
Iron and Targeted Iron Therapy in Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
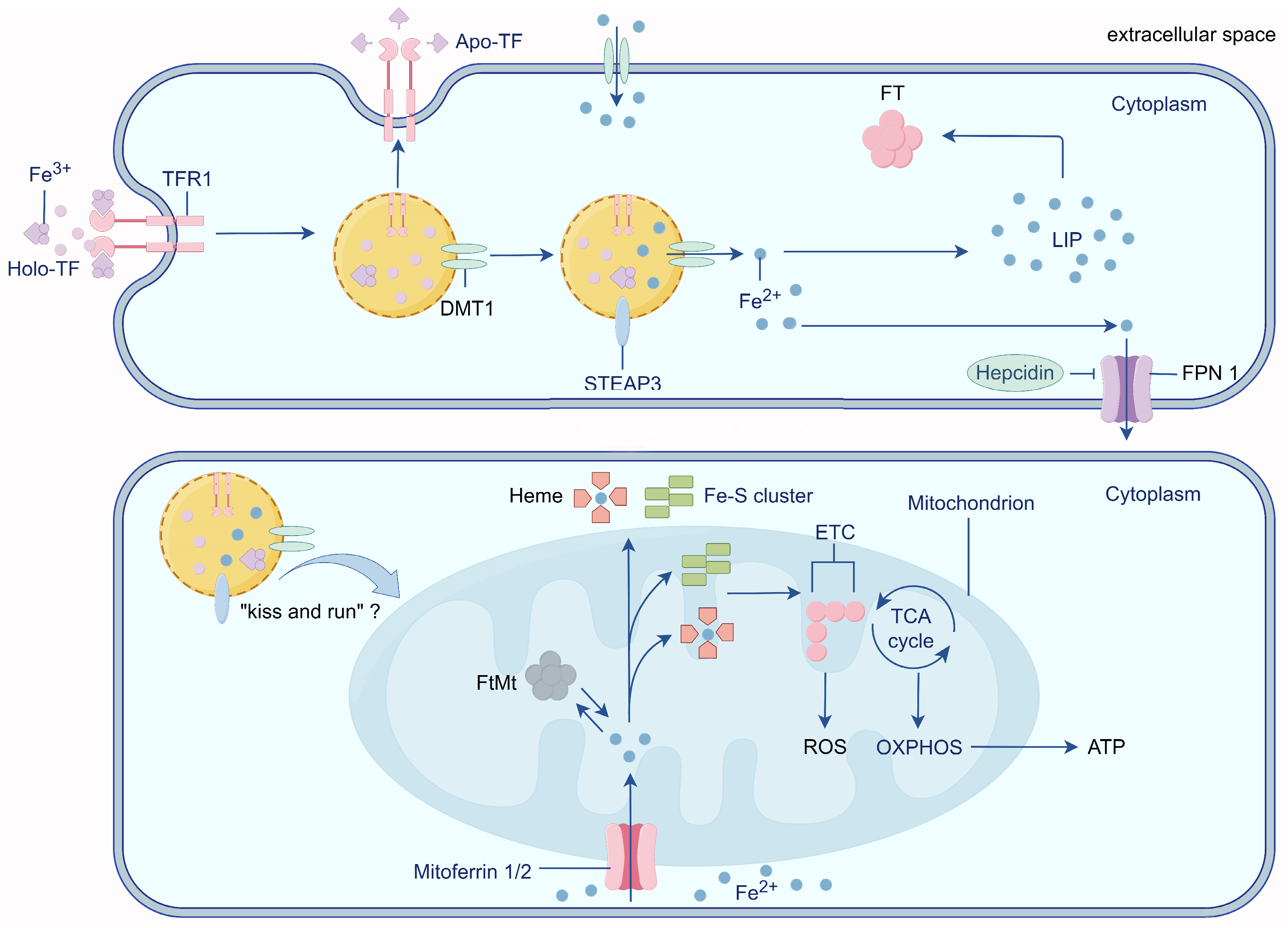
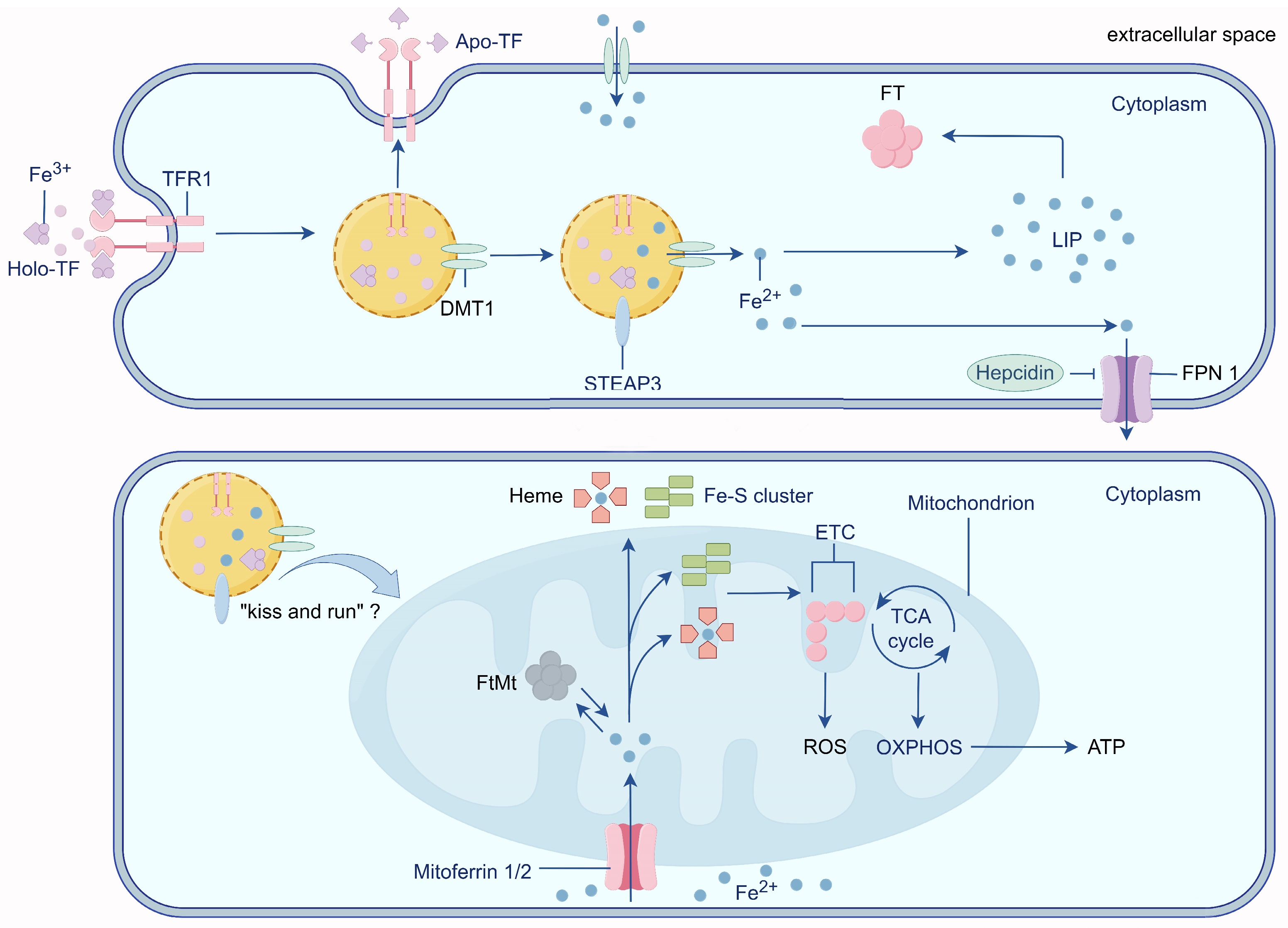
2. Physiological Iron Transport
3. Mitochondria: Intracellular Iron Stores
4. Impairment of the Blood–Brain Barrier (BBB) Is an Important Prerequisite for Iron Accumulation in the Brain
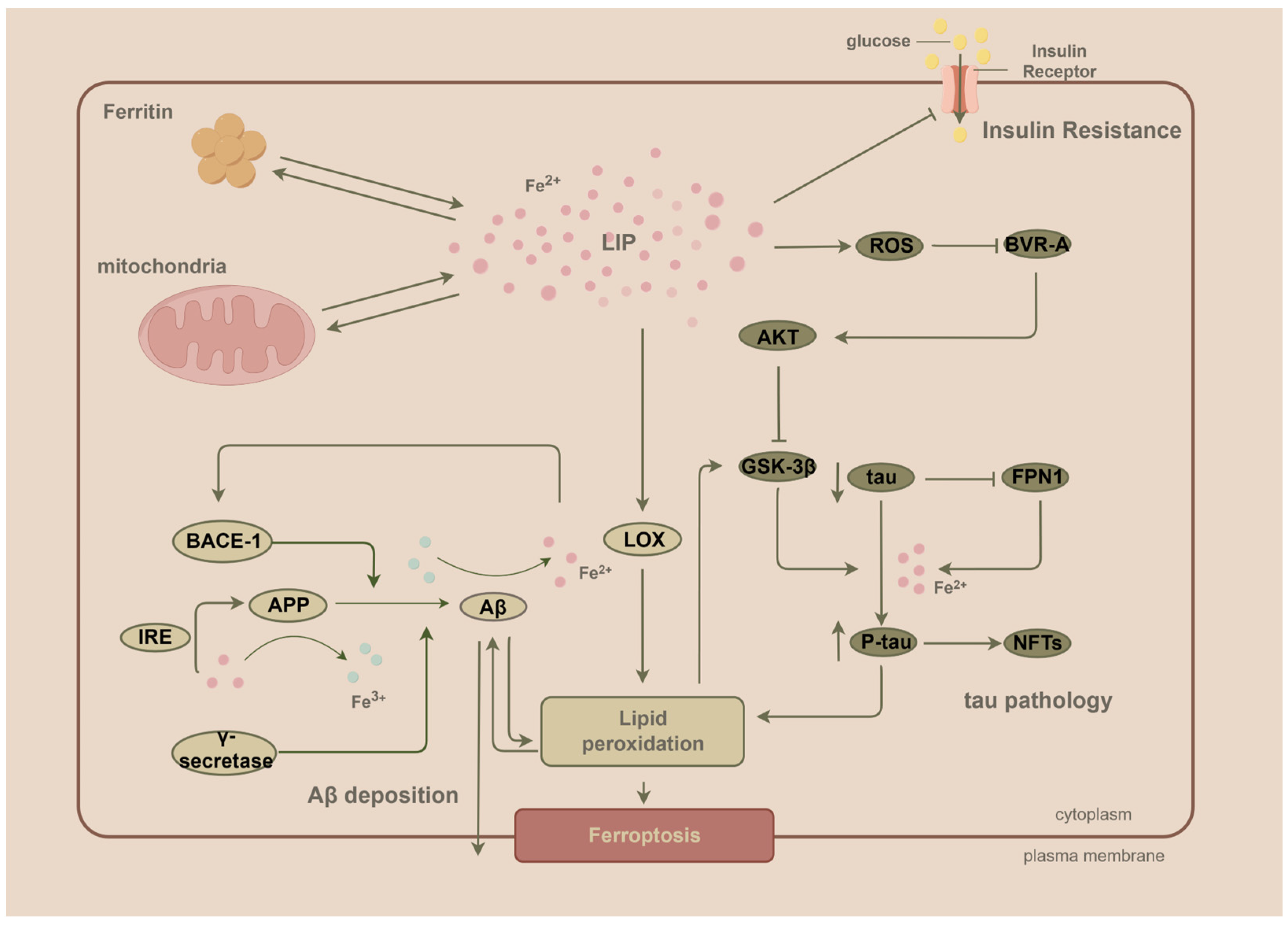
5. Iron and Aβ
5.1. Iron Promotes the Expression of the Aβ Precursor APP and the Abnormal Cleavage Process of APP
5.2. For Aβ to Exert Its Toxicity, Iron Is a Key Factor
6. Iron and Phosphorylation of Tau
Iron Promotes Tau Phosphorylation through Multiple Pathways
7. Ferroptosis and AD
8. Iron and Dysregulated Energy Metabolism in the Brain
8.1. Elevated Iron Levels Induce Brain Insulin Resistance (IR)
8.2. Dysregulated Insulin Signaling Precedes and Contributes to Aβ and Tau Pathology
9. Iron Accumulation throughout AD: From IR-Induced Impairment of Energy Metabolism to Aβ Deposition and Tau Phosphorylation
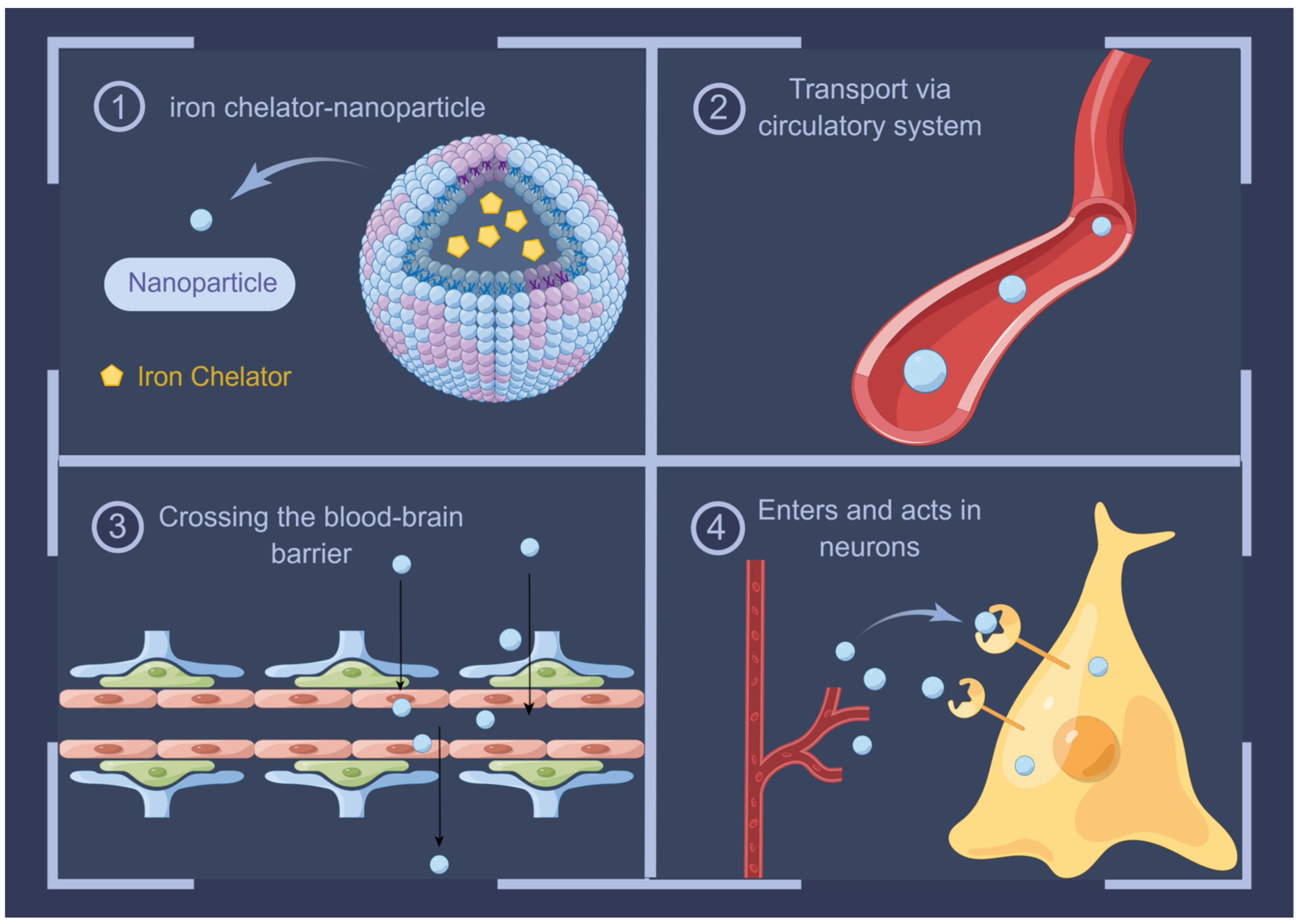
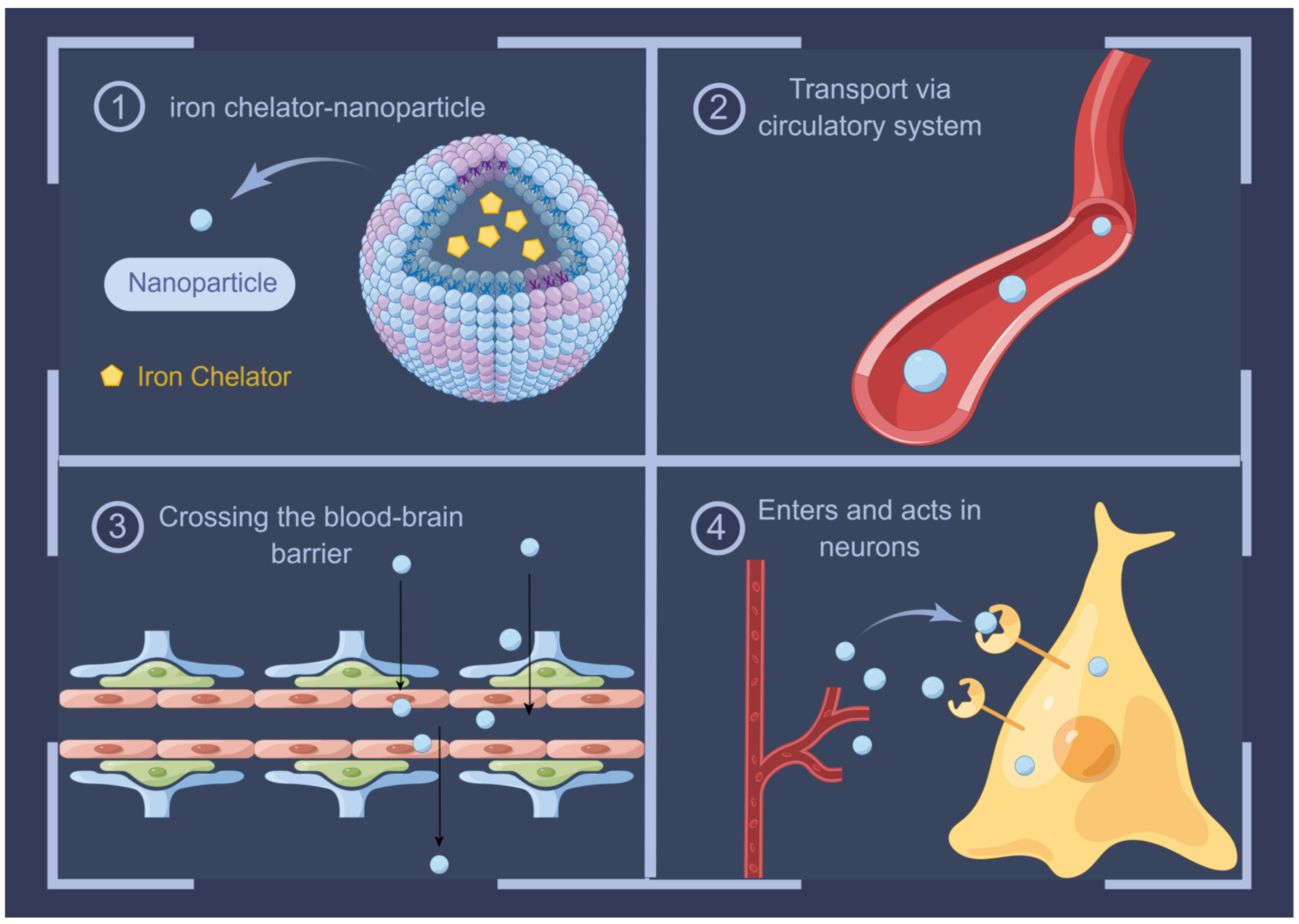
10. Advances in Iron Chelators in AD Therapy
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.D.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef]
- Drayer, B.; Burger, P.; Darwin, R.; Riederer, S.; Herfkens, R.; Johnson, G.A. MRI of brain iron. AJR Am. J. Roentgenol. 1986, 147, 103–110. [Google Scholar] [CrossRef]
- Bartzokis, G.; Tishler, T.A.; Lu, P.H.; Villablanca, P.; Altshuler, L.L.; Carter, M.; Huang, D.; Edwards, N.; Mintz, J. Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiol. Aging 2007, 28, 414–423. [Google Scholar] [CrossRef]
- Burgetova, R.; Dusek, P.; Burgetova, A.; Pudlac, A.; Vaneckova, M.; Horakova, D.; Krasensky, J.; Varga, Z.; Lambert, L. Age-related magnetic susceptibility changes in deep grey matter and cerebral cortex of normal young and middle-aged adults depicted by whole brain analysis. Quant. Imaging Med. Surg. 2021, 11, 3906–3919. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, H.; Cronin, M.J.; He, N.; Yan, F.; Liu, C. Longitudinal atlas for normative human brain development and aging over the lifespan using quantitative susceptibility mapping. Neuroimage 2018, 171, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Cabronero, J.; Betts, M.J.; Cardenas-Blanco, A.; Yang, S.; Nestor, P.J. In Vivo MRI Mapping of Brain Iron Deposition across the Adult Lifespan. J. Neurosci. 2016, 36, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Cerchiaro, G.; Rani, I.; Ventriglia, M.; Rongioletti, M.; Longobardi, A.; Squitti, R. Iron in Alzheimer’s Disease: From Physiology to Disease Disabilities. Biomolecules 2022, 12, 1248. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 (Suppl. S1), 179–197. [Google Scholar] [CrossRef]
- Zhao, Z. Iron and oxidizing species in oxidative stress and Alzheimer’s disease. Aging Med. (Milton) 2019, 2, 82–87. [Google Scholar] [CrossRef]
