
Ensemble Learning with Supervised Methods Based on Large-Scale Protein Language Models for Protein Mutation Effects Prediction
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Prediction of Protein Mutation Effects
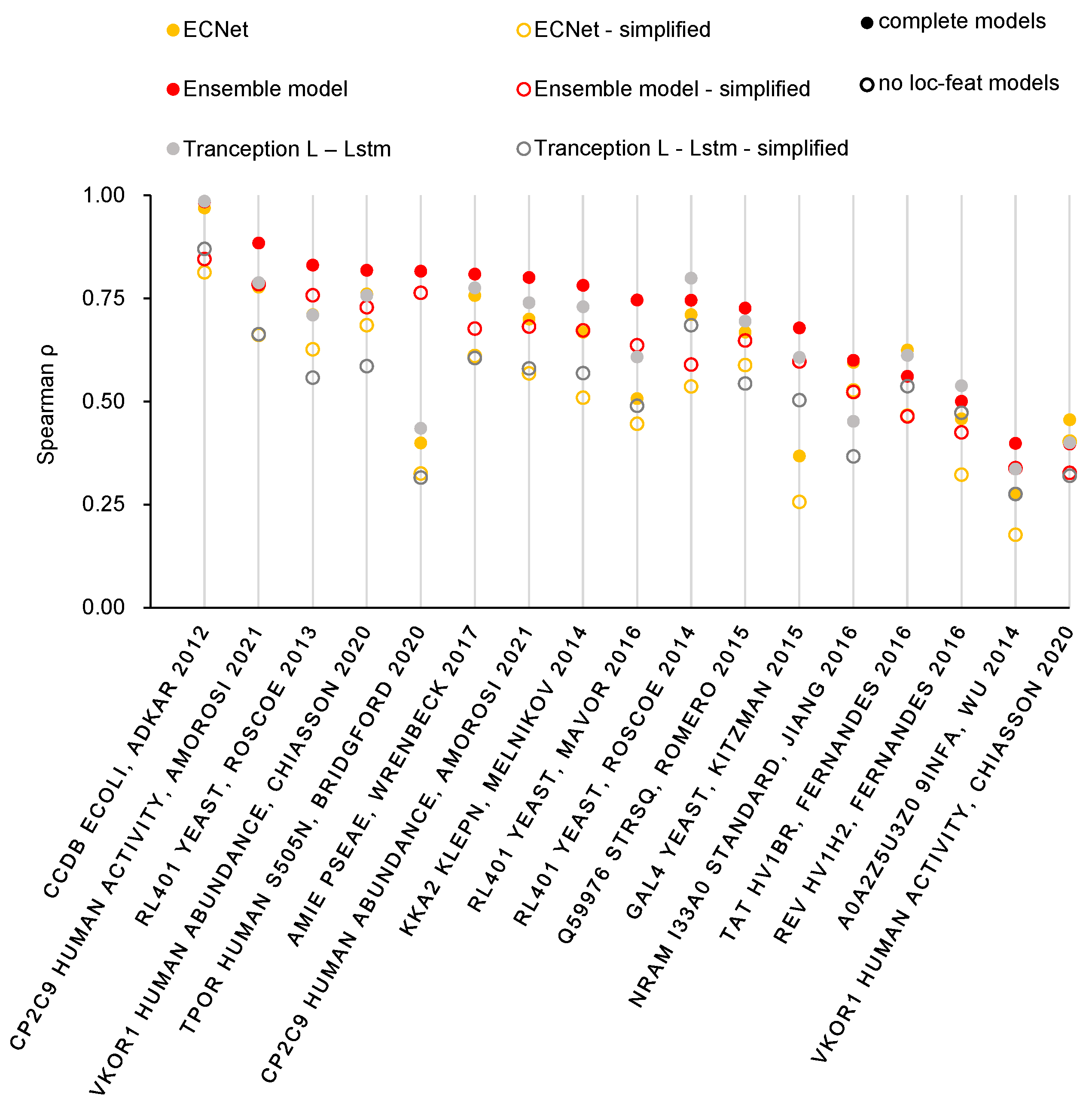
2.2. Improved Prediction Performance of Mutation Effects Benefited from the Ensemble of Multiple Global Features
