
Whole-Transcriptome RNA Sequencing Uncovers the Global Expression Changes and RNA Regulatory Networks in Duck Embryonic Myogenesis


Abstract
:1. Introduction
2. Results
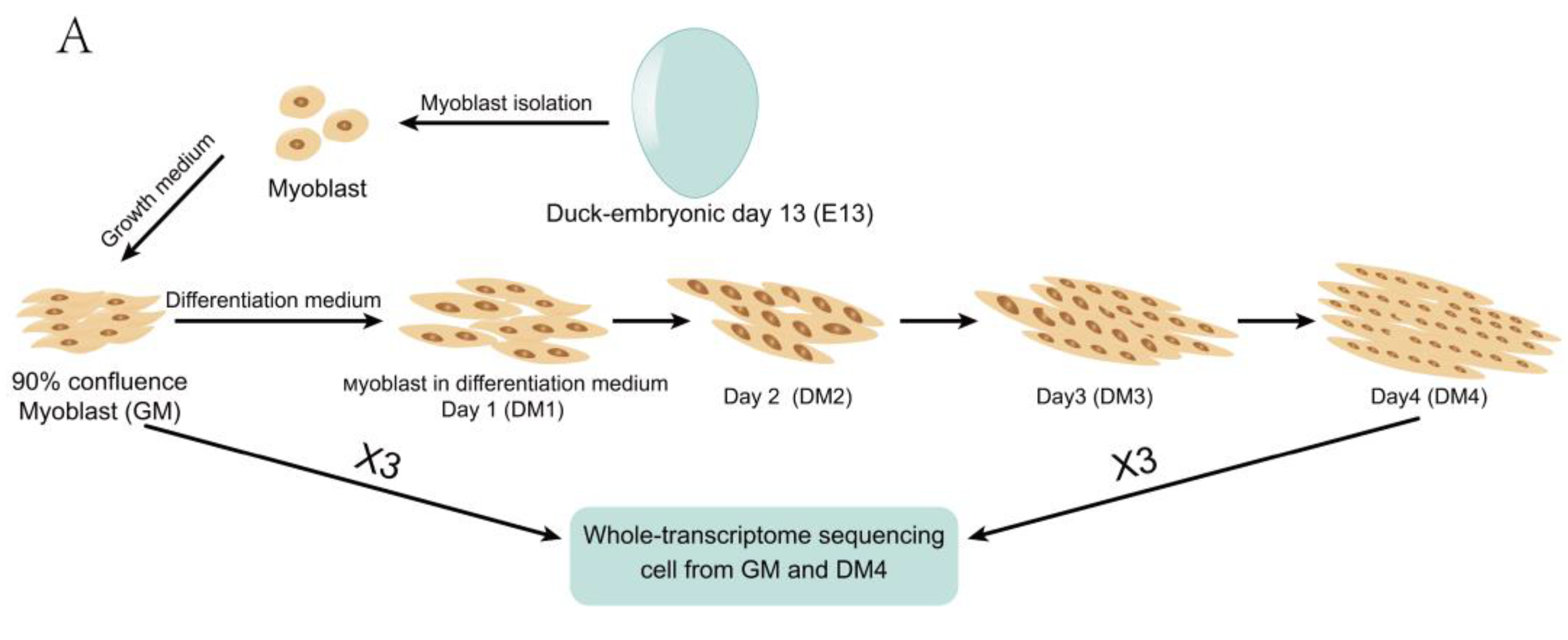
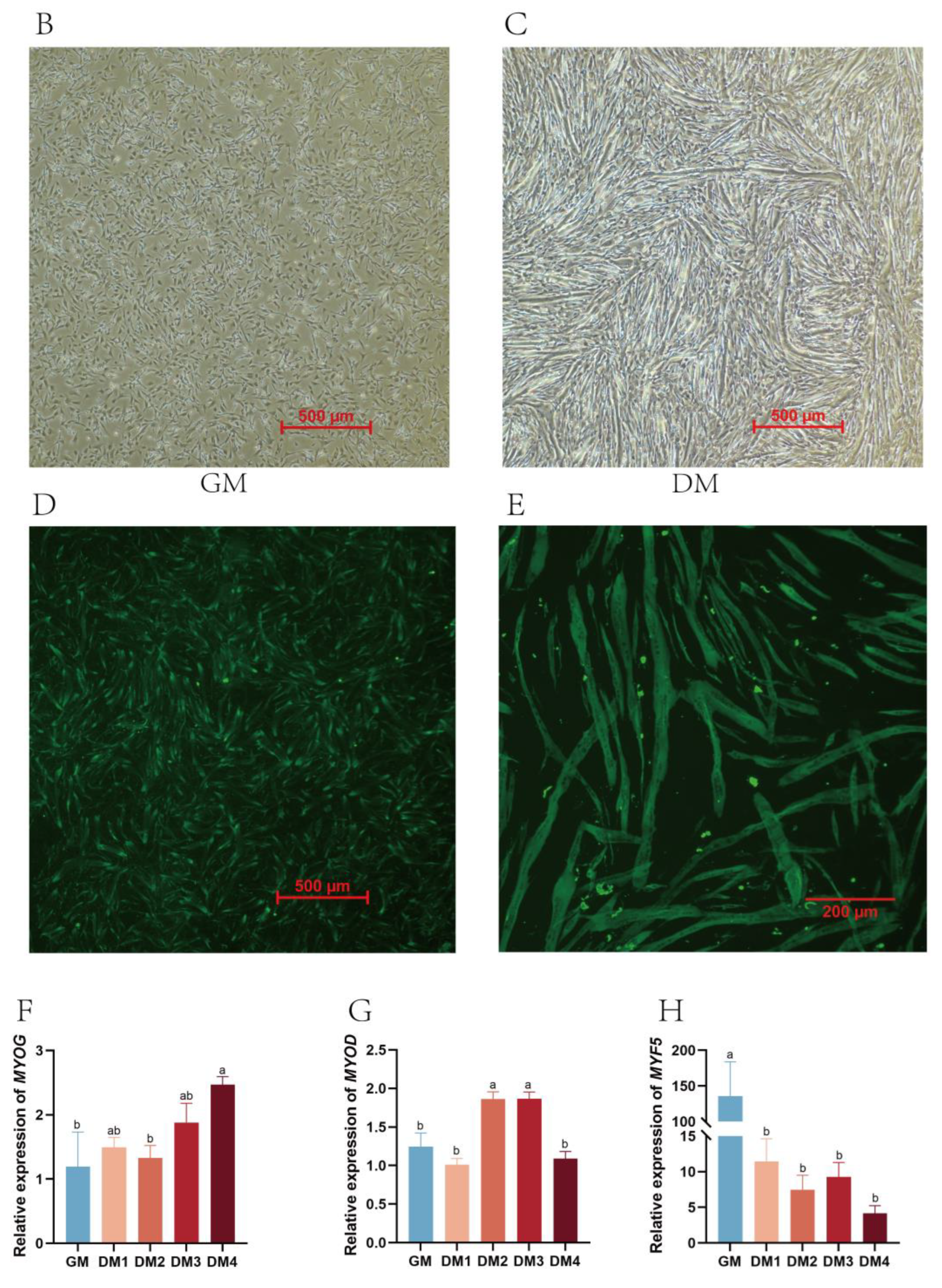
2.1. Duck Primary Myoblast Differentiation
2.2. Expression Patterns of mRNAs, lncRNAs, and circRNAs in Duck Myogenesis
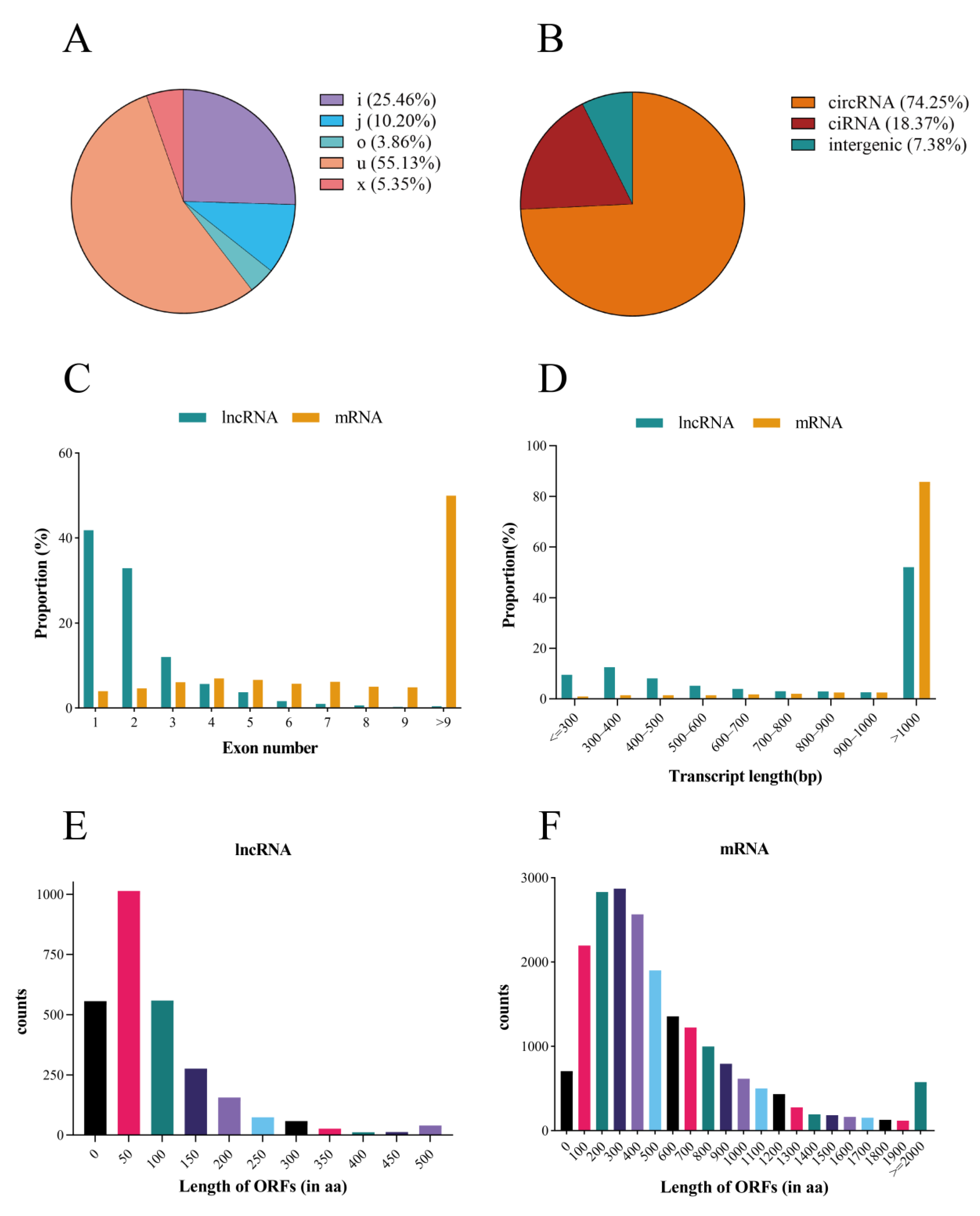
2.3. Characteristics of Duck lncRNAs and mRNAs
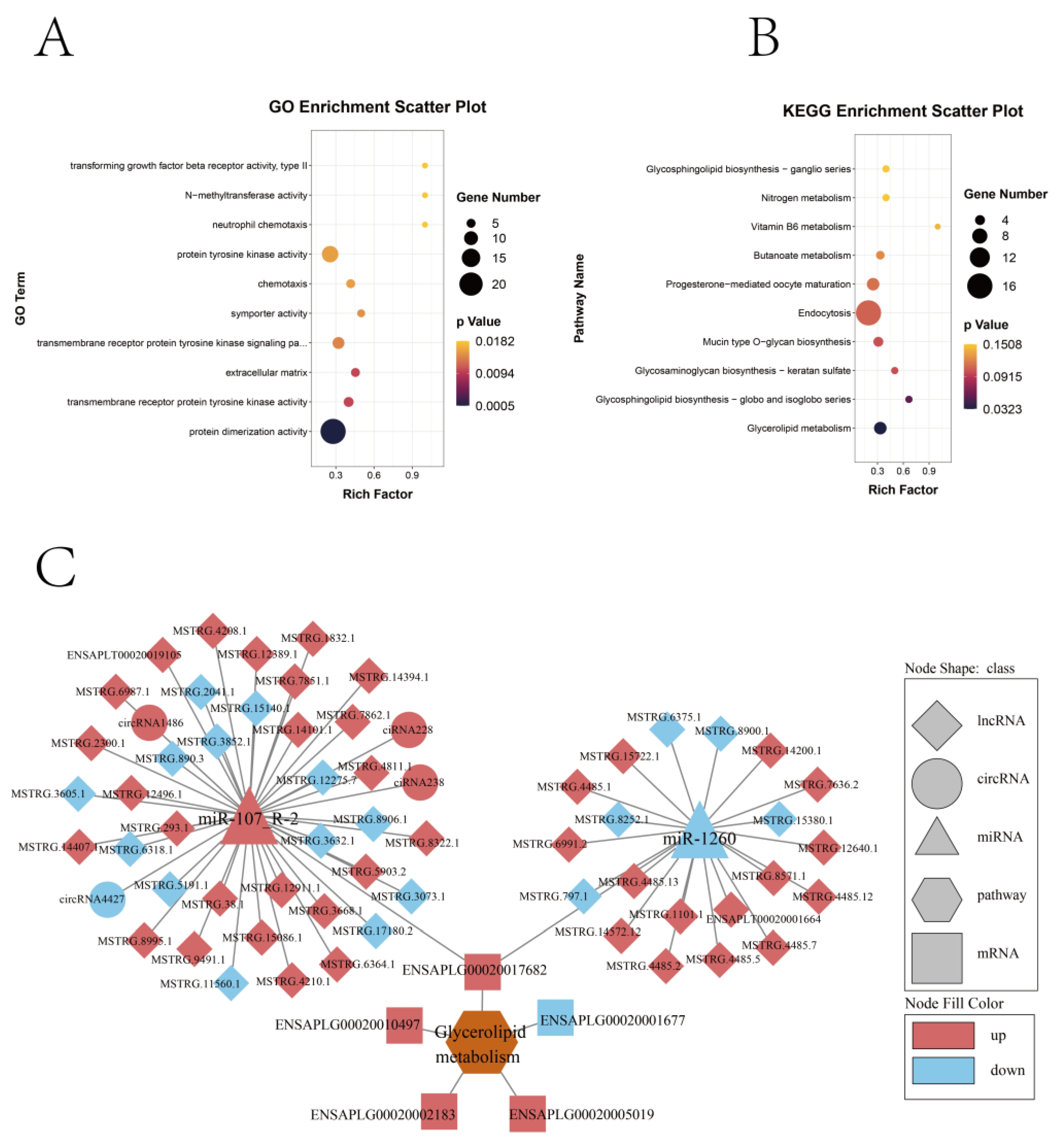
2.4. Differential Expression and Enrichment Analysis of mRNAs, lncRNAs, and circRNAs
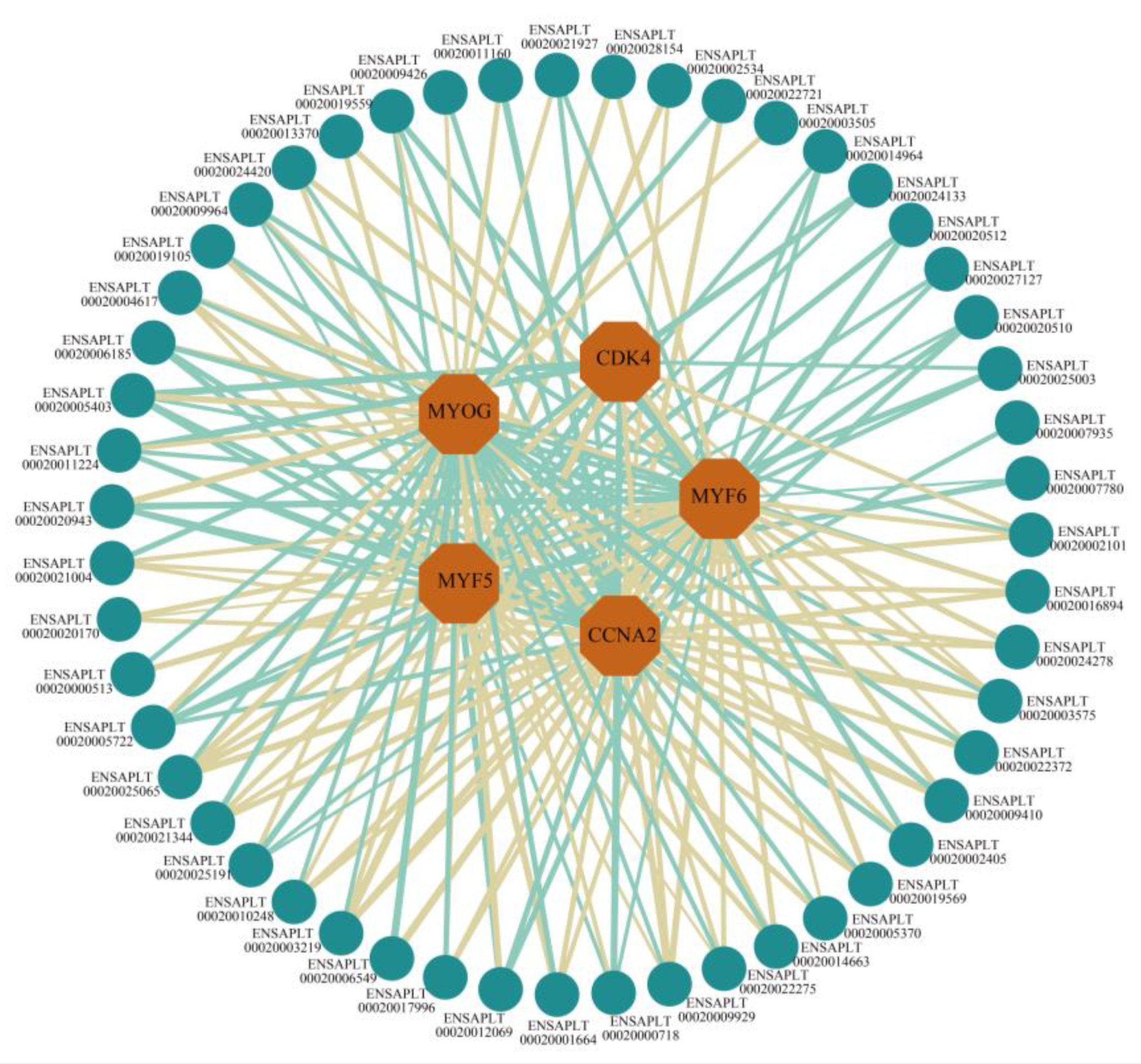
2.5. Analysis of the Interaction between Trans-Acting lncRNAs and mRNAs
2.6. Expression Profiling and Differential Expression of miRNAs
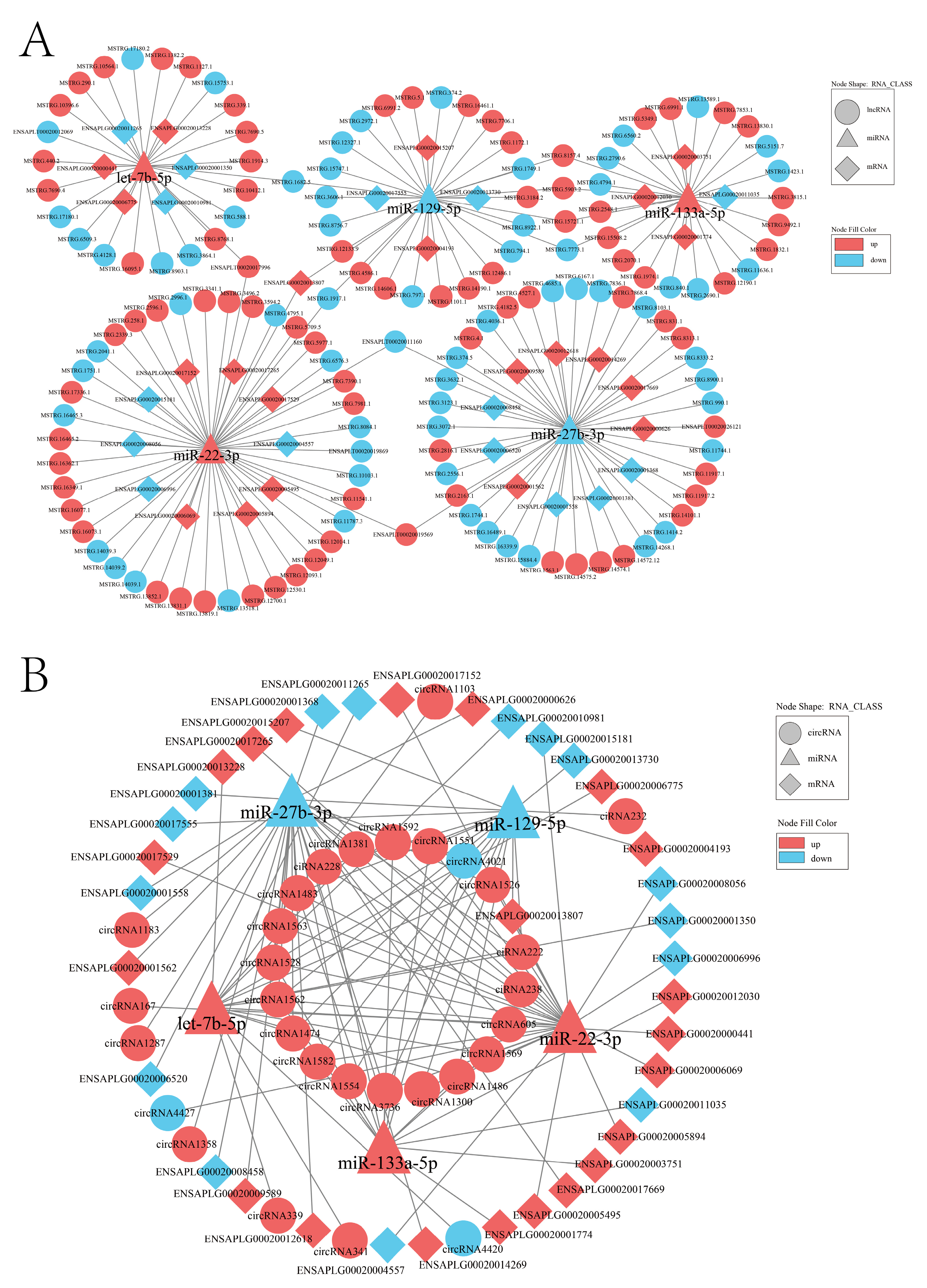
2.7. Construction of ceRNA Interaction Regulatory Network
2.8. qRT-PCR Validation
3. Discussion
4. Materials and Methods
4.1. Isolation, Culture, and Differentiation of Primary Myoblasts from Duck Embryos
4.2. Immunofluorescence Analysis
4.3. RNA Isolation, Library Preparation, and Sequencing
4.4. Identification of lncRNA, circRNA, and miRNA
4.5. Analysis of Differentially Expressed (DE) Genes
4.6. Screening Potential Target Genes of Differentially Expressed circRNA/lncRNA/miRNA and Functional Enrichment Analysis of GO and KEGG
4.7. Analysis of the Interaction between Trans-Acting lncRNA and mRNA
4.8. Construction of lncRNA/circRNA-miRNA-mRNA ceRNA Network
4.9. qRT-PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.; Liu, S.; Zhang, W.; Hu, X.; Mao, H.; Liu, S.; Chen, B. Transcriptome RNA Sequencing Reveals That Circular RNAs Are Abundantly Expressed in Embryonic Breast Muscle of Duck. Vet. Sci. 2023, 10, 75. [Google Scholar] [CrossRef]
- Khalid, T.; Hdaifeh, A.; Federighi, M.; Cummins, E.; Boué, G.; Guillou, S.; Tesson, V. Review of Quantitative Microbial Risk Assessment in Poultry Meat: The Central Position of Consumer Behavior. Foods 2020, 9, 1661. [Google Scholar] [CrossRef]
- Saxena, V.K.; Sachdev, A.K.; Gopal, R.; Pramod, A.B. PRAMOD. Roles of Important Candidate Genes on Broiler Meat Quality. World’s Poult. Sci. J. 2009, 65, 37–50. [Google Scholar] [CrossRef]