- Bradley-Whitman, M.A.; Lovell, M.A. Biomarkers of lipid peroxidation in Alzheimer disease (AD): An update. Arch. Toxicol. 2015, 89, 1035–1044. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Iron transport across the blood-brain barrier: Development, neurovascular regulation and cerebral amyloid angiopathy. Cell Mol. Life Sci. 2015, 72, 709–727. [Google Scholar] [CrossRef]
- Nelson, N.; Harvey, W.R. Vacuolar and plasma membrane proton-adenosinetriphosphatases. Physiol. Rev. 1999, 79, 361–385. [Google Scholar] [CrossRef]
- Yanatori, I.; Kishi, F. DMT1 and iron transport. Free Radic. Biol. Med. 2019, 133, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Patel, S.J.; Protchenko, O. Management versus miscues in the cytosolic labile iron pool: The varied functions of iron chaperones. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yu, X.; Xie, J.; Xu, H. New Insights into the Role of Ferritin in Iron Homeostasis and Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Hadzhieva, M.; Kirches, E.; Mawrin, C. Review: Iron metabolism and the role of iron in neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2014, 40, 240–257. [Google Scholar] [CrossRef]
- Mehlenbacher, M.; Poli, M.; Arosio, P.; Santambrogio, P.; Levi, S.; Chasteen, N.D.; Bou-Abdallah, F. Iron Oxidation and Core Formation in Recombinant Heteropolymeric Human Ferritins. Biochemistry 2017, 56, 3900–3912. [Google Scholar] [CrossRef]
- Connor, J.R.; Snyder, B.S.; Arosio, P.; Loeffler, D.A.; LeWitt, P. A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer’s diseased brains. J. Neurochem. 1995, 65, 717–724. [Google Scholar] [CrossRef]
- De Domenico, I.; McVey Ward, D.; Kaplan, J. Regulation of iron acquisition and storage: Consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81. [Google Scholar] [CrossRef]
- Qian, Z.M.; Chang, Y.Z.; Zhu, L.; Yang, L.; Du, J.R.; Ho, K.P.; Wang, Q.; Li, L.Z.; Wang, C.Y.; Ge, X.; et al. Development and iron-dependent expression of hephaestin in different brain regions of rats. J. Cell Biochem. 2007, 102, 1225–1233. [Google Scholar] [CrossRef]
- Moos, T.; Rosengren Nielsen, T.; Skjørringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Lill, R. The power plant of the cell is also a smithy: The emerging role of mitochondria in cellular iron homeostasis. Ann. Med. 2009, 41, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Christenson, E.T.; Gallegos, A.S.; Banerjee, A. In vitro reconstitution, functional dissection, and mutational analysis of metal ion transport by mitoferrin-1. J. Biol. Chem. 2018, 293, 3819–3828. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, S.; Hu, L.; Niu, H.; Sun, Q.; Li, W.; Tan, G.; Li, J.; Jin, L.; Lyu, J.; et al. Mitoferrin-1 Is Involved in the Progression of Alzheimer’s Disease Through Targeting Mitochondrial Iron Metabolism in a Caenorhabditis elegans Model of Alzheimer’s Disease. Neuroscience 2018, 385, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, L.; Endres, T.; Scholz, J.; Kirches, E.; Ward, D.M.; Lessmann, V.; Borucki, K.; Mawrin, C. Mitoferrin-1 is required for brain energy metabolism and hippocampus-dependent memory. Neurosci. Lett. 2019, 713, 134521. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta 2016, 1863, 2859–2867. [Google Scholar] [CrossRef]
