
Cancer Associated PRDM9: Implications for Linking Genomic Instability and Meiotic Recombination
 ,
,  , , ,
, , , 
Abstract
:1. Introduction
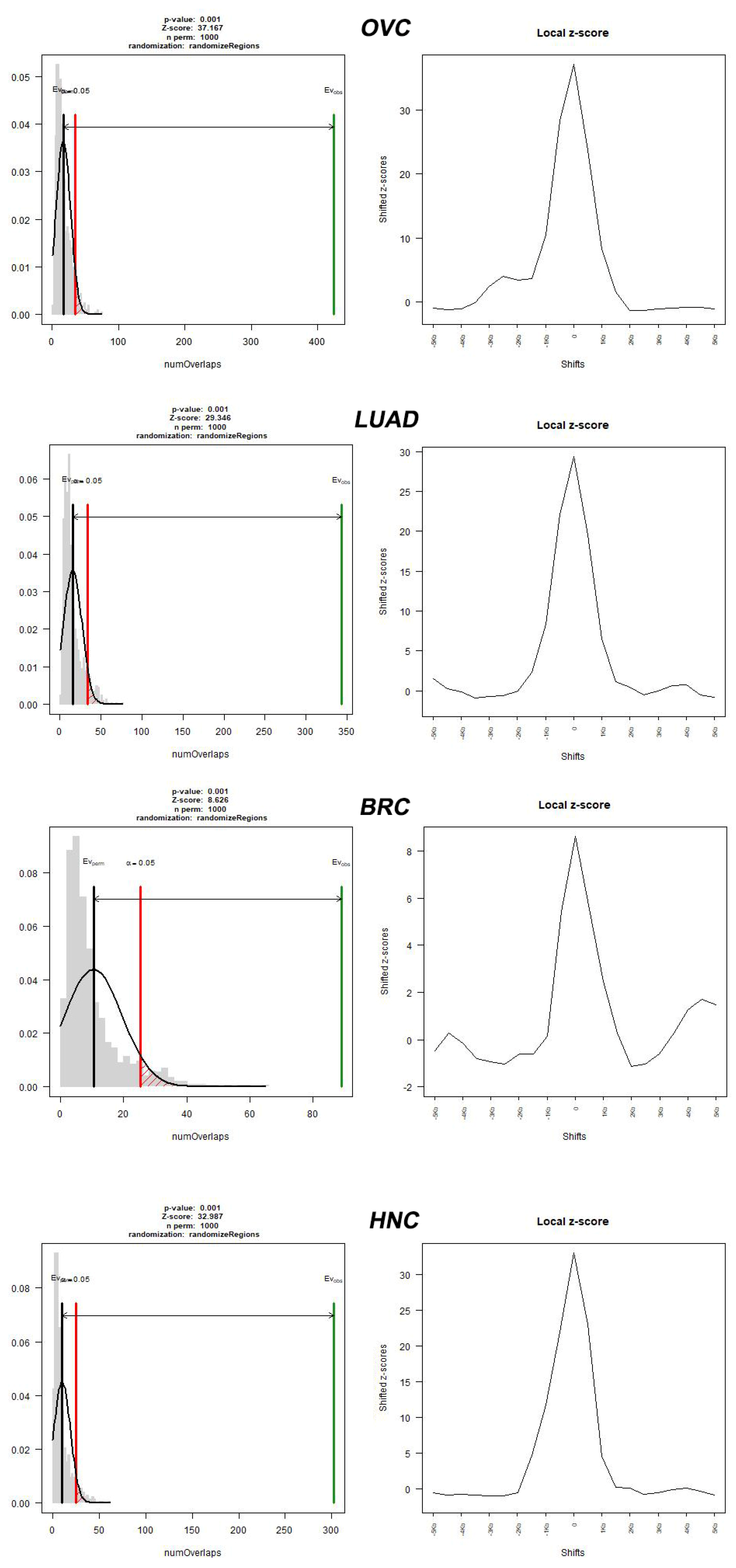
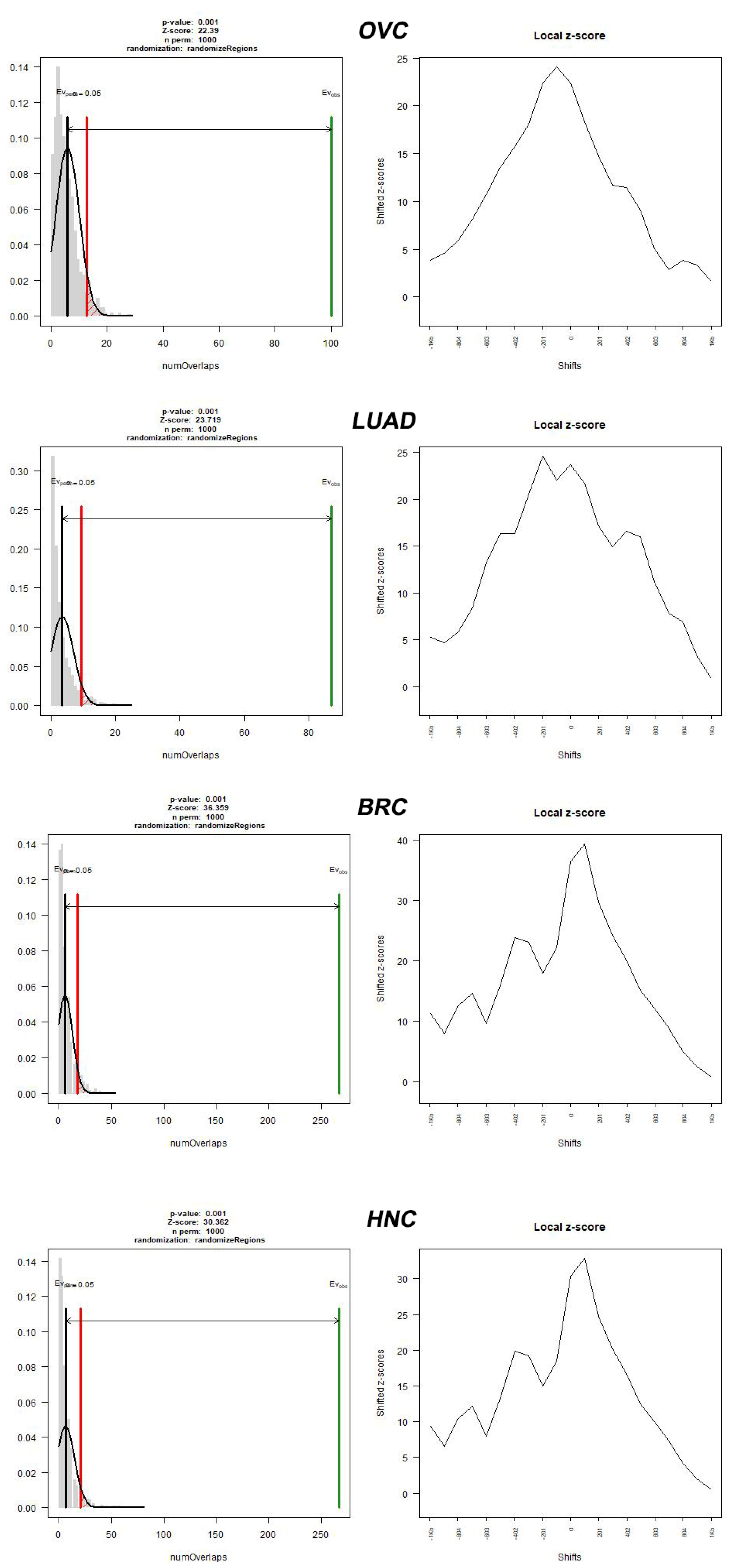
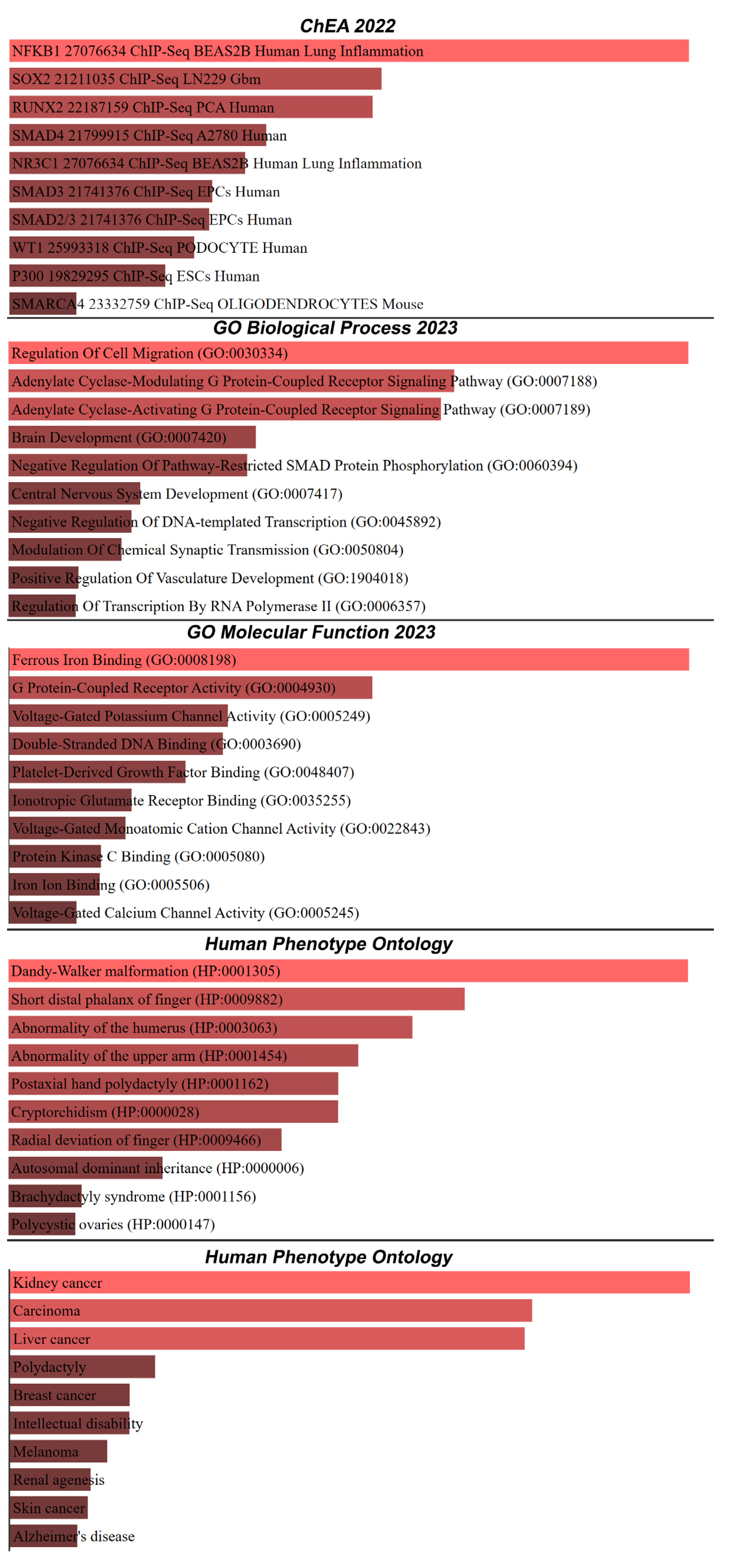
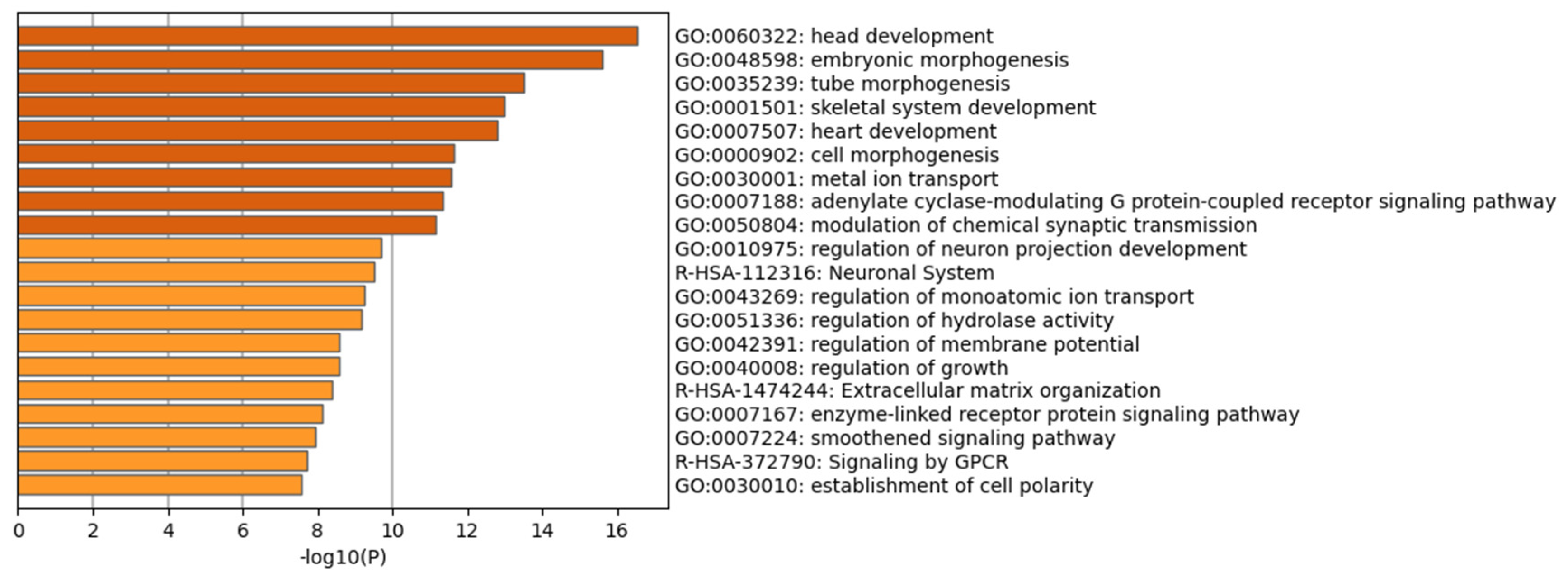
2. Results
3. Discussion
4. Materials and Methods
4.1. Bioinformatic Mining Tools for the Identification of PRDM9 Residing within the Boundaries of a Flanking Region that Contains Cancer Breakpoints
4.2. Genomic Annotation and Analysis of Genomic Distribution
4.3. Enrichment Analysis with Chip-Seq Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paigen, K.; Petkov, P.M. PRDM9 and Its Role in Genetic Recombination. Trends Genet. 2018, 34, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.; Freeman, C.; Auton, A.; Donnelly, P.; McVean, G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008, 40, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Grey, C.; Baudat, F.; de Massy, B. PRDM9, a driver of the genetic map. PLoS Genet. 2018, 14, e1007479. [Google Scholar] [CrossRef] [PubMed]
- Jen, J.; Wang, Y.C. Zinc finger proteins in cancer progression. J. Biomed. Sci. 2016, 23, 53. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Federico, A.; Rienzo, M.; Gazzerro, P.; Bifulco, M.; Ciccodicola, A.; Casamassimi, A.; Abbondanza, C. PR/SET domain family and cancer: Novel insights from the cancer genome atlas. Int. J. Mol. Sci. 2018, 19, 3250. [Google Scholar] [CrossRef] [PubMed]
- Hanquier, J.N.; Sanders, K.; Berryhill, C.A.; Sahoo, F.K.; Hudmon, A.; Vilseck, J.Z.; Cornett, E.M. Identification of nonhistone substrates of the lysine methyltransferase PRDM9. J. Biol. Chem. 2023, 299, 104651. [Google Scholar] [CrossRef] [PubMed]