- Lehka, L.; Rędowicz, M.J. Mechanisms Regulating Myoblast Fusion: A Multilevel Interplay. Semin. Cell Dev. Biol. 2020, 104, 81–92. [Google Scholar] [CrossRef]
- Allen, R.E.; Boxhorn, L.K. Regulation of Skeletal Muscle Satellite Cell Proliferation and Differentiation by Transforming Growth Factor-Beta, Insulin-like Growth Factor I, and Fibroblast Growth Factor. J. Cell. Physiol. 1989, 138, 311–315. [Google Scholar] [CrossRef]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The Formation of Skeletal Muscle: From Somite to Limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Sanchez, J.C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with Proteome Projects: Why All Proteins Expressed by a Genome Should Be Identified and How to Do It. Biotechnol. Genet. Eng. 1996, 13, 19–50. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of Transcription in Human Cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Wu, T.; Wang, S.; Wang, L.; Zhang, W.; Chen, W.; Lv, X.; Li, Y.; Hussain, Z.; Sun, W. Long Noncoding RNA (lncRNA) CTTN-IT1 Elevates Skeletal Muscle Satellite Cell Proliferation and Differentiation by Acting as ceRNA for YAP1 Through Absorbing miR-29a in Hu Sheep. Front. Genet. 2020, 11, 843. [Google Scholar] [CrossRef]
- Cai, B.; Ma, M.; Zhou, Z.; Kong, S.; Zhang, J.; Zhang, X.; Nie, Q. circPTPN4 Regulates Myogenesis via the miR-499-3p/NAMPT Axis. J. Anim. Sci. Biotechnol. 2022, 13, 2. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, Y.; Zhang, Y.; Yan, C.; Yang, M.; Wang, Z.; Li, W.; Li, F.; Wang, W.; Yang, Y.; et al. MicroRNA-200c-5p Regulates Migration and Differentiation of Myoblasts via Targeting Adamts5 in Skeletal Muscle Regeneration and Myogenesis. Int. J. Mol. Sci. 2023, 24, 4995. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xiong, T.; Zhou, M.; Wan, L.; Xi, S.; Liu, Q.; Chen, Y.; Mao, H.; Liu, S.; Chen, B. Characterization of microRNAs during Embryonic Skeletal Muscle Development in the Shan Ma Duck. Animals 2020, 10, E1417. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Cao, J.; Ge, L.; Zhang, J.; Zhang, H.; Liu, X. Characterization and Comparative Transcriptomic Analysis of Skeletal Muscle in Pekin Duck at Different Growth Stages Using RNA-Seq. Animals 2021, 11, 834. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Gu, T.; Lu, L.; Cao, Z.; Song, Q.; Wang, Z.; Zhang, Y.; Chang, G.; Xu, Q.; Chen, G. Roles of miRNA-1 and miRNA-133 in the Proliferation and Differentiation of Myoblasts in Duck Skeletal Muscle. J. Cell. Physiol. 2019, 234, 3490–3499. [Google Scholar] [CrossRef]
- Zhou, T.; Wu, Y.; Bi, Y.; Bai, H.; Jiang, Y.; Chen, G.; Chang, G.; Wang, Z. MYOZ1 Gene Promotes Muscle Growth and Development in Meat Ducks. Genes 2022, 13, 1574. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Sun, T.; Yang, Z.; Zou, L.; Deng, J.; Yang, X. Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and circRNA/lncRNA-miRNA-mRNA ceRNA Regulatory Network in Chicken Fat Deposition. Poult. Sci. 2022, 101, 102121. [Google Scholar] [CrossRef]
- Pan, Z.; Yang, C.; Zhao, R.; Jiang, X.; Yu, C.; Li, Z. Characterization of lncRNA/circRNA-miRNA-mRNA Network to Reveal Potential Functional ceRNAs in the Skeletal Muscle of Chicken. Front. Physiol. 2022, 13, 969854. [Google Scholar] [CrossRef]
- Hu, Z.; Cao, J.; Zhang, J.; Ge, L.; Zhang, H.; Liu, X. Skeletal Muscle Transcriptome Analysis of Hanzhong Ma Duck at Different Growth Stages Using RNA-Seq. Biomolecules 2021, 11, 315. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, S.; Chen, G.; Deng, X.; Zhang, D.; Wen, H.; Yin, Y.; Lin, Z.; Zhang, X.; Luo, W. Transcriptome Sequencing Reveals Pathways Related to Proliferation and Differentiation of Shitou Goose Myoblasts. Animals 2022, 12, 2956. [Google Scholar] [CrossRef]
- Wang, J.; Ren, Q.; Hua, L.; Chen, J.; Zhang, J.; Bai, H.; Li, H.; Xu, B.; Shi, Z.; Cao, H.; et al. Comprehensive Analysis of Differentially Expressed mRNA, lncRNA and circRNA and Their ceRNA Networks in the Longissimus Dorsi Muscle of Two Different Pig Breeds. Int. J. Mol. Sci. 2019, 20, 1107. [Google Scholar] [CrossRef]
- Yang, X.; Ma, X.; Mei, C.; Zan, L. A Genome-Wide Landscape of mRNAs, lncRNAs, circRNAs and miRNAs during Intramuscular Adipogenesis in Cattle. BMC Genom. 2022, 23, 691. [Google Scholar] [CrossRef]