2.3. Localized Features Further Enrich the Feature Information
3. Discussion
4. Materials and Methods
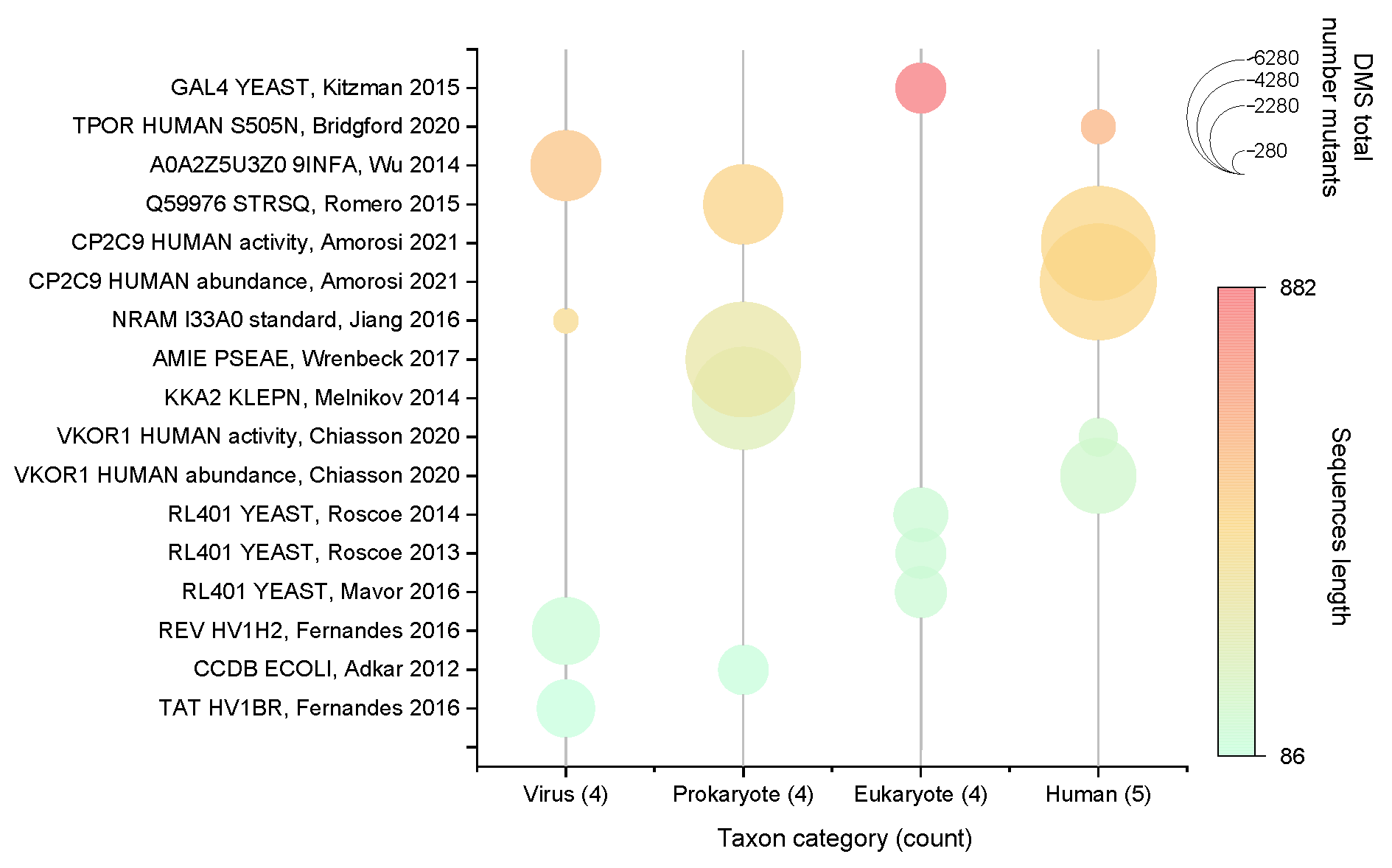
4.1. Datasets
- Inclusion of all taxa present in the full dataset (e.g., humans, other eukaryotes, prokaryotes, viruses);
- Even distribution of protein sequence lengths (100aa–1000aa);
- A wide range of sample sizes (ranging from several hundred to several thousand).