- Houle, A.A.; Gibling, H.; Lamaze, F.C.; Edgington, H.A.; Soave, D.; Fave, M.J.; Agbessi, M.; Bruat, V.; Stein, L.D.; Awadalla, P. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res. 2018, 28, 1611–1620. [Google Scholar] [CrossRef]
- Casamassimi, A.; Rienzo, M.; Di Zazzo, E.; Sorrentino, A.; Fiore, D.; Proto, M.C.; Moncharmont, B.; Gazzerro, P.; Bifulco, M.; Abbondanza, C. Multifaceted Role of PRDM Proteins in Human Cancer. Int. J. Mol. Sci. 2020, 21, 2648. [Google Scholar] [CrossRef]
- Thibault-Sennett, S.; Yu, Q.; Smagulova, F.; Cloutier, J.; Brick, K.; Camerini-Otero, R.D.; Petukhova, G.V. Interrogating the Functions of PRDM9 Domains in Meiosis. Genetics 2018, 209, 475–487. [Google Scholar] [CrossRef]
- Diagouraga, B.; Clément, J.A.J.; Duret, L.; Kadlec, J.; de Massy, B.; Baudat, F. PRDM9 Methyltransferase Activity Is Essential for Meiotic DNA Double-Strand Break Formation at Its Binding Sites. Mol. Cell 2018, 69, 853–865.e856. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Ladias, P.; Markopoulos, G.S.; Kostoulas, C.; Bouba, I.; Georgiou, A.; Markoula, S.; Georgiou, I. Sequence Motif Analysis of PRDM9 and Short Inverted Repeats Suggests Their Contribution to Human Microdeletion and Microduplication Syndromes. BioMedInformatics 2023, 3, 267–279. [Google Scholar] [CrossRef]
- Stefanou, K.; Bellos, C.; Stergios, G.; Fyraridis, A.; Ladias, P.; Sakaloglou, P.; Kostoulas, C.; Markoula, S.; Georgiou, I. An intelligent web-based system for the detection and visualization of biomarkers in Microdeletion and Microduplication Syndromes. In Proceedings of the 2020 IEEE 20th International Conference on Bioinformatics and Bioengineering (BIBE), Cincinnati, OH, USA, 26–28 October 2020; pp. 217–221. [Google Scholar]