- Moreno-Sánchez, N.; Rueda, J.; Reverter, A.; Carabaño, M.J.; Díaz, C. Muscle-specific gene expression is underscored by differ-ential stressor responses and coexpression changes. Funct. Integr. Genom. 2012, 12, 93–103. [Google Scholar] [CrossRef]
- Braun, T.; Gautel, M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell. Biol. 2011, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle Deficiency and Neonatal Death in Mice with a Targeted Mutation in the Myogenin Gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.A.; Schnegelsberg, P.N.; Stead, R.H.; Braun, T.; Arnold, H.H.; Jaenisch, R. MyoD or Myf-5 Is Required for the Formation of Skeletal Muscle. Cell 1993, 75, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Gajda, A.M.; Storch, J. Enterocyte Fatty Acid-Binding Proteins (FABPs): Different Functions of Liver and Intestinal FABPs in the Intestine. Prostaglandins Leukot. Essent. Fat. Acids 2015, 93, 9–16. [Google Scholar] [CrossRef]
- Tao, Q.; Chen, Y.; Bai, D.-P.; Mai, L.; Fan, Q.-M.; Shi, Y.-Z.; Chen, C.; Li, A. Differential Expression of MSTN, IGF2BP1, and FABP2 across Different Embryonic Ages and Sexes in White Muscovy Ducks. Gene 2022, 829, 146479. [Google Scholar] [CrossRef]
- Hitachi, K.; Honda, M.; Tsuchida, K. The Functional Role of Long Non-Coding RNA in Myogenesis and Skeletal Muscle Atrophy. Cells 2022, 11, 2291. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Ma, M.; Zhang, J.; Wang, Z.; Kong, S.; Zhou, Z.; Lian, L.; Zhang, J.; Li, J.; Wang, Y.; et al. LncEDCH1 Improves Mitochondrial Function to Reduce Muscle Atrophy by Interacting with SERCA2. Molecular Therapy. Nucleic Acids 2022, 27, 319–334. [Google Scholar] [CrossRef]
- Li, Z.; Cai, B.; Abdalla, B.A.; Zhu, X.; Zheng, M.; Han, P.; Nie, Q.; Zhang, X. LncIRS1 Controls Muscle Atrophy via Sponging miR-15 Family to Activate IGF1-PI3K/AKT Pathway. J. Cachexia Sarcopenia Muscle 2019, 10, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.; Wankell, M.; Saxena, P.; McFarlane, C.; Hebbard, L. Cell Adhesion an Important Determinant of Myogenesis and Satellite Cell Activity. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119170. [Google Scholar] [CrossRef]