4.2. Pre-Training Protein Language Models
4.2.1. Tranception
4.2.2. Tape-Transformer
4.2.3. Details of Model Modifications
4.3. Ensemble Methods for Predicting
4.4. CCMPred and Local Representation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Young, V.R.; Steffee, W.P.; Pencharz, P.B.; Winterer, J.C.; Scrimshaw, N.S. Total human body protein synthesis in relation to protein requirements at various ages. Nature 1975, 253, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.; Crump, M.P. The structural role of the carrier protein–active controller or passive carrier. Nat. Prod. Rep. 2012, 29, 1111–1137. [Google Scholar] [CrossRef]
- Cummings, C.; Murata, H.; Koepsel, R.; Russell, A.J. Tailoring enzyme activity and stability using polymer-based protein engineering. Biomaterials 2013, 34, 7437–7443. [Google Scholar] [CrossRef]
- Diaz, D.J.; Kulikova, A.V.; Ellington, A.D.; Wilke, C.O. Using machine learning to predict the effects and consequences of mutations in proteins. Curr. Opin. Struct. Biol. 2023, 78, 102518. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.A.; Arnold, F.H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Raven, S.A.; Payne, B.; Bruce, M.; Filipovska, A.; Rackham, O. In silico evolution of nucleic acid-binding proteins from a nonfunctional scaffold. Nat. Chem. Biol. 2022, 18, 403–411. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Tunyasuvunakool, K.; Ronneberger, O.; Bates, R.; Žídek, A.; Bridgland, A.; et al. AlphaFold 2. Fourteenth Critical Assessment of Techniques for Protein Structure Prediction; DeepMind: London, UK, 2020. [Google Scholar]
- You, R.; Yao, S.; Xiong, Y.; Huang, X.; Sun, F.; Mamitsuka, H.; Zhu, S. NetGO: Improving large-scale protein function prediction with massive network information. Nucleic Acids Res. 2019, 47, W379–W387. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; You, R.; Wang, S.; Xiong, Y.; Huang, X.; Zhu, S. NetGO 2.0: Improving large-scale protein function prediction with massive sequence, text, domain, family and network information. Nucleic Acids Res. 2021, 49, W469–W475. [Google Scholar] [CrossRef]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Khimulya, G.; Alley, E.C.; Esvelt, K.M.; Church, G.M. Low-N protein engineering with data-efficient deep learning. Nat. Methods 2021, 18, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.; Rao, R.; Verkuil, R.; Liu, J.; Sercu, T.; Rives, A. Language models enable zero-shot prediction of the effects of mutations on protein function. In Proceedings of the Advances in Neural Information Processing Systems, Virtual, 6–14 December 2021; Volume 34, pp. 29287–29303. [Google Scholar]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef]
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: NLP, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar]
- Wittmund, M.; Cadet, F.; Davari, M.D. Learning epistasis and residue coevolution patterns: Current trends and future perspectives for advancing enzyme engineering. Acs Catal. 2022, 12, 14243–14263. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Bepler, T.; Berger, B. Learning the protein language: Evolution, structure, and function. Cell Syst. 2021, 12, 654–669. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos Jr, J.L.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, S.; Pratyush, P.; Heinzinger, M.; Newman, R.H.; Kc, D.B. Improving protein succinylation sites prediction using embeddings from protein language model. Sci. Rep. 2022, 12, 16933. [Google Scholar] [CrossRef]
