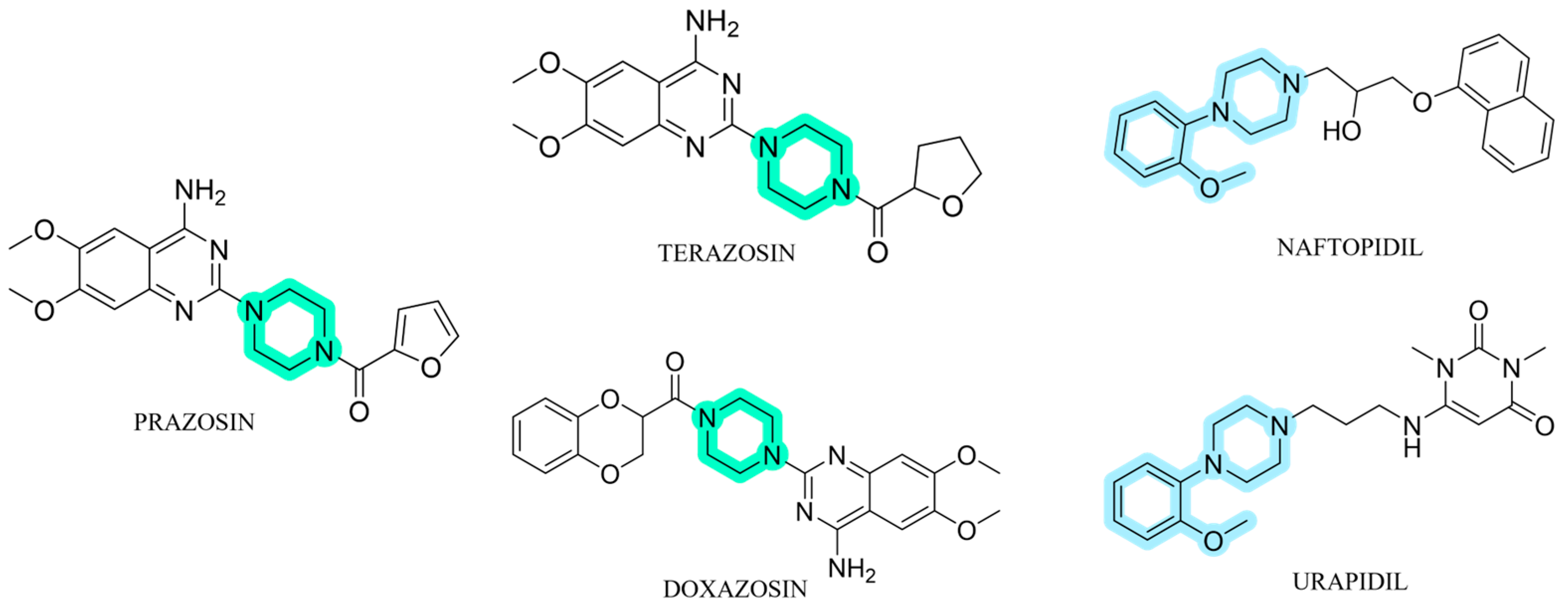
1. Introduction
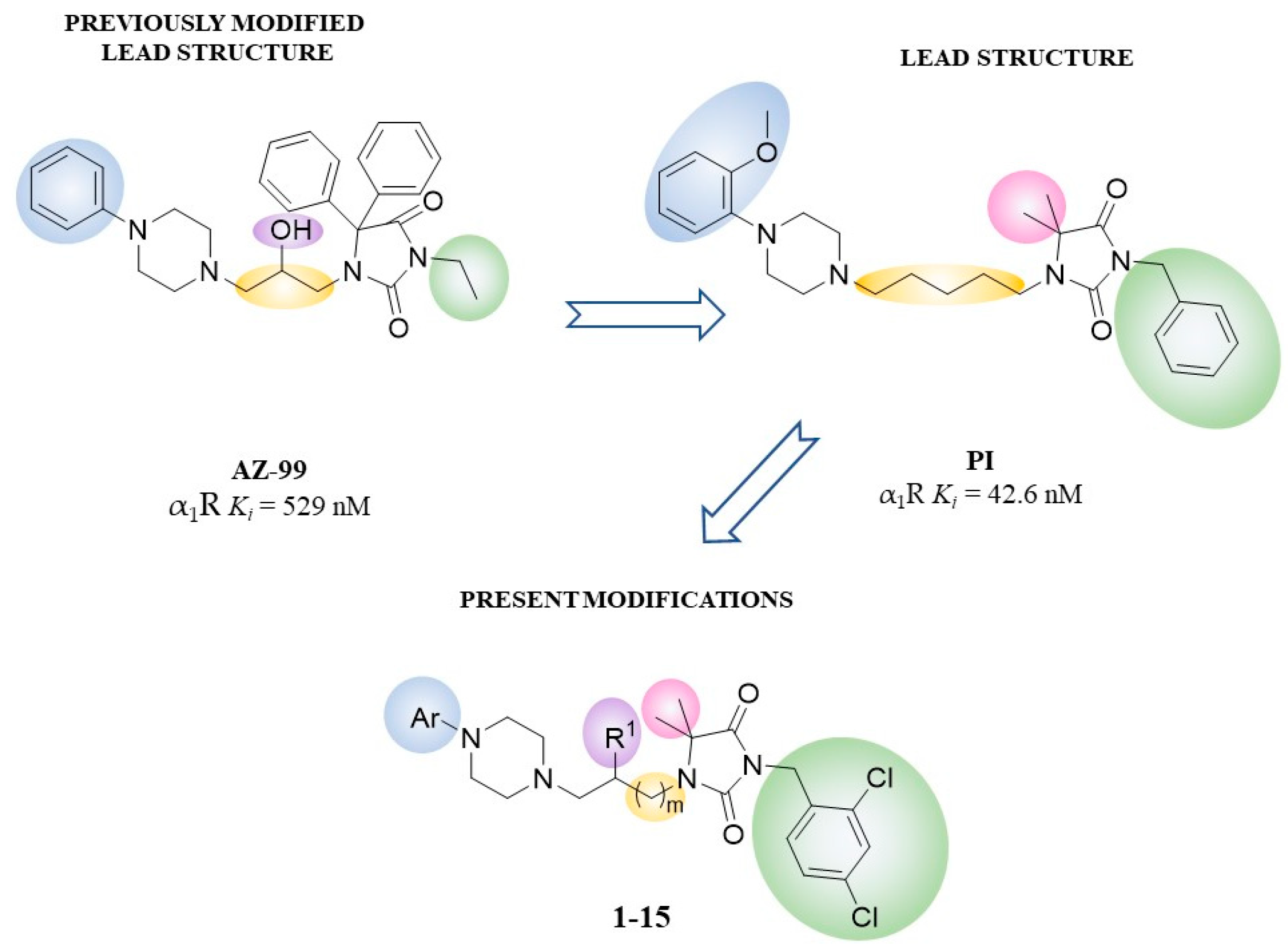
2. Results
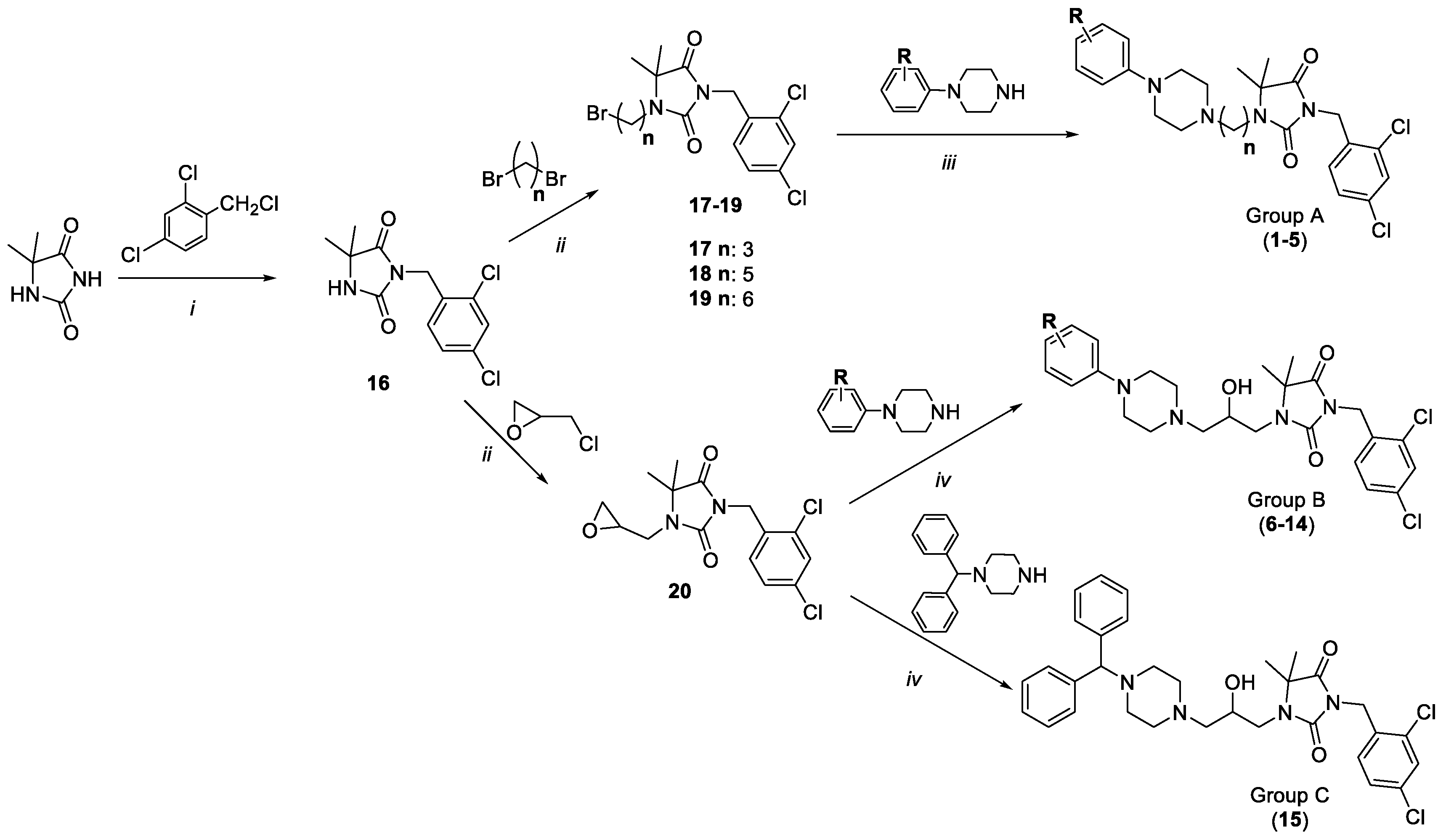
2.1. Chemistry
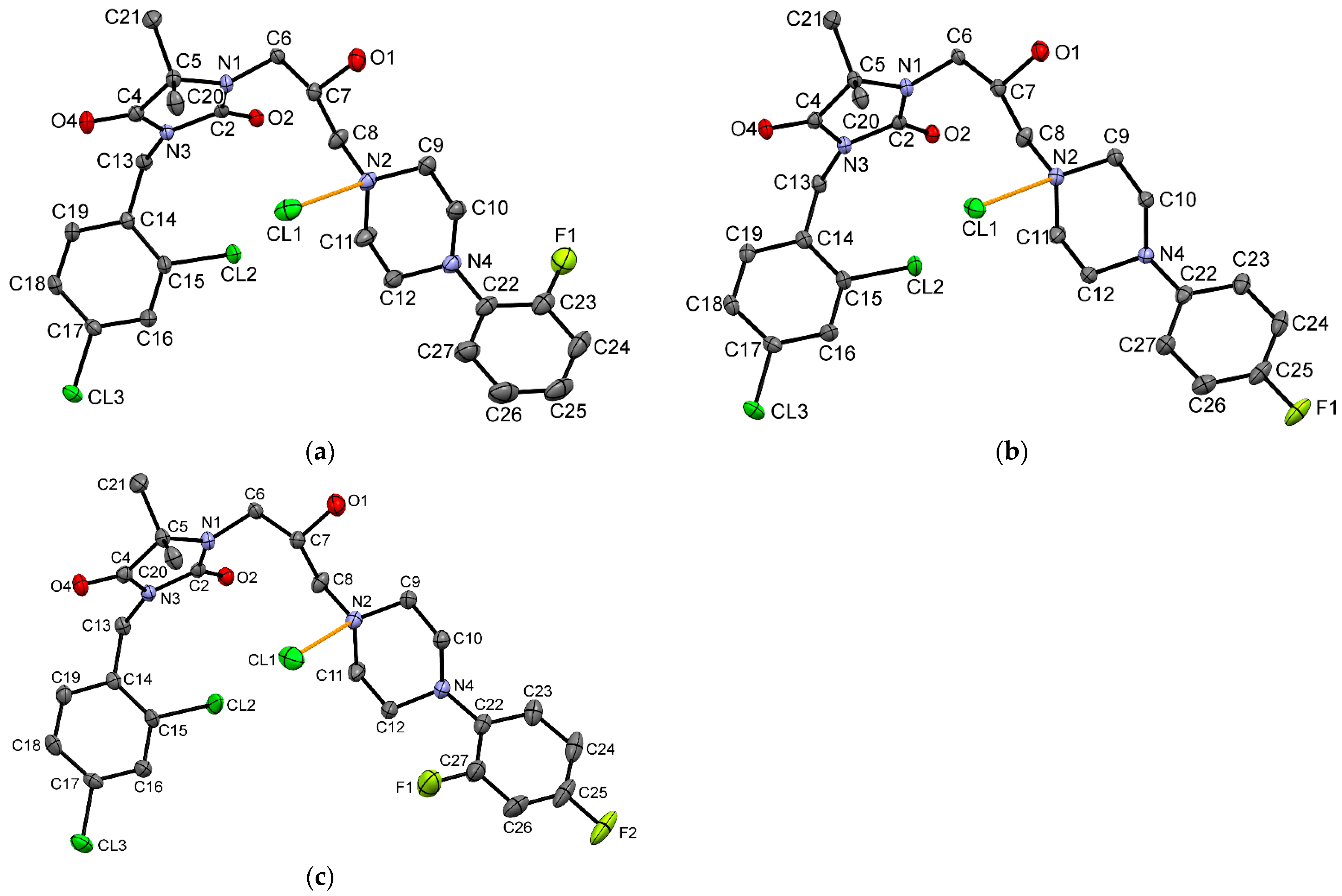
2.2. X-ray Crystallographic Studies
2.3. Affinity for α1-Adrenoceptors
2.4. Intrinsic Activity
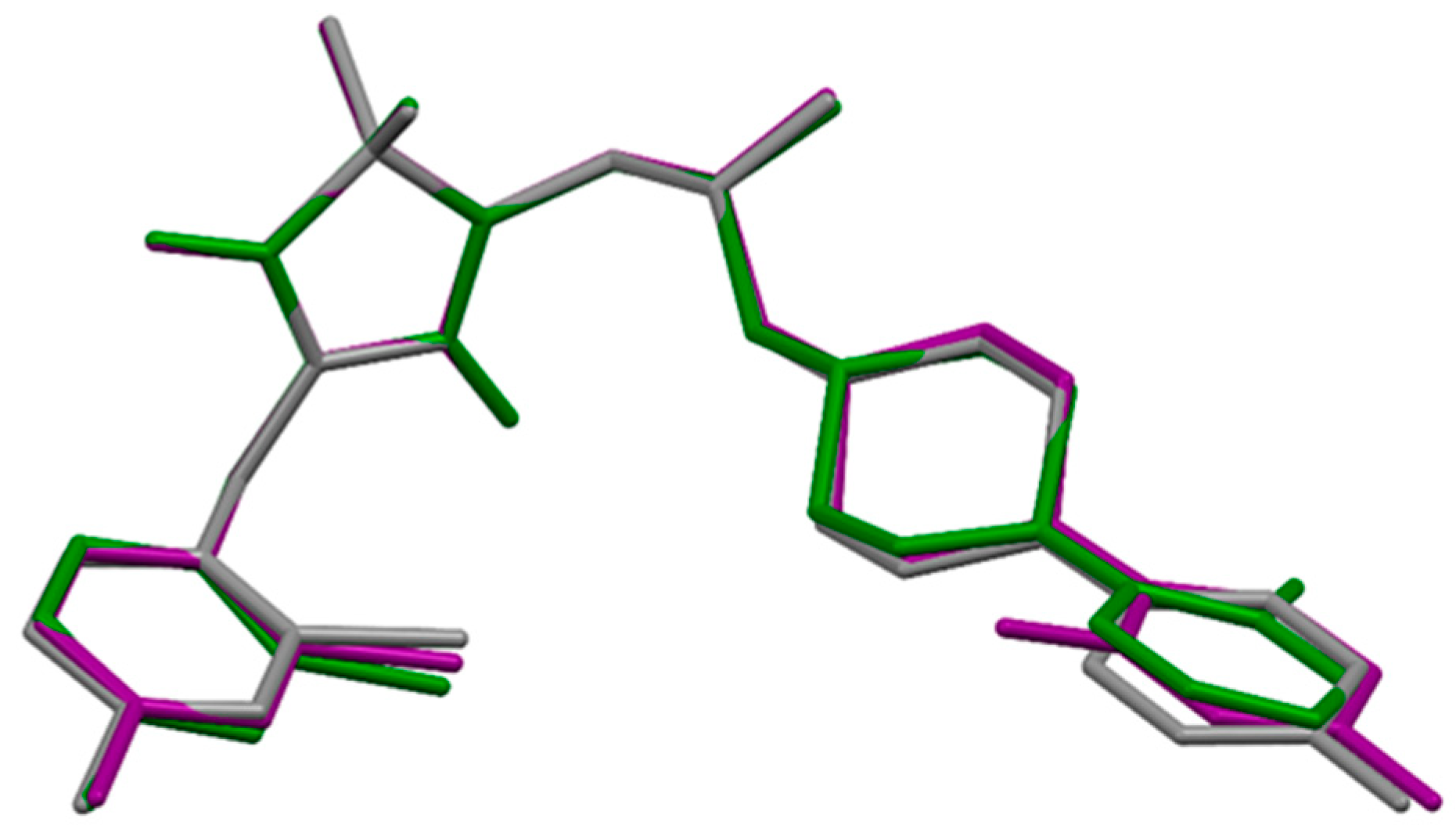
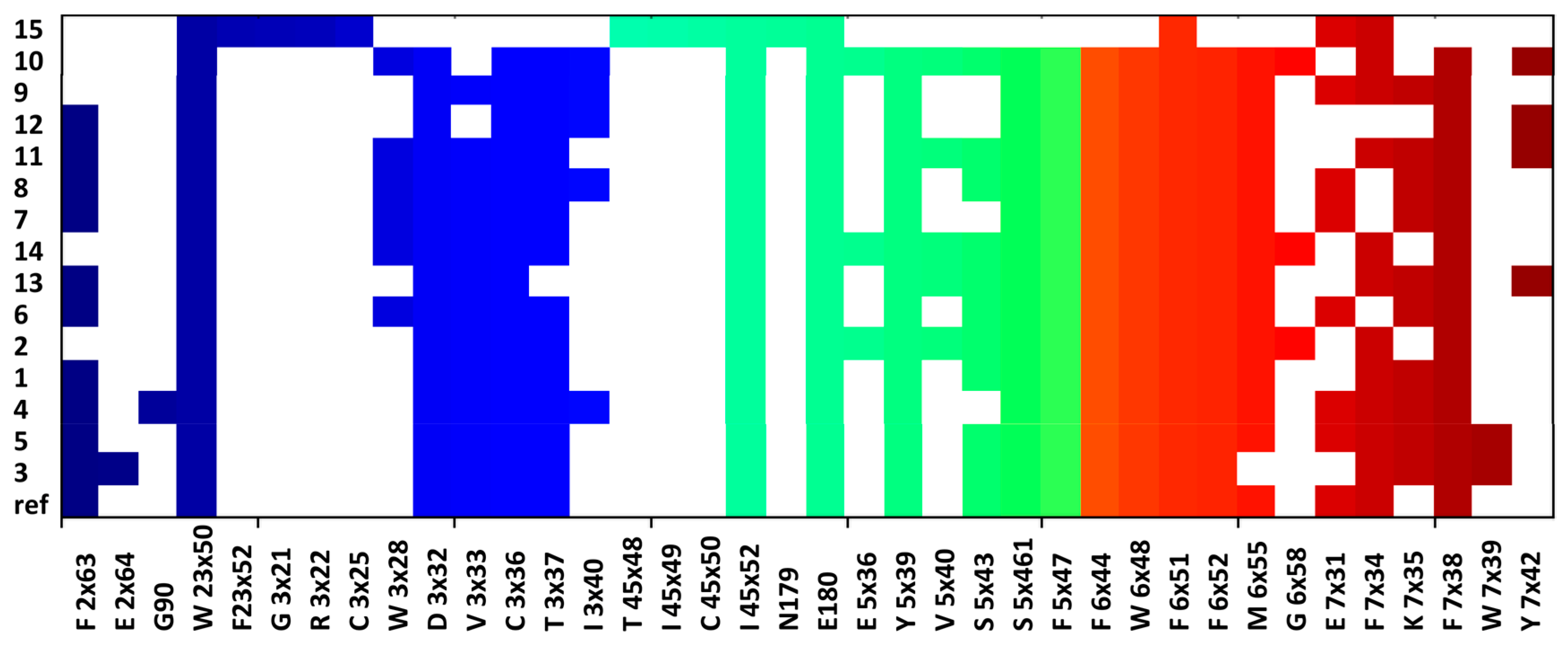
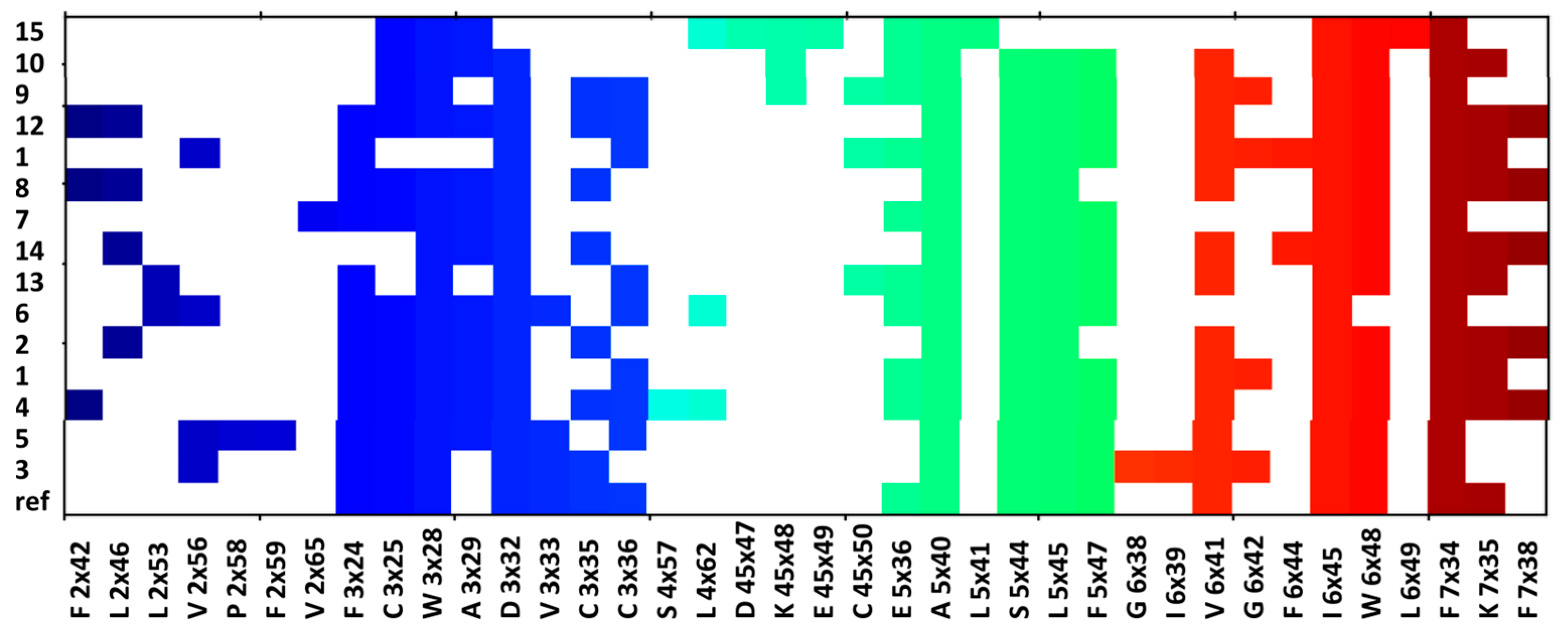
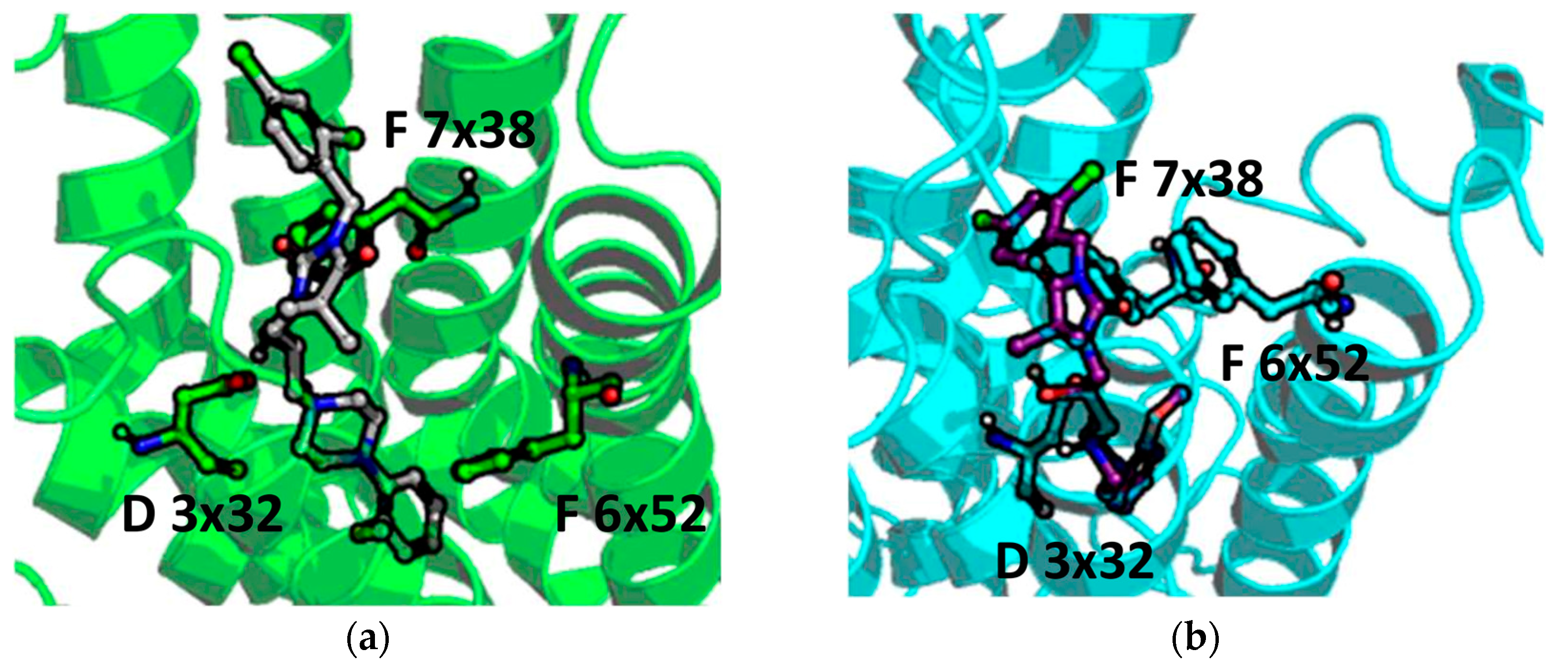
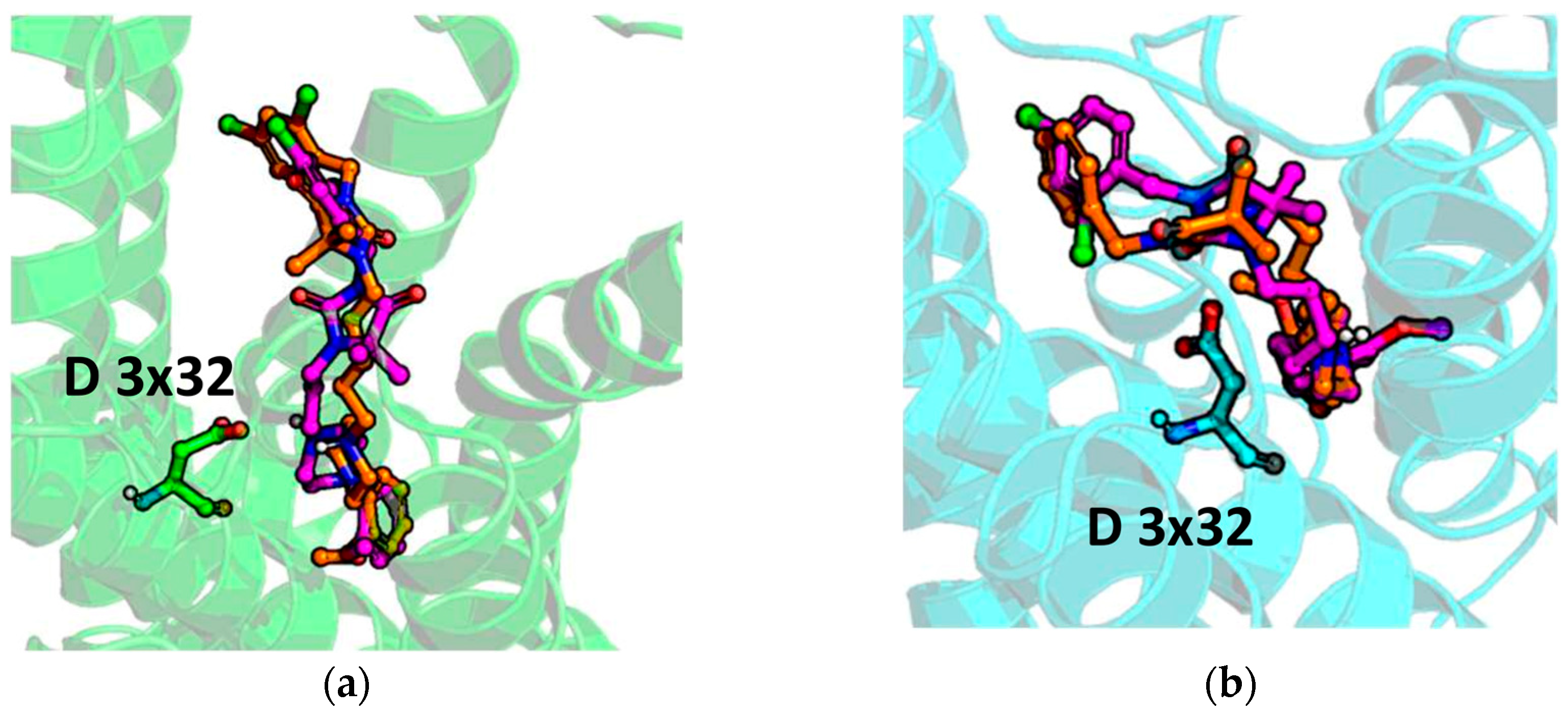
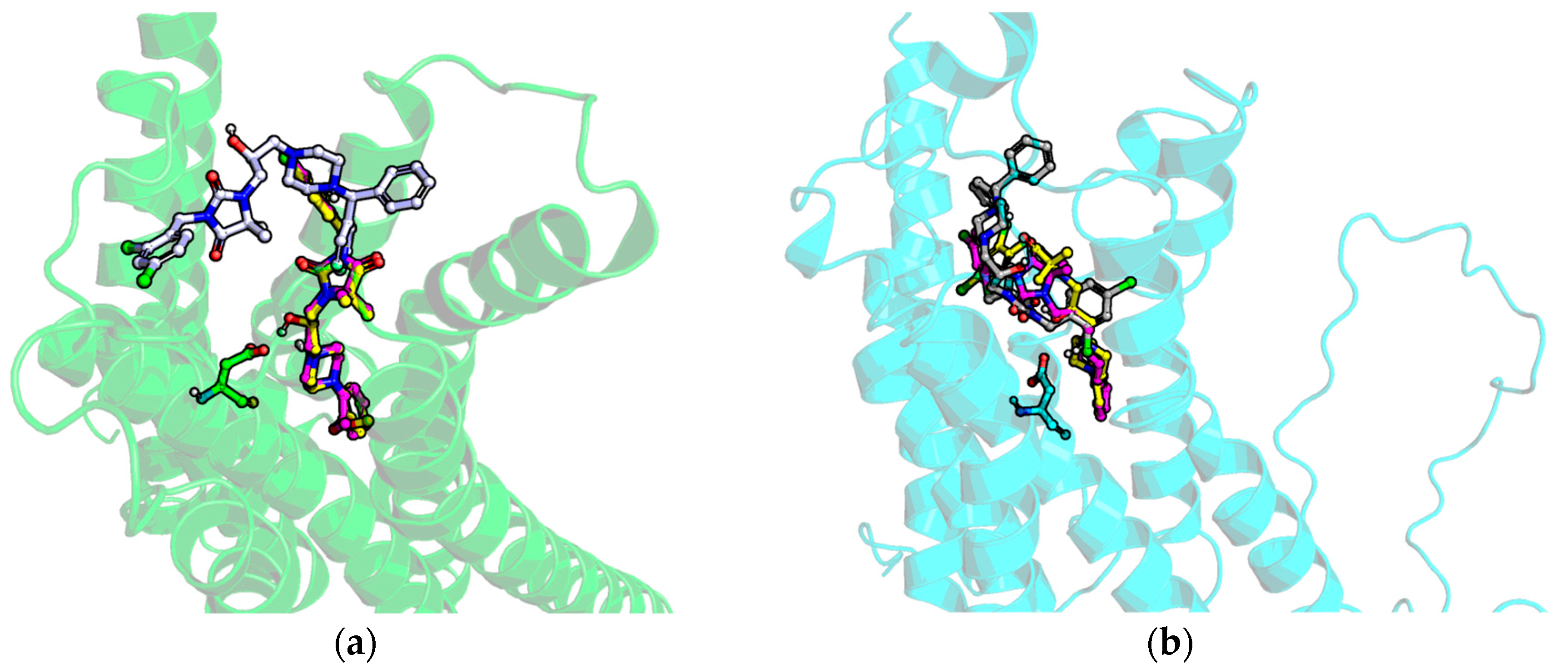
2.5. Molecular Modelling
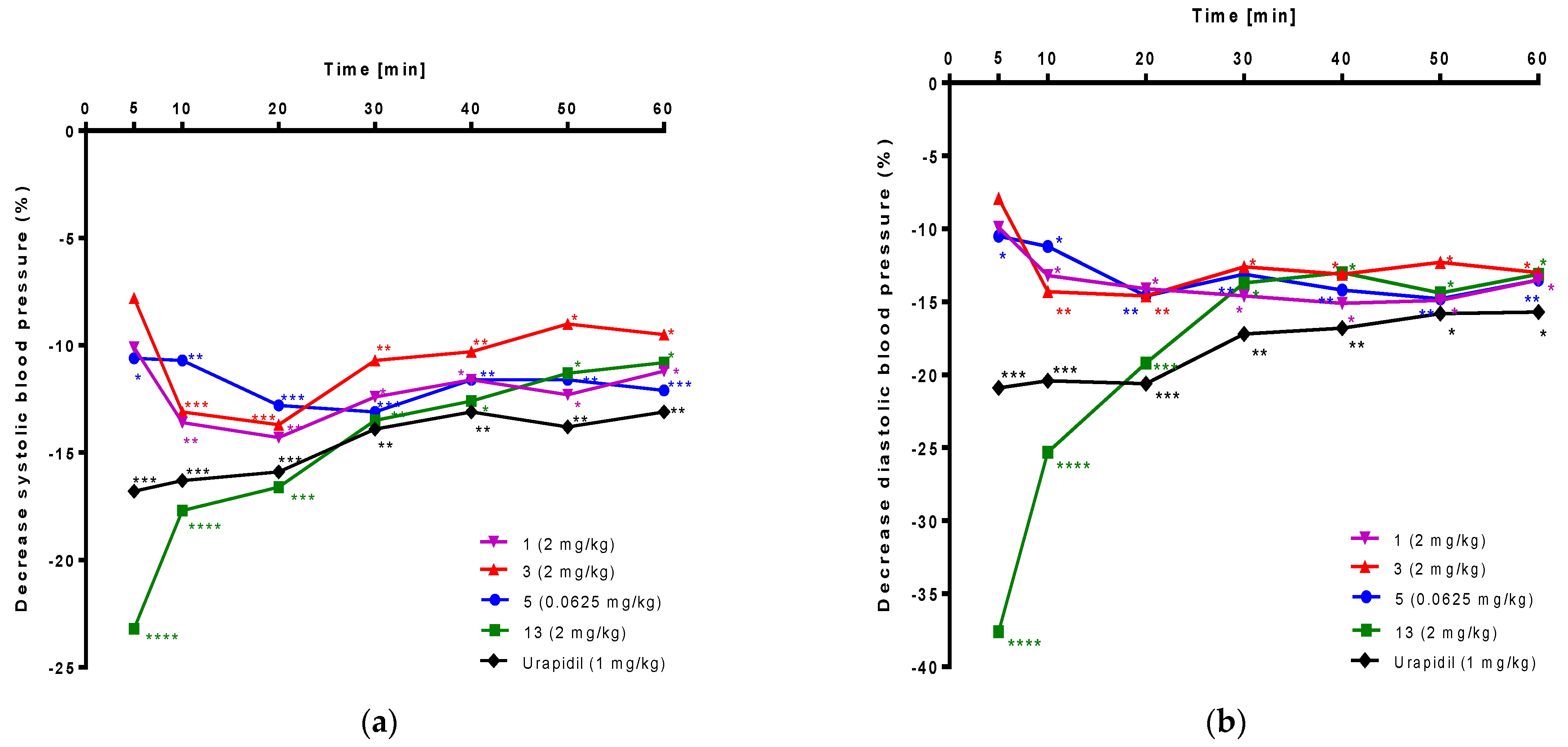
2.6. The Influence on Blood Pressure in Rats
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure to Obtain Final Products from Group A (1–5)
4.1.2. General Procedure to Obtain Final Products from Group B (6, 10–14)
4.2. Crystallography
4.3. Pharmacology
4.3.1. Chemicals
4.3.2. Animals
4.3.3. In Vitro Binding Assay—Determination of Affinity for α1-ARs
4.3.4. In Vitro Functional Assays—Determination of Intrinsic Activity at the α1A-ARs and the α1B-ARs
4.3.5. Influence on Blood Pressure in Normotensive Rats
4.3.6. Statistical Analysis
4.4. Molecular Modelling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Docherty, J.R. Subtypes of functional alpha1-adrenoceptor. Cell. Mol. Life Sci. 2010, 67, 405–417. [Google Scholar] [CrossRef]
- Civantos Calzada, B.; Aleixandre de Artiñano, A. Alpha-adrenoceptor subtypes. Pharmacol. Res. 2001, 44, 195–208. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef]
- Sica, D.A. Alpha1-adrenergic blockers: Current usage considerations. J. Clin. Hypertens. 2005, 7, 757–762. [Google Scholar] [CrossRef]
- Jain, K.S.; Bariwal, J.B.; Kathiravan, M.K.; Phoujdar, M.S.; Sahne, R.S.; Chauhan, B.S.; Shah, A.K.; Yadav, M.R. Recent advances in selective alpha1-adrenoreceptor antagonists as antihypertensive agents. Bioorganic Med. Chem. 2008, 16, 4759–4800. [Google Scholar] [CrossRef]
- Chapman, N.; Chen, C.Y.; Fujita, T.; Hobbs, F.D.; Kim, S.J.; Staessen, J.A.; Tanomsup, S.; Wang, J.G.; Williams, B. Time to re-appraise the role of alpha-1 adrenoceptor antagonists in the management of hypertension? J. Hypertens. 2010, 28, 1796–1803. [Google Scholar] [CrossRef]
- Lepor, H.; Kazzazi, A.; Djavan, B. α-Blockers for benign prostatic hyperplasia: The new era. Curr. Opin. Urol. 2012, 22, 7–15. [Google Scholar] [CrossRef]