- Ladias, P.; Markopoulos, G.; Lazaros, L.; Markoula, S.; Tzavaras, T.; Georgiou, I. Holliday Junctions Are Associated with Transposable Element Sequences in the Human Genome. J. Mol. Biol. 2016, 428, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Narang, P.; Dhapola, P.; Chowdhury, S. BreCAN-DB: A repository cum browser of personalized DNA breakpoint profiles of cancer genomes. Nucleic Acids Res. 2016, 44, D952–D958. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, J.L. Using R and Bioconductor in Clinical Genomics and Transcriptomics. J. Mol. Diagn. 2020, 22, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Gel, B.; Díez-Villanueva, A.; Serra, E.; Buschbeck, M.; Peinado, M.A.; Malinverni, R. regioneR: An R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 2016, 32, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Mukaj, A.; Petkov, P.M.; Resnick, I.; Tonchev, A.; Taniguchi, H.; Parvanov, E.D. Emerging evidence for clinical significance of histone methyltransferase PRDM9 in reproductive system and cancer development. Anim. Sci. Pap. Rep. 2022, 40, 239–252. [Google Scholar]
- Suszynska, M.; Ratajska, M.; Galka-Marciniak, P.; Ryszkowska, A.; Wydra, D.; Debniak, J.; Jasiak, A.; Wasag, B.; Cybulski, C.; Kozlowski, P. Variant Identification in BARD1, PRDM9, RCC1, and RECQL in Patients with Ovarian Cancer by Targeted Next-generation Sequencing of DNA Pools. Cancer Prev. Res. 2022, 15, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, V.B.; Semple, C.A. Chromatin loop anchors are associated with genome instability in cancer and recombination hotspots in the germline. Genome Biol. 2018, 19, 101. [Google Scholar] [CrossRef] [PubMed]