- Goh, Q.; Dearth, C.L.; Corbett, J.T.; Pierre, P.; Chadee, D.N.; Pizza, F.X. Intercellular Adhesion Molecule-1 Expression by Skeletal Muscle Cells Augments Myogenesis. Exp. Cell Res. 2015, 331, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Cichewicz, M.A.; Kiran, M.; Przanowska, R.K.; Sobierajska, E.; Shibata, Y.; Dutta, A. MUNC, an Enhancer RNA Upstream from the MYOD Gene, Induces a Subgroup of Myogenic Transcripts in Trans Independently of MyoD. Mol. Cell. Biol. 2018, 38, e00655-17. [Google Scholar] [CrossRef]
- So, K.K.H.; Huang, Y.; Zhang, S.; Qiao, Y.; He, L.; Li, Y.; Chen, X.; Sham, M.H.; Sun, H.; Wang, H. seRNA PAM Controls Skeletal Muscle Satellite Cell Proliferation and Aging through Trans Regulation of Timp2 Expression Synergistically with Ddx5. Aging Cell 2022, 21, e13673. [Google Scholar] [CrossRef]
- Chen, R.; Lei, S.; Jiang, T.; Zeng, J.; Zhou, S.; She, Y. Roles of lncRNAs and circRNAs in Regulating Skeletal Muscle Development. Acta Physiol. 2020, 228, e13356. [Google Scholar] [CrossRef]
- Chen, B.; Yu, J.; Guo, L.; Byers, M.S.; Wang, Z.; Chen, X.; Xu, H.; Nie, Q. Circular RNA circHIPK3 Promotes the Proliferation and Differentiation of Chicken Myoblast Cells by Sponging miR-30a-3p. Cells 2019, 8, 177. [Google Scholar] [CrossRef]
- Chen, M.; Wei, X.; Song, M.; Jiang, R.; Huang, K.; Deng, Y.; Liu, Q.; Shi, D.; Li, H. Circular RNA circMYBPC1 Promotes Skeletal Muscle Differentiation by Targeting MyHC. Molecular Therapy. Nucleic Acids 2021, 24, 352–368. [Google Scholar] [CrossRef]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA That Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37.e9. [Google Scholar] [CrossRef]
- Hu, Z.; Cao, J.; Liu, G.; Zhang, H.; Liu, X. Comparative Transcriptome Profiling of Skeletal Muscle from Black Muscovy Duck at Different Growth Stages Using RNA-Seq. Genes 2020, 11, 1228. [Google Scholar] [CrossRef]