- Kadekawa, K.; Sugaya, K.; Ashitomi, K.; Nishijima, S. Clinical Efficacy of α1-Adrenargic Receptor Antagonist Naftopidil 75 mg/day in Patients with Benign Prostatic Hyperplasia. Low. Urin. Tract Symptoms 2010, 2, 106–112. [Google Scholar] [CrossRef]
- Manohar, C.M.S.; Nagabhushana, M.; Karthikeyan, V.S.; Sanjay, R.P.; Kamath, A.J.; Keshavamurthy, R. Safety and efficacy of tamsulosin, alfuzosin or silodosin as monotherapy for LUTS in BPH-a double-blind randomized trial. Cent. Eur. J. Urol. 2017, 70, 148–153. [Google Scholar] [CrossRef]
- Rudner, X.L.; Berkowitz, D.E.; Booth, J.V.; Funk, B.L.; Cozart, K.L.; D’Amico, E.B.; El-Moalem, H.; Page, S.O.; Richardson, C.D.; Winters, B.; et al. Subtype specific regulation of human vascular alpha(1)-adrenergic receptors by vessel bed and age. Circulation 1999, 100, 2336–2343. [Google Scholar] [CrossRef]
- Adefurin, A.; Ghimire, L.V.; Kohli, U.; Muszkat, M.; Sofowora, G.G.; Li, C.; Levinson, R.T.; Paranjape, S.Y.; Stein, C.M.; Kurnik, D. Genetic variation in the alpha1B-adrenergic receptor and vascular response. Pharm. J. 2017, 17, 366–371. [Google Scholar] [CrossRef]
- Hatano, A.; Takahashi, H.; Tamaki, M.; Komeyama, T.; Koizumi, T.; Takeda, M. Pharmacological evidence of distinct alpha 1-adrenoceptor subtypes mediating the contraction of human prostatic urethra and peripheral artery. Br. J. Pharmacol. 1994, 113, 723–728. [Google Scholar] [CrossRef]
- Harada, K.; Ohmori, M.; Kitoh, Y.; Sugimoto, K.; Fujimura, A. A comparison of the antagonistic activities of tamsulosin and terazosin against human vascular alpha1-adrenoceptors. Jpn. J. Pharmacol. 1999, 80, 209–215. [Google Scholar] [CrossRef][Green Version]
- Jensen, B.C.; Swigart, P.M.; Montgomery, M.D.; Simpson, P.C. Functional alpha-1B adrenergic receptors on human epicardial coronary artery endothelial cells. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 382, 475–482. [Google Scholar] [CrossRef]
- Oliver, E.; Montó, F.; Rovira, E.; Valldecabres, C.; Muedra, V.; D’Ocon, P. Changes in the expression of α1B-adrenoceptor in peripheral mononuclear cells correlates with blood pressure and plasmatic homocysteine. Biomed. Pharmacother. 2017, 88, 721–727. [Google Scholar] [CrossRef]
- Grimm, R.H., Jr.; Flack, J.M. Alpha 1 adrenoreceptor antagonists. J. Clin. Hypertens. 2011, 13, 654–657. [Google Scholar] [CrossRef]
- Black, H.R. Doxazosin as combination therapy for patients with stage 1 and stage 2 hypertension. J. Cardiovasc. Pharmacol. 2003, 41, 866–869. [Google Scholar] [CrossRef]