- Wadhawan, A.; Smith, C.; Nicholson, R.I.; Barrett-Lee, P.; Hiscox, S. Src-mediated regulation of homotypic cell adhesion: Implications for cancer progression and opportunities for therapeutic intervention. Cancer Treat. Rev. 2011, 37, 234–241. [Google Scholar] [CrossRef]
- García-Higuera, I.; Manchado, E.; Dubus, P.; Cañamero, M.; Méndez, J.; Moreno, S.; Malumbres, M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 2008, 10, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C., Jr.; Squire, J.A. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Keum, S.; Kim, D.; You, E.; Ko, P.; Lee, J.; Kim, J.; Kim, J.-W.; Rhee, S. Spindle pole body component 25 homolog expressed by ECM stiffening is required for lung cancer cell proliferation. Biochem. Biophys. Res. Commun. 2018, 500, 937–943. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F.; Frenette, P.S.; Garzia, L.; Gutmann, D.H.; Hanahan, D. Roadmap for the emerging field of cancer neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef]
- Tryka, K.A.; Hao, L.; Sturcke, A.; Jin, Y.; Wang, Z.Y.; Ziyabari, L.; Lee, M.; Popova, N.; Sharopova, N.; Kimura, M.; et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2013, 42, D975–D979. [Google Scholar] [CrossRef]
- Xian, S.; Feng, X. Meerkat optimization algorithm: A new meta-heuristic optimization algorithm for solving constrained engineering problems. Expert. Syst. Appl. 2023, 231, 120482. [Google Scholar] [CrossRef]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.T.