- Zhang, A.S.; Sheftel, A.D.; Ponka, P. Intracellular kinetics of iron in reticulocytes: Evidence for endosome involvement in iron targeting to mitochondria. Blood 2005, 105, 368–375. [Google Scholar] [CrossRef]
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome-mitochondria interactions are modulated by iron release from transferrin. J. Cell Biol. 2016, 214, 831–845. [Google Scholar] [CrossRef]
- Gao, G.; Chang, Y.Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharmacol. 2014, 5, 19. [Google Scholar] [CrossRef]
- Shi, Z.H.; Shi, F.F.; Wang, Y.Q.; Sheftel, A.D.; Nie, G.; Zhao, Y.S.; You, L.H.; Gou, Y.J.; Duan, X.L.; Zhao, B.L.; et al. Mitochondrial ferritin, a new target for inhibiting neuronal tumor cell proliferation. Cell Mol. Life Sci. 2015, 72, 983–997. [Google Scholar] [CrossRef]
- Shi, Z.H.; Nie, G.; Duan, X.L.; Rouault, T.; Wu, W.S.; Ning, B.; Zhang, N.; Chang, Y.Z.; Zhao, B.L. Neuroprotective mechanism of mitochondrial ferritin on 6-hydroxydopamine-induced dopaminergic cell damage: Implication for neuroprotection in Parkinson’s disease. Antioxid. Redox Signal 2010, 13, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S.; Zhao, Y.S.; Shi, Z.H.; Chang, S.Y.; Nie, G.J.; Duan, X.L.; Zhao, S.M.; Wu, Q.; Yang, Z.L.; Zhao, B.L.; et al. Mitochondrial ferritin attenuates β-amyloid-induced neurotoxicity: Reduction in oxidative damage through the Erk/P38 mitogen-activated protein kinase pathways. Antioxid. Redox Signal 2013, 18, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, H.; Zhao, S.; Sato, H.; Konishi, Y.; Beach, T.G.; Abdelalim, E.M.; Bisem, N.J.; Tooyama, I. Expression and localization of mitochondrial ferritin mRNA in Alzheimer’s disease cerebral cortex. PLoS ONE 2011, 6, e22325. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Nie, G.; Li, Y.; Soe-Lin, S.; Tao, Y.; Cao, Y.; Zhang, Z.; Liu, N.; Ponka, P.; Zhao, B. Overexpression of mitochondrial ferritin sensitizes cells to oxidative stress via an iron-mediated mechanism. Antioxid. Redox Signal 2009, 11, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Vessey, D.A.; Lee, K.H.; Blacker, K.L. Characterization of the oxidative stress initiated in cultured human keratinocytes by treatment with peroxides. J. Investig. Dermatol. 1992, 99, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gan, L.; Ren, L.; Lin, Y.; Ma, C.; Lin, X. Factors influencing the blood-brain barrier permeability. Brain Res. 2022, 1788, 147937. [Google Scholar] [CrossRef]
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The blood-brain barrier: Structure, regulation, and drug delivery. Signal Transduct. Target. Ther. 2023, 8, 217. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Mechanistic analysis of iron accumulation by endothelial cells of the BBB. Biometals 2012, 25, 665–675. [Google Scholar] [CrossRef]