- Buch, J. Urapidil, a dual-acting antihypertensive agent: Current usage considerations. Adv. Ther. 2010, 27, 426–443. [Google Scholar] [CrossRef]
- Itskovitz, H.D. Alpha 1-blockade for the treatment of hypertension: A megastudy of terazosin in 2214 clinical practice settings. Clin. Ther. 1994, 16, 490–504. [Google Scholar]
- Levy, D.; Walmsley, P.; Levenstein, M. Principal results of the Hypertension and Lipid Trial (HALT): A multicenter study of doxazosin in patients with hypertension. Am. Heart J. 1996, 131, 966–973. [Google Scholar] [CrossRef]
- Dell’Omo, G.; Penno, G.; Del Prato, S.; Pedrinelli, R. Doxazosin in metabolically complicated hypertension. Expert Rev. Cardiovasc. Ther. 2007, 5, 1027–1035. [Google Scholar] [CrossRef]
- Lehtonen, A. Doxazosin effects on insulin and glucose in hypertensive patients. The Finnish Multicenter Study Group. Am. Heart J. 1991, 121 Pt 2, 1307–1311. [Google Scholar] [CrossRef]
- Maheux, P.; Facchini, F.; Jeppesen, J.; Greenfield, M.S.; Clinkingbeard, C.; Chen, Y.D.; Reaven, G.M. Changes in glucose, insulin, lipid, lipoprotein, and apoprotein concentrations and insulin action in doxazosin-treated patients with hypertension. Comparison between nondiabetic individuals and patients with non-insulin-dependent diabetes mellitus. Am. J. Hypertens. 1994, 7, 416–424. [Google Scholar] [CrossRef]
- Grimm, R.H., Jr.; Flack, J.M.; Grandits, G.A.; Elmer, P.J.; Neaton, J.D.; Cutler, J.A.; Lewis, C.; McDonald, R.; Schoenberger, J.; Stamler, J. Long-term effects on plasma lipids of diet and drugs to treat hypertension. Treatment of Mild Hypertension Study (TOMHS) Research Group. JAMA 1996, 275, 1549–1556. [Google Scholar] [CrossRef]
- Hirano, T.; Yoshino, G.; Kashiwazaki, K.; Adachi, M. Doxazosin reduces prevalence of small dense low density lipoprotein and remnant-like particle cholesterol levels in nondiabetic and diabetic hypertensive patients. Am. J. Hypertens. 2001, 14 Pt 1, 908–913. [Google Scholar] [CrossRef][Green Version]
- Kinoshita, M.; Shimazu, N.; Fujita, M.; Fujimaki, Y.; Kojima, K.; Mikuni, Y.; Horie, E.; Teramoto, T. Doxazosin, an alpha1-adrenergic antihypertensive agent, decreases serum oxidized LDL. Am. J. Hypertens. 2001, 14, 267–270. [Google Scholar] [CrossRef]
- Derosa, G.; Cicero, A.F.; D’Angelo, A.; Ragonesi, P.D.; Ciccarelli, L.; Fogari, E.; Salvadeo, S.A.; Ferrari, I.; Gravina, A.; Fassi, R.; et al. Synergistic effect of doxazosin and acarbose in improving metabolic control in patients with impaired glucose tolerance. Clin. Drug Investig. 2006, 26, 529–539. [Google Scholar] [CrossRef]