- Bai, Y.; Ding, X.; Liu, Z.; Shen, J.; Huang, Y. Identification and Functional Analysis of circRNAs in the Skeletal Muscle of Juvenile and Adult Largemouth Bass (Micropterus Salmoides). Comp. Biochem. Physiol. Part D Genom. Proteom. 2022, 42, 100969. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Jiang, T.; Lei, S.; She, Y.; Shi, H.; Zhou, S.; Ou, J.; Liu, Y. Expression of Circular RNAs during C2C12 Myoblast Differentiation and Prediction of Coding Potential Based on the Number of Open Reading Frames and N6-Methyladenosine Motifs. Cell Cycle 2018, 17, 1832–1845. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and Cross-Talk between Non-Coding RNAs. Cell. Mol. Life Sci. 2018, 75, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Xu, M.; Dou, M.; Shi, X.; Yang, G.; Li, X. MicroRNA-129-5p Inhibits C2C12 Myogenesis and Represses Slow Fiber Gene Expression in Vitro. Am. J. Physiol.-Cell Physiol. 2021, 320, C1031–C1041. [Google Scholar] [CrossRef]
- Chen, X.; Ouyang, H.; Wang, Z.; Chen, B.; Nie, Q. A Novel Circular RNA Generated by FGFR2 Gene Promotes Myoblast Proliferation and Differentiation by Sponging miR-133a-5p and miR-29b-1-5p. Cells 2018, 7, 199. [Google Scholar] [CrossRef]
- Wang, S.; Cao, X.; Ge, L.; Gu, Y.; Lv, X.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Sun, W. MiR-22-3p Inhibits Proliferation and Promotes Differentiation of Skeletal Muscle Cells by Targeting IGFBP3 in Hu Sheep. Animals 2022, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; He, M.; Wu, P.; Zhang, X.; Zhou, K.; Li, T.; Zhang, T.; Xie, K.; Dai, G.; Wang, J. MicroRNA-27b-3p Targets the Myostatin Gene to Regulate Myoblast Proliferation and Is Involved in Myoblast Differentiation. Cells 2021, 10, 423. [Google Scholar] [CrossRef]
- Araujo, H.N.; Lima, T.I.; Guimarães, D.S.P.S.F.; Oliveira, A.G.; Favero-Santos, B.C.; Branco, R.C.S.; da Silva Araújo, R.M.; Dantas, A.F.B.; Castro, A.; Chacon-Mikahil, M.P.T.; et al. Regulation of Lin28a-miRNA Let-7b-5p Pathway in Skeletal Muscle Cells by Peroxisome Proliferator-Activated Receptor Delta. American Journal of Physiology. Cell Physiol. 2020, 319, C541–C551. [Google Scholar] [CrossRef]
- Zhang, P.; Reue, K. Lipin Proteins and Glycerolipid Metabolism: Roles at the ER Membrane and Beyond. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1583–1595. [Google Scholar] [CrossRef]