- Damulina, A.; Pirpamer, L.; Soellradl, M.; Sackl, M.; Tinauer, C.; Hofer, E.; Enzinger, C.; Gesierich, B.; Duering, M.; Ropele, S.; et al. Cross-sectional and Longitudinal Assessment of Brain Iron Level in Alzheimer Disease Using 3-T MRI. Radiology 2020, 296, 619–626. [Google Scholar] [CrossRef]
- De Reuck, J.L.; Deramecourt, V.; Auger, F.; Durieux, N.; Cordonnier, C.; Devos, D.; Defebvre, L.; Moreau, C.; Caparros-Lefebvre, D.; Leys, D.; et al. Iron deposits in post-mortem brains of patients with neurodegenerative and cerebrovascular diseases: A semi-quantitative 7.0 T magnetic resonance imaging study. Eur. J. Neurol. 2014, 21, 1026–1031. [Google Scholar] [CrossRef]
- Du, L.; Zhao, Z.; Cui, A.; Zhu, Y.; Zhang, L.; Liu, J.; Shi, S.; Fu, C.; Han, X.; Gao, W.; et al. Increased Iron Deposition on Brain Quantitative Susceptibility Mapping Correlates with Decreased Cognitive Function in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Chiou, B.; Neal, E.H.; Bowman, A.B.; Lippmann, E.S.; Simpson, I.A.; Connor, J.R. Endothelial cells are critical regulators of iron transport in a model of the human blood-brain barrier. J. Cereb. Blood Flow. Metab. 2019, 39, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Verheggen, I.C.M.; de Jong, J.J.A.; van Boxtel, M.P.J.; Gronenschild, E.; Palm, W.M.; Postma, A.A.; Jansen, J.F.A.; Verhey, F.R.J.; Backes, W.H. Increase in blood-brain barrier leakage in healthy, older adults. Geroscience 2020, 42, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, M.; Ge, Y.; Chen, J.; Ma, J.; Wang, C.; Sun, M.; Wang, L.; Yao, S.; Yao, C. β-amyloid protein induces mitophagy-dependent ferroptosis through the CD36/PINK/PARKIN pathway leading to blood-brain barrier destruction in Alzheimer’s disease. Cell Biosci. 2022, 12, 69. [Google Scholar] [CrossRef]
- Takechi, R.; Lam, V.; Brook, E.; Giles, C.; Fimognari, N.; Mooranian, A.; Al-Salami, H.; Coulson, S.H.; Nesbit, M.; Mamo, J.C.L. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front. Aging Neurosci. 2017, 9, 399. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Mockett, B.G.; Richter, M.; Abraham, W.C.; Müller, U.C. Therapeutic Potential of Secreted Amyloid Precursor Protein APPsα. Front. Mol. Neurosci. 2017, 10, 30. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.; Moore, S.; Daria, A.; et al. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447. [Google Scholar] [CrossRef]
- Ward, J.; Wang, H.; Saunders, A.J.; Tanzi, R.E.; Zhang, C. Mechanisms that synergistically regulate η-secretase processing of APP and Aη-α protein levels: Relevance to pathogenesis and treatment of Alzheimer’s disease. Discov. Med. 2017, 23, 121–128. [Google Scholar]