- Jackevicius, C.A.; Ghaznavi, Z.; Lu, L.; Warner, A.L. Safety of Alpha-Adrenergic Receptor Antagonists in Heart Failure. JACC Heart Fail. 2018, 6, 917–925. [Google Scholar] [CrossRef]
- Perez, D.M.; Doze, V.A. Cardiac and neuroprotection regulated by α(1)-adrenergic receptor subtypes. J. Recept. Signal Transduct. Res. 2011, 31, 98–110. [Google Scholar] [CrossRef]
- Shannon, R.; Chaudhry, M. Effect of alpha1-adrenergic receptors in cardiac pathophysiology. Am. Heart J. 2006, 152, 842–850. [Google Scholar] [CrossRef]
- Anyukhovsky, E.P.; Rosen, M.R. Abnormal automatic rhythms in ischemic Purkinje fibers are modulated by a specific alpha 1-adrenergic receptor subtype. Circulation 1991, 83, 2076–2082. [Google Scholar] [CrossRef]
- Geller, J.C.; Cua, M.; Prieto, L.; Guo, S.D.; Danilo, P., Jr.; Rosen, M.R. Chloroethylclonidine increases the incidence of lethal arrhythmias during coronary occlusion in anesthetized dogs. Eur. J. Pharmacol. 1995, 294, 423–428. [Google Scholar] [CrossRef]
- Yasutake, M.; Avkiran, M. Effects of selective alpha 1A-adrenoceptor antagonists on reperfusion arrhythmias in isolated rat hearts. Mol. Cell. Biochem. 1995, 147, 173–180. [Google Scholar] [CrossRef]
- Suita, K.; Fujita, T.; Hasegawa, N.; Cai, W.; Jin, H.; Hidaka, Y.; Prajapati, R.; Umemura, M.; Yokoyama, U.; Sato, M.; et al. Norepinephrine-Induced Adrenergic Activation Strikingly Increased the Atrial Fibrillation Duration through β1- and α1-Adrenergic Receptor-Mediated Signaling in Mice. PLoS ONE 2015, 10, e0133664. [Google Scholar] [CrossRef]
- Luo, D.L.; Gao, J.; Fan, L.L.; Tang, Y.; Zhang, Y.Y.; Han, Q.D. Receptor subtype involved in alpha 1-adrenergic receptor-mediated Ca2+ signaling in cardiomyocytes. Acta Pharmacol. Sin. 2007, 28, 968–974. [Google Scholar] [CrossRef]
- Yasutake, M.; Avkiran, M. Exacerbation of reperfusion arrhythmias by alpha 1 adrenergic stimulation: A potential role for receptor mediated activation of sarcolemmal sodium-hydrogen exchange. Cardiovasc. Res. 1995, 29, 222–230. [Google Scholar]
- Desiniotis, A.; Kyprianou, N. Advances in the design and synthesis of prazosin derivatives over the last ten years. Expert Opin. Ther. Targets 2011, 15, 1405–1418. [Google Scholar] [CrossRef]
- Li, H.; Xu, T.Y.; Li, Y.; Chia, Y.C.; Buranakitjaroen, P.; Cheng, H.M.; Van Huynh, M.; Sogunuru, G.P.; Tay, J.C.; Wang, T.D.; et al. Role of α1-blockers in the current management of hypertension. J. Clin. Hypertens. 2022, 24, 1180–1186. [Google Scholar] [CrossRef]