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S. A Systems Biology Approach on the Regulatory Footprint of Human Endogenous Retroviruses (HERVs). Diseases 2022, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.; Noutsopoulos, D.; Mantziou, S.; Gerogiannis, D.; Thrasyvoulou, S.; Vartholomatos, G.; Kolettas, E.; Tzavaras, T. Genomic analysis of mouse VL30 retrotransposons. Mob. DNA 2016, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Mantziou, S.; Markopoulos, G.S. Origins and Function of VL30 lncRNA Packaging in Small Extracellular Vesicles: Implications for Cellular Physiology and Pathology. Biomedicines 2021, 9, 1742. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC genome browser and associated tools. Brief. Bioinform. 2013, 14, 144–161. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Source | Number of Cancer-Normal Pairs | Number of Breakpoints | Number of Breakpoints per Cancer Genome |
|---|---|---|---|---|
| Breast invasive carcinoma (BRC) | Banerji et al. | 15 pairs | 22,077 | 1471 |
| Lung adenocarcinoma (LUAD) | TCGA | 17 pairs | 36,086 | 2122 |
| Ovarian serous cystadenocarcinoma (OV) | TCGA | 23 pairs | 56,435 | 2453 |
| Head and neck squamous cell carcinoma (HN) | TCGA | 17 pairs | 22,154 | 1303 |
| Total | 72 pairs | 136,752 | 7349 |
| Pathway Name | Found | Total | p Value | FDR | Found | Ratio |
|---|---|---|---|---|---|---|
| Regulation of CDH11 Expression and Function | 10/35 | 2 × 10−3 | 4 × 10−4 | 4.69 × 10−3 | 25/28 | 2 × 10−3 |
| Regulation of Expression and Function of Type II Classical Cadherins | 10/39 | 3 × 10−3 | 1 × 10−3 | 4.69 × 10−3 | 26/33 | 2 × 10−3 |
| Regulation of Homotypic Cell-Cell Adhesion | 10/39 | 3 × 10−3 | 1 × 10−3 | 4.69 × 10−3 | 26/33 | 2 × 10−3 |
| Ca2+ activated K+ channels | 5/10 | 6.57 × 10−4 | 1 × 10−3 | 4.69 × 10−3 | 2/3 | 2.10 × 10−4 |