- Bandyopadhyay, S.; Huang, X.; Cho, H.; Greig, N.H.; Youdim, M.B.; Rogers, J.T. Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5’untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J. Neural Transm. Suppl. 2006, 71, 237–247. [Google Scholar]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5’-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Xin, N.; Chi, Z.H.; Zhao, B.L.; Zhang, J.; Li, J.Y.; Wang, Z.Y. Divalent metal transporter 1 is involved in amyloid precursor protein processing and Abeta generation. Faseb j 2009, 23, 4207–4217. [Google Scholar] [CrossRef] [PubMed]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Zheng, Q.; Yao, G.; Cheng, P.; Bu, G.; Xu, H.; Zhang, Y.W. Ferritin light chain interacts with PEN-2 and affects γ-secretase activity. Neurosci. Lett. 2013, 548, 90–94. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chen, W.Y.; Huang, X.T.; Xu, Y.C.; Zhang, H.Y. Iron dysregulates APP processing accompanying with sAPPα cellular retention and β-secretase inhibition in rat cortical neurons. Acta Pharmacol. Sin. 2018, 39, 177–183. [Google Scholar] [CrossRef]
- Xiong, K.; Cai, H.; Luo, X.G.; Struble, R.G.; Clough, R.W.; Yan, X.X. Mitochondrial respiratory inhibition and oxidative stress elevate beta-secretase (BACE1) proteins and activity in vivo in the rat retina. Exp. Brain Res. 2007, 181, 435–446. [Google Scholar] [CrossRef]
- Raha, A.A.; Vaishnav, R.A.; Friedland, R.P.; Bomford, A.; Raha-Chowdhury, R. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 55. [Google Scholar] [CrossRef]
- Lovell, M.A.; Soman, S.; Bradley, M.A. Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech. Ageing Dev. 2011, 132, 443–448. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Markesbery, W.R.; Butterfield, D.A. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging 2006, 27, 1564–1576. [Google Scholar] [CrossRef]
- Benseny-Cases, N.; Klementieva, O.; Cotte, M.; Ferrer, I.; Cladera, J. Microspectroscopy (μFTIR) reveals co-localization of lipid oxidation and amyloid plaques in human Alzheimer disease brains. Anal. Chem. 2014, 86, 12047–12054. [Google Scholar] [CrossRef]
- De Ricco, R.; Valensin, D.; Dell’Acqua, S.; Casella, L.; Hureau, C.; Faller, P. Copper(I/II), α/β-Synuclein and Amyloid-β: Menage à Trois? Chembiochem 2015, 16, 2319–2328. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-β peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Faller, P.; Hureau, C.; Granzotto, A.; White, A.R.; Kepp, K.P. Copper Imbalance in Alzheimer’s Disease and Its Link with the Amyloid Hypothesis: Towards a Combined Clinical, Chemical, and Genetic Etiology. J. Alzheimers Dis. 2021, 83, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimers Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef]
- Everett, J.; Céspedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide β-amyloid (1-42). J. R. Soc. Interface 2014, 11, 20140165. [Google Scholar] [CrossRef]