- Marona, H.; Kubacka, M.; Filipek, B.; Siwek, A.; Dybała, M.; Szneler, E.; Pociecha, T.; Gunia, A.; Waszkielewicz, A.M. Synthesis, alpha-adrenoceptors affinity and alpha 1-adrenoceptor antagonistic properties of some 1,4-substituted piperazine derivatives. Pharmazie 2011, 66, 733–739. [Google Scholar]
- Wu, Y.; Zeng, L.; Zhao, S. Ligands of Adrenergic Receptors: A Structural Point of View. Biomolecules 2021, 11, 936. [Google Scholar] [CrossRef]
- Handzlik, J.; Bajda, M.; Zygmunt, M.; Maciąg, D.; Dybała, M.; Bednarski, M.; Filipek, B.; Malawska, B.; Kieć-Kononowicz, K. Antiarrhythmic properties of phenylpiperazine derivatives of phenytoin with α1-adrenoceptor affinities. Bioorganic Med. Chem. 2012, 20, 2290–2303. [Google Scholar] [CrossRef]
- Waszkielewicz, A.M.; Kubacka, M.; Pańczyk, K.; Mogilski, S.; Siwek, A.; Głuch-Lutwin, M.; Gryboś, A.; Filipek, B. Synthesis and activity of newly designed aroxyalkyl or aroxyethoxyethyl derivatives of piperazine on the cardiovascular and the central nervous systems. Bioorganic Med. Chem. Lett. 2016, 26, 5315–5321. [Google Scholar] [CrossRef]
- Bopp, C.; Auger, C.; Diemunsch, P.; Schini-Kerth, V. The effect of urapidil, an alpha-1 adrenoceptor antagonist and a 5-HT1A agonist, on the vascular tone of the porcine coronary and pulmonary arteries, the rat aorta and the human pulmonary artery. Eur. J. Pharmacol. 2016, 779, 53–58. [Google Scholar] [CrossRef]
- Masumori, N. Naftopidil for the treatment of urinary symptoms in patients with benign prostatic hyperplasia. Ther. Clin. Risk Manag. 2011, 7, 227–238. [Google Scholar] [CrossRef]
- Handzlik, J.; Maciag, D.; Kubacka, M.; Mogilski, S.; Filipek, B.; Stadnicka, K.; Kieć-Kononowicz, K. Synthesis, alpha 1-adrenoceptor antagonist activity, and SAR study of novel arylpiperazine derivatives of phenytoin. Bioorganic Med. Chem. 2008, 16, 5982–5998. [Google Scholar] [CrossRef]
- Handzlik, J.; Pertz, H.H.; Görnemann, T.; Jähnichen, S.; Kieć-Kononowicz, K. Search for influence of spatial properties on affinity at α1-adrenoceptor subtypes for phenylpiperazine derivatives of phenytoin. Bioorganic Med. Chem. Lett. 2010, 20, 6152–6156. [Google Scholar] [CrossRef]