- Dunham, A.S.; Beltrao, P.; AlQuraishi, M. High-throughput deep learning variant effect prediction with Sequence UNET. Genome Biol. 2023, 24, 1–19. [Google Scholar] [CrossRef]
- Thirunavukarasu, A.J.; Ting, D.S.J.; Elangovan, K.; Gutierrez, L.; Tan, T.F.; Ting, D.S.W. Large language models in medicine. Nat. Med. 2023, 29, 1930–1940. [Google Scholar] [CrossRef]
- Li, H.L.; Pang, Y.H.; Liu, B. BioSeq-BLM: A platform for analyzing DNA, RNA and protein sequences based on biological language models. Nucleic Acids Res. 2021, 49, e129. [Google Scholar] [CrossRef] [PubMed]
- So, D.R.; Mańke, W.; Liu, H.; Dai, Z.; Shazeer, N.; Le, Q.V. Primer: Searching for efficient transformers for language modeling. arXiv 2021, arXiv:2109.08668. [Google Scholar]
- Notin, P.; Dias, M.; Frazer, J.; Hurtado, J.M.; Gomez, A.N.; Marks, D.; Gal, Y. Tranception: Protein fitness prediction with autoregressive transformers and inference-time retrieval. In Proceedings of the International Conference on Machine Learning. PMLR, Baltimore, MD, USA, 17–23 July 2022; pp. 16990–17017. [Google Scholar]
- Hie, B.L.; Shanker, V.R.; Xu, D.; Bruun, T.U.; Weidenbacher, P.A.; Tang, S.; Wu, W.; Pak, J.E.; Kim, P.S. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 2023. Available online: https://www.nature.com/articles/s41587-023-01763-2#citeas (accessed on 17 November 2023).
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Hopf, T.A.; Ingraham, J.B.; Poelwijk, F.J.; Schärfe, C.P.; Springer, M.; Sander, C.; Marks, D.S. Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 2017, 35, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.E.; Riesselman, A.J.; Kollasch, A.W.; McMahon, C.; Simon, E.; Sander, C.; Manglik, A.; Kruse, A.C.; Marks, D.S. Protein design and variant prediction using autoregressive generative models. Nat. Commun. 2021, 12, 2403. [Google Scholar] [CrossRef] [PubMed]
- Seemayer, S.; Gruber, M.; Söding, J. CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics 2014, 30, 3128–3130. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Jiang, G.; Yu, T.; Liu, Y.; Vo, L.; Ding, H.; Su, Y.; Qian, W.W.; Zhao, H.; Peng, J. ECNet is an evolutionary context-integrated deep learning framework for protein engineering. Nat. Commun. 2021, 12, 5743. [Google Scholar] [CrossRef]
- Zar, J.H. Spearman rank correlation. Encycl. Biostat. 2005, 7. [Google Scholar] [CrossRef]
- Frazer, J.; Notin, P.; Dias, M.; Gomez, A.; Min, J.K.; Brock, K.; Gal, Y.; Marks, D.S. Disease variant prediction with deep generative models of evolutionary data. Nature 2021, 599, 91–95. [Google Scholar] [CrossRef]
- Fernandes, J.D.; Faust, T.B.; Strauli, N.B.; Smith, C.; Crosby, D.C.; Nakamura, R.L.; Hernandez, R.D.; Frankel, A.D. Functional segregation of overlapping genes in HIV. Cell 2016, 167, 1762–1773. [Google Scholar] [CrossRef]
- Adkar, B.V.; Tripathi, A.; Sahoo, A.; Bajaj, K.; Goswami, D.; Chakrabarti, P.; Swarnkar, M.K.; Gokhale, R.S.; Varadarajan, R. Protein model discrimination using mutational sensitivity derived from deep sequencing. Structure 2012, 20, 371–381. [Google Scholar] [CrossRef]
- Mavor, D.; Barlow, K.; Thompson, S.; Barad, B.A.; Bonny, A.R.; Cario, C.L.; Gaskins, G.; Liu, Z.; Deming, L.; Axen, S.D.; et al. Determination of ubiquitin fitness landscapes under different chemical stresses in a classroom setting. Elife 2016, 5, e15802. [Google Scholar] [CrossRef]