- Everett, J.; Céspedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Evidence of redox-active iron formation following aggregation of ferrihydrite and the Alzheimer’s disease peptide β-amyloid. Inorg. Chem. 2014, 53, 2803–2809. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Ponce, A.; Collingwood, J.F.; Arellano-Jiménez, M.J.; Zhu, X.; Rogers, J.T.; Betancourt, I.; José-Yacamán, M.; Perry, G. High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer’s disease. Sci. Rep. 2016, 6, 24873. [Google Scholar] [CrossRef] [PubMed]
- Dingwall, C. A copper-binding site in the cytoplasmic domain of BACE1 identifies a possible link to metal homoeostasis and oxidative stress in Alzheimer’s disease. Biochem. Soc. Trans. 2007, 35 Pt 3, 571–573. [Google Scholar] [CrossRef]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimers Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Dorostkar, M.M.; Zou, C.; Blazquez-Llorca, L.; Herms, J. Analyzing dendritic spine pathology in Alzheimer’s disease: Problems and opportunities. Acta Neuropathol. 2015, 130, 1–19. [Google Scholar] [CrossRef]
- Chu, D.; Liu, F. Pathological Changes of Tau Related to Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 931–944. [Google Scholar] [CrossRef]
- Losev, Y.; Frenkel-Pinter, M.; Abu-Hussien, M.; Viswanathan, G.K.; Elyashiv-Revivo, D.; Geries, R.; Khalaila, I.; Gazit, E.; Segal, D. Differential effects of putative N-glycosylation sites in human Tau on Alzheimer’s disease-related neurodegeneration. Cell Mol. Life Sci. 2021, 78, 2231–2245. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef]
- de Wit, J.; Ghosh, A. Specification of synaptic connectivity by cell surface interactions. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef]
- Alonso Adel, C.; Mederlyova, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 2004, 279, 34873–34881. [Google Scholar] [CrossRef]
- Wallin, C.; Hiruma, Y.; Wärmländer, S.; Huvent, I.; Jarvet, J.; Abrahams, J.P.; Gräslund, A.; Lippens, G.; Luo, J. The Neuronal Tau Protein Blocks In Vitro Fibrillation of the Amyloid-β (Aβ) Peptide at the Oligomeric Stage. J. Am. Chem. Soc. 2018, 140, 8138–8146. [Google Scholar] [CrossRef]
- Nübling, G.; Bader, B.; Levin, J.; Hildebrandt, J.; Kretzschmar, H.; Giese, A. Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with α-synuclein at the single molecule level. Mol. Neurodegener. 2012, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Bader, B.; Nübling, G.; Mehle, A.; Nobile, S.; Kretzschmar, H.; Giese, A. Single particle analysis of tau oligomer formation induced by metal ions and organic solvents. Biochem. Biophys. Res. Commun. 2011, 411, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Ebralidze, I.I.; She, Z.; Kraatz, H.B. Electrochemical studies of tau protein-iron interactions-Potential implications for Alzheimer’s Disease. Electrochim. Acta 2017, 236, 374–383. [Google Scholar] [CrossRef]
- Nuebling, G.S.; Plesch, E.; Ruf, V.C.; Högen, T.; Lorenzl, S.; Kamp, F.; Giese, A.; Levin, J. Binding of Metal-Ion-Induced Tau Oligomers to Lipid Surfaces Is Enhanced by GSK-3β-Mediated Phosphorylation. ACS Chem. Neurosci. 2020, 11, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Stankowski, J.N.; Dawson, V.L.; Dawson, T.M. Ironing out tau’s role in parkinsonism. Nat. Med. 2012, 18, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef]