- Handzlik, J.; Szymańska, E.; Wójcik, R.; Dela, A.; Jastrzębska-Więsek, M.; Karolak-Wojciechowska, J.; Fruziński, A.; Siwek, A.; Filipek, B.; Kieć-Kononowicz, K. Synthesis and SAR-study for novel arylpiperazine derivatives of 5-arylidenehydantoin with α1-adrenoceptor antagonistic properties. Bioorganic Med. Chem. 2012, 20, 4245–4257. [Google Scholar] [CrossRef]
- Handzlik, J.; Bojarski, A.J.; Satała, G.; Kubacka, M.; Sadek, B.; Ashoor, A.; Siwek, A.; Więcek, M.; Kucwaj, K.; Filipek, B.; et al. SAR-studies on the importance of aromatic ring topologies in search for selective 5-HT(7) receptor ligands among phenylpiperazine hydantoin derivatives. Eur. J. Med. Chem. 2014, 78, 324–339. [Google Scholar] [CrossRef]
- Barbaro, R.; Betti, L.; Botta, M.; Corelli, F.; Giannaccini, G.; Maccari, L.; Manetti, F.; Strappaghetti, G.; Corsano, S. Synthesis, biological evaluation, and pharmacophore generation of new pyridazinone derivatives with affinity toward alpha(1)- and alpha(2)-adrenoceptors. J. Med. Chem. 2001, 44, 2118–2132. [Google Scholar] [CrossRef]
- Witek, K.; Latacz, G.; Kaczor, A.; Czekajewska, J.; Żesławska, E.; Chudzik, A.; Karczewska, E.; Nitek, W.; Kieć-Kononowicz, K.; Handzlik, J. Phenylpiperazine 5,5-Dimethylhydantoin Derivatives as First Synthetic Inhibitors of Msr(A) Efflux Pump in Staphylococcus epidermidis. Molecules 2020, 25, 3788. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Foglar, R.; Shibata, K.; Horie, K.; Hirasawa, A.; Tsujimoto, G. Use of recombinant alpha 1-adrenoceptors to characterize subtype selectivity of drugs for the treatment of prostatic hypertrophy. Eur. J. Pharmacol. 1995, 288, 201–207. [Google Scholar] [CrossRef]
- Takei, R.; Ikegaki, I.; Shibata, K.; Tsujimoto, G.; Asano, T. Naftopidil, a novel alpha1-adrenoceptor antagonist, displays selective inhibition of canine prostatic pressure and high affinity binding to cloned human alpha1-adrenoceptors. Jpn. J. Pharmacol. 1999, 79, 447–454. [Google Scholar]
- Szkaradek, N.; Rapacz, A.; Pytka, K.; Filipek, B.; Siwek, A.; Cegła, M.; Marona, H. Synthesis and preliminary evaluation of pharmacological properties of some piperazine derivatives of xanthone. Bioorganic Med. Chem. 2013, 21, 514–522. [Google Scholar] [CrossRef]
- Szkaradek, N.; Rapacz, A.; Pytka, K.; Filipek, B.; Żelaszczyk, D.; Szafrański, P.; Słoczyńska, K.; Marona, H. Cardiovascular activity of the chiral xanthone derivatives. Bioorganic Med. Chem. 2015, 23, 6714–6724. [Google Scholar] [CrossRef]
- Manetti, F.; Corelli, F.; Strappaghetti, G.; Botta, M. Arylpiperazines with affinity toward alpha(1)-adrenergic receptors. Curr. Med. Chem. 2002, 9, 1303–1321. [Google Scholar] [CrossRef]