- Roscoe, B.P.; Thayer, K.M.; Zeldovich, K.B.; Fushman, D.; Bolon, D.N. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J. Mol. Biol. 2013, 425, 1363–1377. [Google Scholar] [CrossRef]
- Roscoe, B.P.; Bolon, D.N. Systematic exploration of ubiquitin sequence, E1 activation efficiency, and experimental fitness in yeast. J. Mol. Biol. 2014, 426, 2854–2870. [Google Scholar] [CrossRef] [PubMed]
- Chiasson, M.A.; Rollins, N.J.; Stephany, J.J.; Sitko, K.A.; Matreyek, K.A.; Verby, M.; Sun, S.; Roth, F.P.; DeSloover, D.; Marks, D.S.; et al. Multiplexed measurement of variant abundance and activity reveals VKOR topology, active site and human variant impact. elife 2020, 9, e58026. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.; Rogov, P.; Wang, L.; Gnirke, A.; Mikkelsen, T.S. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014, 42, e112. [Google Scholar] [CrossRef] [PubMed]
- Wrenbeck, E.E.; Azouz, L.R.; Whitehead, T.A. Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded. Nature Commun. 2017, 8, 15695. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, P.; Bank, C.; Renzette, N.; Prachanronarong, K.; Yilmaz, L.S.; Caffrey, D.R.; Zeldovich, K.B.; Schiffer, C.A.; Kowalik, T.F.; et al. A balance between inhibitor binding and substrate processing confers influenza drug resistance. J. Mol. Biol. 2016, 428, 538–553. [Google Scholar] [CrossRef]
- Amorosi, C.J.; Chiasson, M.A.; McDonald, M.G.; Wong, L.H.; Sitko, K.A.; Boyle, G.; Kowalski, J.P.; Rettie, A.E.; Fowler, D.M.; Dunham, M.J. Massively parallel characterization of CYP2C9 variant enzyme activity and abundance. Am. J. Hum. Genet. 2021, 108, 1735–1751. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.A.; Tran, T.M.; Abate, A.R. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. USA 2015, 112, 7159–7164. [Google Scholar] [CrossRef]
- Kitzman, J.O.; Starita, L.M.; Lo, R.S.; Fields, S.; Shendure, J. Massively parallel single-amino-acid mutagenesis. Nat. Methods 2015, 12, 203–206. [Google Scholar] [CrossRef]
- Bridgford, J.L.; Lee, S.M.; Lee, C.M.; Guglielmelli, P.; Rumi, E.; Pietra, D.; Wilcox, S.; Chhabra, Y.; Rubin, A.F.; Cazzola, M.; et al. Novel drivers and modifiers of MPL-dependent oncogenic transformation identified by deep mutational scanning. Blood J. Am. Soc. Hematol. 2020, 135, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Young, A.P.; Al-Mawsawi, L.Q.; Olson, C.A.; Feng, J.; Qi, H.; Chen, S.H.; Lu, I.H.; Lin, C.Y.; Chin, R.G.; et al. High-throughput profiling of influenza A virus hemagglutinin gene at single-nucleotide resolution. Sci. Rep. 2014, 4, 4942. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Volume 30. [Google Scholar]
- Araya, C.L.; Fowler, D.M.; Chen, W.; Muniez, I.; Kelly, J.W.; Fields, S. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc. Natl. Acad. Sci. USA 2012, 109, 16858–16863. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Bhattacharya, N.; Thomas, N.; Duan, Y.; Chen, P.; Canny, J.; Abbeel, P.; Song, Y. Evaluating protein transfer learning with TAPE. In Proceedings of the Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 8–14 December 2019; Volume 32. [Google Scholar]
- McDonald, G.C. Ridge regression. Wiley Interdiscip. Rev. Comput. Stat. 2009, 1, 93–100. [Google Scholar] [CrossRef]