- Uranga, R.M.; Giusto, N.M.; Salvador, G.A. Iron-induced oxidative injury differentially regulates PI3K/Akt/GSK3beta pathway in synaptic endings from adult and aged rats. Toxicol. Sci. 2009, 111, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Apopa, P.L.; Qian, Y.; Shao, R.; Guo, N.L.; Schwegler-Berry, D.; Pacurari, M.; Porter, D.; Shi, X.; Vallyathan, V.; Castranova, V.; et al. Iron oxide nanoparticles induce human microvascular endothelial cell permeability through reactive oxygen species production and microtubule remodeling. Part. Fibre Toxicol. 2009, 6, 1. [Google Scholar] [CrossRef]
- Zhang, S.; Lachance, B.B.; Mattson, M.P.; Jia, X. Glucose metabolic crosstalk and regulation in brain function and diseases. Prog. Neurobiol. 2021, 204, 102089. [Google Scholar] [CrossRef]
- Zhang, X.D.; Liu, Z.Y.; Wang, M.S.; Guo, Y.X.; Wang, X.K.; Luo, K.; Huang, S.; Li, R.F. Mechanisms and regulations of ferroptosis. Front. Immunol. 2023, 14, 1269451. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal 2018, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Riano, C.; Garcia, A.; Barbas, C. Metabolomics studies in brain tissue: A review. J. Pharm. Biomed. Anal. 2016, 130, 141–168. [Google Scholar] [CrossRef] [PubMed]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, W.; Li, J.; Qiu, Z.; Wang, X.; Xu, H.; Wang, H.; Lu, D.; Qi, R. Senegenin Rescues PC12 Cells with Oxidative Damage through Inhibition of Ferroptosis. Mol. Neurobiol. 2022, 59, 6983–6992. [Google Scholar] [CrossRef]
- Kuwata, H.; Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019, 144, 106363. [Google Scholar] [CrossRef]
- Gwon, A.R.; Park, J.S.; Arumugam, T.V.; Kwon, Y.K.; Chan, S.L.; Kim, S.H.; Baik, S.H.; Yang, S.; Yun, Y.K.; Choi, Y.; et al. Oxidative lipid modification of nicastrin enhances amyloidogenic γ-secretase activity in Alzheimer’s disease. Aging Cell 2012, 11, 559–568. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, Y.; Liu, Y.; Liu, Q.; Sun, H.; Mei, M.; Liao, X. Ferroptosis promotes microtubule-associated protein tau aggregation via GSK-3β activation and proteasome inhibition. Mol. Neurobiol. 2022, 59, 1486–1501. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, N.; Shi, F.X.; Xu, W.Q.; Cao, Y.; Lei, Y.; Wang, J.Z.; Tian, Q.; Zhou, X.W. Upregulation of AMPK Ameliorates Alzheimer’s Disease-like Tau Pathology and Memory Impairment. Mol. Neurobiol. 2020, 57, 3349–3361. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ma, C.; Sun, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. J. Diabetes Res. 2020, 2020, 4981814. [Google Scholar] [CrossRef] [PubMed]