- Tantawy, M.; Collins, J.M.; Wang, D. Genome-Wide microRNA Profiles Identify miR-107 as a Top miRNA Associating with Expression of the CYP3As and Other Drug Metabolizing Cytochrome P450 Enzymes in the Liver. Front. Pharmacol. 2022, 13, 943538. [Google Scholar] [CrossRef]
- Li, H.; Wei, X.; Yang, J.; Dong, D.; Hao, D.; Huang, Y.; Lan, X.; Plath, M.; Lei, C.; Ma, Y.; et al. circFGFR4 Promotes Differentiation of Myoblasts via Binding miR-107 to Relieve Its Inhibition of Wnt3a. Molecular Therapy. Nucleic Acids 2018, 11, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, L.; Chen, X.; Cao, W.; Zhang, R.; Yu, H.; Xu, F.; He, H.; Wang, J. Characterization of in Vitro Cultured Myoblasts Isolated from Duck (Anas Platyrhynchos) Embryo. Cytotechnology 2011, 63, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the Protein-Coding Potential of Transcripts Using Sequence Features and Support Vector Machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing Sequence Intrinsic Composition to Classify Protein-Coding and Long Non-Coding Transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Zhang, X.-O.; Dong, R.; Zhang, Y.; Zhang, J.-L.; Luo, Z.; Zhang, J.; Chen, L.-L.; Yang, L. Diverse Alternative Back-Splicing and Alternative Splicing Landscape of Circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef]
- Zhang, X.-O.; Wang, H.-B.; Zhang, Y.; Lu, X.; Chen, L.-L.; Yang, L. Complementary Sequence-Mediated Exon Circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An Efficient and Unbiased Algorithm for de Novo Circular RNA Identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Kim, D.; Salzberg, S.L. TopHat-Fusion: An Algorithm for Discovery of Novel Fusion Transcripts. Genome Biol. 2011, 12, R72. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching during Cell Differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-Level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice. Int. J. Mol. Sci. 2016, 17, 499. [Google Scholar] [CrossRef]