| NOTCH2 intracellular domain regulates transcription | 6/16 | 1 × 10−3 | 2 × 10−3 | 5.35 × 10−3 | 9/9 | 2.10 × 10−4 |
| Adherens junctions interactions | 13/66 | 4 × 10−3 | 2 × 10−3 | 5.63 × 10−3 | 31/49 | 6.99 × 10−4 |
| Regulation of CDH11 function | 5/13 | 8.53 × 10−4 | 4 × 10−3 | 5.63 × 10−3 | 10/10 | 3.49 × 10−4 |
| Formation of intermediate mesoderm | 5/13 | 8.53 × 10−4 | 4 × 10−3 | 5.63 × 10−3 | 4/5 | 9.79 × 10−4 |
| Formation of lateral plate mesoderm | 4/8 | 5.25 × 10−4 | 4 × 10−3 | 5.63 × 10−3 | 4/5 | 6.29 × 10−4 |
| HS-GAG biosynthesis | 9/39 | 3 × 10−3 | 4 × 10−3 | 5.63 × 10−3 | 13/14 | 2.80 × 10−4 |
| Other semaphorin interactions | 6/19 | 1 × 10−3 | 4 × 10−3 | 5.63 × 10−3 | 5/9 | 3.49 × 10−4 |
| Regulation of CDH11 mRNA translation by microRNAs | 5/14 | 9.19 × 10−4 | 5 × 10−3 | 6.53 × 10−3 | 4/4 | 9.79 × 10−4 |
| RUNX3 regulates WNT signaling | 4/10 | 6.57 × 10−4 | 8 × 10−3 | 7.61 × 10−3 | 5/5 | 6.29 × 10−4 |
| MECP2 regulates transcription factors | 4/10 | 6.57 × 10−4 | 8 × 10−3 | 7.61 × 10−3 | 4/8 | 2.80 × 10−4 |
| Defective EXT1 causes exostoses 1, TRPS2 and CHDS | 5/16 | 1 × 10−3 | 8 × 10−3 | 7.61 × 10−3 | 4/4 | 3.49 × 10−4 |
| Defective EXT2 causes exostoses | 5/16 | 1 × 10−3 | 8 × 10−3 | 7.61 × 10−3 | 4/4 | 5.59 × 10−4 |
| Gastrulation | 20/143 | 9 × 10−3 | 8 × 10−3 | 7.61 × 10−3 | 43/72 | 2.80 × 10−4 |
| Signaling by NOTCH2 | 8/38 | 2 × 10−3 | 1 × 10−2 | 7.61 × 10−3 | 14/20 | 2.80 × 10−4 |
| Cristae formation | 7/31 | 2 × 10−3 | 1.1 × 10−2 | 7.61 × 10−3 | 2/2 | 5 × 10−3 |
| Germ layer formation at gastrulation | 6/24 | 2 × 10−3 | 1.1 × 10−2 | 7.61 × 10−3 | 11/11 | 1 × 10−3 |