- Betti, L.; Zanelli, M.; Giannaccini, G.; Manetti, F.; Schenone, S.; Strappaghetti, G. Synthesis of new piperazine-pyridazinone derivatives and their binding affinity toward alpha1-, alpha2-adrenergic and 5-HT1A serotoninergic receptors. Bioorganic Med. Chem. 2006, 14, 2828–2836. [Google Scholar] [CrossRef]
- Fukiyama, K.; Omae, T.; Iimura, O.; Yoshinaga, K.; Yagi, S.; Inagaki, Y.; Ishii, M.; Kaneko, Y.; Yamada, K.; Ijichi, H.; et al. A double-blind comparative study of doxazosin and prazosin in the treatment of essential hypertension. Am. Heart J. 1991, 121 Pt 2, 317–322. [Google Scholar] [CrossRef]
- Joglekar, S.J.; Nanivadekar, A.S. A randomized, controlled, multicenter study to compare prazosin GITS with enalapril in hypertensive patients with diabetes mellitus. Bombay Hypertension Study Group. J. Assoc. Physicians India 1998, Suppl 1, 52–62. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Ciazovazzo, C.; Mallamo, M.; Mazzone, A.G.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- LigPrep. Schrödinger Release 2019-4; Schrödinger LLC: New York, NY, USA, 2019. [Google Scholar]
- Schrödinger Release 2019-4: Glide; Schrödinger LLC: New York, NY, USA, 2019.
- Pandy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.3.2; Schrödinger LLC: New York, NY, USA, 2019.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cpd | Group | n | R | Cpd | Group | n | R |
|---|---|---|---|---|---|---|---|
| 1 | A | 3 | 2-MeO | 9 | B | - | 2,4-diF |
| 2 | A | 3 | 2,3-diCl | 10 | B | - | 3,4-diCl |
| 3 | A | 5 | H | 11 | B | - | 2-CN |
| 4 | A | 6 | 3,4-diCl | 12 | B | - | 3,4-diMe |
| 5 | A | 6 | 2-MeO | 13 | B | - | 2-MeO |
| 6 | B | - | H | 14 | B | - | 3-MeO |
| 7 | B | - | 2-F | 15 | C | - | - |
| 8 | B | - | 4-F |
| Compound | Ki (nM) |
|---|---|
| 1 | 31.0 |
| 2 | 535.0 |
| 3 | 43.0 |
| 4 | 255.0 |
| 5 | 12.7 |
| 6 | 53.0 |
| 7 | 100.0 |
| 8 | 44.0 |
| 9 | 42.0 |
| 10 | 778.0 |
| 11 | 26.0 |
| 12 | 270.0 |
| 13 | 29.0 |
| 14 | 289.0 |
| 15 | 2443.0 |
| AZ-99 | 529.0 a |
| Prazosin | 0.24 a |
| Urapidil | 127.9 b |
| Compound | α1A-AR | α1B-AR | Selectivity Ratio | |||
|---|---|---|---|---|---|---|
| IC50 a (nM) | Profile | IC50 a (nM) | Profile | α1A/α1B b | α1B/α1A c | |
| 1 | 90.62 | Antagonist | 10.71 | Antagonist | 8.46 | 0.12 |
| 2 | 1730.00 | Antagonist | 1146.00 | Antagonist | 1.51 | 0.66 |
| 3 | 290.00 | Antagonist | 3.27 | Antagonist | 88.73 | 0.01 |
| 5 | 0.02 | Antagonist | 0.002 | Antagonist | 10.00 | 0.10 |