- Nam, J.-W.; Rissland, O.S.; Koppstein, D.; Abreu-Goodger, C.; Jan, C.H.; Agarwal, V.; Yildirim, M.A.; Rodriguez, A.; Bartel, D.P. Global Analyses of the Effect of Different Cellular Contexts on microRNA Targeting. Mol. Cell 2014, 53, 1031–1043. [Google Scholar] [CrossRef]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive Modeling of microRNA Targets Predicts Functional Non-Conserved and Non-Canonical Sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef]
- Wenzel, A.; Akbasli, E.; Gorodkin, J. RIsearch: Fast RNA-RNA Interaction Search Using a Simplified Nearest-Neighbor Energy Model. Bioinformatics 2012, 28, 2738–2746. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terms | mRNAs | lncRNAs | circRNAs | |||
|---|---|---|---|---|---|---|
| up | down | up | down | up | down | |
| DM vs. GM | 1065 | 668 | 653 | 463 | 44 | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Wu, J.; Zhang, W.; Jiang, H.; Zhou, Y.; Liu, J.; Mao, H.; Liu, S.; Chen, B. Whole-Transcriptome RNA Sequencing Uncovers the Global Expression Changes and RNA Regulatory Networks in Duck Embryonic Myogenesis. Int. J. Mol. Sci. 2023, 24, 16387. https://doi.org/10.3390/ijms242216387
Liu S, Wu J, Zhang W, Jiang H, Zhou Y, Liu J, Mao H, Liu S, Chen B. Whole-Transcriptome RNA Sequencing Uncovers the Global Expression Changes and RNA Regulatory Networks in Duck Embryonic Myogenesis. International Journal of Molecular Sciences. 2023; 24(22):16387. https://doi.org/10.3390/ijms242216387
Chicago/Turabian StyleLiu, Shuibing, Jintao Wu, Wentao Zhang, Hongxia Jiang, Yanan Zhou, Jing Liu, Huirong Mao, Sanfeng Liu, and Biao Chen. 2023. "Whole-Transcriptome RNA Sequencing Uncovers the Global Expression Changes and RNA Regulatory Networks in Duck Embryonic Myogenesis" International Journal of Molecular Sciences 24, no. 22: 16387. https://doi.org/10.3390/ijms242216387
APA StyleLiu, S., Wu, J., Zhang, W., Jiang, H., Zhou, Y., Liu, J., Mao, H., Liu, S., & Chen, B. (2023). Whole-Transcriptome RNA Sequencing Uncovers the Global Expression Changes and RNA Regulatory Networks in Duck Embryonic Myogenesis. International Journal of Molecular Sciences, 24(22), 16387. https://doi.org/10.3390/ijms242216387