| RUNX3 regulates BCL2L11 (BIM) transcription | 3/6 | 2 × 10−3 | 1.1 × 10−2 | 7.61 × 10−3 | 2/2 | 1.40 × 10−4 |
| Vasopressin-like receptors | 3/6 | 3.94 × 10−4 | 1.1 × 10−2 | 7.61 × 10−3 | 5/7 | 4.89 × 10−4 |
| cGMP effects | 5/18 | 1 × 10−3 | 1.3 × 10−2 | 7.61 × 10−3 | 2/4 | 2.80 × 10−4 |
| Extracellular matrix organization | 37/328 | 2.2 × 10−3 | 1.4 × 10−2 | 7.61 × 10−3 | 150/319 | 2.20 × 10−4 |
| Regulation of CDH11 gene transcription | 4/12 | 7.88 × 10−4 | 1.4 × 10−2 | 7.61 × 10−3 | 11/14 | 9.79 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ladias, P.; Markopoulos, G.S.; Kostoulas, C.; Bouba, I.; Markoula, S.; Georgiou, I. Cancer Associated PRDM9: Implications for Linking Genomic Instability and Meiotic Recombination. Int. J. Mol. Sci. 2023, 24, 16522. https://doi.org/10.3390/ijms242216522
Ladias P, Markopoulos GS, Kostoulas C, Bouba I, Markoula S, Georgiou I. Cancer Associated PRDM9: Implications for Linking Genomic Instability and Meiotic Recombination. International Journal of Molecular Sciences. 2023; 24(22):16522. https://doi.org/10.3390/ijms242216522
Chicago/Turabian StyleLadias, Paris, Georgios S. Markopoulos, Charilaos Kostoulas, Ioanna Bouba, Sofia Markoula, and Ioannis Georgiou. 2023. "Cancer Associated PRDM9: Implications for Linking Genomic Instability and Meiotic Recombination" International Journal of Molecular Sciences 24, no. 22: 16522. https://doi.org/10.3390/ijms242216522
APA StyleLadias, P., Markopoulos, G. S., Kostoulas, C., Bouba, I., Markoula, S., & Georgiou, I. (2023). Cancer Associated PRDM9: Implications for Linking Genomic Instability and Meiotic Recombination. International Journal of Molecular Sciences, 24(22), 16522. https://doi.org/10.3390/ijms242216522