| 6 | 5909.00 | Antagonist | 905.00 | Antagonist | 6.53 | 0.15 |
| 7 | 1415.00 | Antagonist | 483.90 | Antagonist | 2.92 | 0.34 |
| 8 | 816.70 | Antagonist | 170.40 | Antagonist | 4.79 | 0.21 |
| 9 | 162.40 | Antagonist | 147.40 | Antagonist | 1.10 | 0.91 |
| 11 | 162.30 | Antagonist | 359.10 | Antagonist | 0.45 | 2.21 |
| 12 | 3136.00 | Antagonist | 377.30 | Antagonist | 8.31 | 0.12 |
| 13 | 138.70 | Antagonist | 331.10 | Antagonist | 0.42 | 2.39 |
| 14 | 1221.00 | Antagonist | 1366.00 | Antagonist | 0.89 | 1.12 |
| Tamsulosin | 0.03 | Antagonist | 0.48 | Antagonist | 0.06 | 16.00 |
| Terazosin | 4.52 | Antagonist | 0.67 | Antagonist | 6.70 | 0.15 |
| Prazosin | 0.72 | Antagonist | 0.17 | Antagonist | 4.25 | 0.24 |
| Phenylephrine | 23.64 | Agonist | 2.31 | Agonist | 10.25 | 0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczor, A.; Knutelska, J.; Kucwaj-Brysz, K.; Zygmunt, M.; Żesławska, E.; Siwek, A.; Bednarski, M.; Podlewska, S.; Jastrzębska-Więsek, M.; Nitek, W.; et al. The Subtype Selectivity in Search of Potent Hypotensive Agents among 5,5-Dimethylhydantoin Derived α1-Adrenoceptors Antagonists. Int. J. Mol. Sci. 2023, 24, 16609. https://doi.org/10.3390/ijms242316609
Kaczor A, Knutelska J, Kucwaj-Brysz K, Zygmunt M, Żesławska E, Siwek A, Bednarski M, Podlewska S, Jastrzębska-Więsek M, Nitek W, et al. The Subtype Selectivity in Search of Potent Hypotensive Agents among 5,5-Dimethylhydantoin Derived α1-Adrenoceptors Antagonists. International Journal of Molecular Sciences. 2023; 24(23):16609. https://doi.org/10.3390/ijms242316609
Chicago/Turabian StyleKaczor, Aneta, Joanna Knutelska, Katarzyna Kucwaj-Brysz, Małgorzata Zygmunt, Ewa Żesławska, Agata Siwek, Marek Bednarski, Sabina Podlewska, Magdalena Jastrzębska-Więsek, Wojciech Nitek, and et al. 2023. "The Subtype Selectivity in Search of Potent Hypotensive Agents among 5,5-Dimethylhydantoin Derived α1-Adrenoceptors Antagonists" International Journal of Molecular Sciences 24, no. 23: 16609. https://doi.org/10.3390/ijms242316609
APA StyleKaczor, A., Knutelska, J., Kucwaj-Brysz, K., Zygmunt, M., Żesławska, E., Siwek, A., Bednarski, M., Podlewska, S., Jastrzębska-Więsek, M., Nitek, W., Sapa, J., & Handzlik, J. (2023). The Subtype Selectivity in Search of Potent Hypotensive Agents among 5,5-Dimethylhydantoin Derived α1-Adrenoceptors Antagonists. International Journal of Molecular Sciences, 24(23), 16609. https://doi.org/10.3390/ijms242316609