- Barbero-Aparicio, J.A.; Olivares-Gil, A.; Rodríguez, J.J.; García-Osorio, C.; Díez-Pastor, J.F. Addressing data scarcity in protein fitness landscape analysis: A study on semi-supervised and deep transfer learning techniques. Inf. Fusion 2024, 102, 102035. [Google Scholar] [CrossRef]
- Chen, Z.; Hou, C.; Wang, L.; Yu, C.; Chen, T.; Shen, B.; Hou, Y.; Li, P.; Li, T. Screening membraneless organelle participants with machine-learning models that integrate multimodal features. Proc. Natl. Acad. Sci. USA 2022, 119, e2115369119. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, X.; Schuurmans, D.; Bosma, M.; Xia, F.; Chi, E.; Le, Q.V.; Zhou, D. Chain-of-thought prompting elicits reasoning in large language models. In Proceedings of the Advances in Neural Information Processing Systems, New Orleans, LA, USA, 28 November–9 December 2022; Volume 35, pp. 24824–24837. [Google Scholar]
- Liu, Y.; Han, T.; Ma, S.; Zhang, J.; Yang, Y.; Tian, J.; He, H.; Li, A.; He, M.; Liu, Z.; et al. Summary of chatgpt/gpt-4 research and perspective towards the future of large language models. arXiv 2023, arXiv:2304.01852. [Google Scholar]
- Han, X.; Zhang, Z.; Ding, N.; Gu, Y.; Liu, X.; Huo, Y.; Qiu, J.; Yao, Y.; Zhang, A.; Zhang, L.; et al. Pre-trained models: Past, present and future. AI Open 2021, 2, 225–250. [Google Scholar] [CrossRef]
- Tenney, I.; Das, D.; Pavlick, E. BERT rediscovers the classical NLP pipeline. arXiv 2019, arXiv:1905.05950. [Google Scholar]
- Press, O.; Smith, N.A.; Lewis, M. Train short, test long: Attention with linear biases enables input length extrapolation. arXiv 2021, arXiv:2108.12409. [Google Scholar]
- Greff, K.; Srivastava, R.K.; Koutník, J.; Steunebrink, B.R.; Schmidhuber, J. LSTM: A search space odyssey. IEEE Trans. Neural Netw. Learn. Syst. 2016, 28, 2222–2232. [Google Scholar] [CrossRef] [PubMed]
- Szegedy, C.; Ioffe, S.; Vanhoucke, V.; Alemi, A. Inception-v4, inception-resnet and the impact of residual connections on learning. In Proceedings of the AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017; Volume 31. [Google Scholar]
- Rue, H.; Held, L. Gaussian Markov Random Fields: Theory and Applications; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, Y.; Niu, Z.; Ding, Q.; Zhao, T.; Kong, T.; Bai, B.; Ma, J.; Zhao, Y.; Zheng, J. Ensemble Learning with Supervised Methods Based on Large-Scale Protein Language Models for Protein Mutation Effects Prediction. Int. J. Mol. Sci. 2023, 24, 16496. https://doi.org/10.3390/ijms242216496
Qu Y, Niu Z, Ding Q, Zhao T, Kong T, Bai B, Ma J, Zhao Y, Zheng J. Ensemble Learning with Supervised Methods Based on Large-Scale Protein Language Models for Protein Mutation Effects Prediction. International Journal of Molecular Sciences. 2023; 24(22):16496. https://doi.org/10.3390/ijms242216496
Chicago/Turabian StyleQu, Yang, Zitong Niu, Qiaojiao Ding, Taowa Zhao, Tong Kong, Bing Bai, Jianwei Ma, Yitian Zhao, and Jianping Zheng. 2023. "Ensemble Learning with Supervised Methods Based on Large-Scale Protein Language Models for Protein Mutation Effects Prediction" International Journal of Molecular Sciences 24, no. 22: 16496. https://doi.org/10.3390/ijms242216496
APA StyleQu, Y., Niu, Z., Ding, Q., Zhao, T., Kong, T., Bai, B., Ma, J., Zhao, Y., & Zheng, J. (2023). Ensemble Learning with Supervised Methods Based on Large-Scale Protein Language Models for Protein Mutation Effects Prediction. International Journal of Molecular Sciences, 24(22), 16496. https://doi.org/10.3390/ijms242216496