- Zilliox, L.A.; Chadrasekaran, K.; Kwan, J.Y.; Russell, J.W. Diabetes and Cognitive Impairment. Curr. Diab Rep. 2016, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.P.; Alves, C.J.; Neto, E.; Lamghari, M. Communication from the periphery to the hypothalamus through the blood-brain barrier: An in vitro platform. Int. J. Pharm. 2016, 499, 119–130. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Reaven, G.M. The insulin resistance syndrome: Definition and dietary approaches to treatment. Annu. Rev. Nutr. 2005, 25, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Heni, M.; Kullmann, S.; Preissl, H.; Fritsche, A.; Häring, H.U. Impaired insulin action in the human brain: Causes and metabolic consequences. Nat. Rev. Endocrinol. 2015, 11, 701–711. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef]
- Chung, J.Y.; Kim, H.S.; Song, J. Iron metabolism in diabetes-induced Alzheimer’s disease: A focus on insulin resistance in the brain. Biometals 2018, 31, 705–714. [Google Scholar] [CrossRef]
- Wan, W.; Cao, L.; Kalionis, B.; Murthi, P.; Xia, S.; Guan, Y. Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling. Front. Neurol. 2019, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Mi, J.; Song, L.; Guo, Y.; Li, Y.; Yin, Y.; Zhang, C. SLC40A1 Mediates Ferroptosis and Cognitive Dysfunction in Type 1 Diabetes. Neuroscience 2021, 463, 216–226. [Google Scholar] [CrossRef]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2015, 11, 504–510.e1. [Google Scholar] [CrossRef]
- Ekblad, L.L.; Johansson, J.; Helin, S.; Viitanen, M.; Laine, H.; Puukka, P.; Jula, A.; Rinne, J.O. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology 2018, 90, e1150–e1157. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; Van Bronswijk, W.; Martins, R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J. Neurosci. 2002, 22, Rc221. [Google Scholar] [CrossRef]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a biological hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef]
- Pandey, M.P.; Sasidharan, S.; Raghunathan, V.A.; Khandelia, H. Molecular Mechanism of Hydrotropic Properties of GTP and ATP. J. Phys. Chem. B 2022, 126, 8486–8494. [Google Scholar] [CrossRef]
- Vandal, M.; Bourassa, P.; Calon, F. Can insulin signaling pathways be targeted to transport Aβ out of the brain? Front. Aging Neurosci. 2015, 7, 114. [Google Scholar] [CrossRef]
- Kim, J.J.; Kim, Y.S.; Kumar, V. Heavy metal toxicity: An update of chelating therapeutic strategies. J. Trace Elem. Med. Biol. 2019, 54, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Lei, P.; Bush, A.I. Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 2015, 12, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Entezari, S.; Haghi, S.M.; Norouzkhani, N.; Sahebnazar, B.; Vosoughian, F.; Akbarzadeh, D.; Islampanah, M.; Naghsh, N.; Abbasalizadeh, M.; Deravi, N. Iron Chelators in Treatment of Iron Overload. J. Toxicol. 2022, 2022, 4911205. [Google Scholar] [CrossRef] [PubMed]
- Farr, A.C.; Xiong, M.P. Challenges and Opportunities of Deferoxamine Delivery for Treatment of Alzheimer’s Disease, Parkinson’s Disease, and Intracerebral Hemorrhage. Mol. Pharm. 2021, 18, 593–609. [Google Scholar] [CrossRef]
- Bayanzay, K.; Alzoebie, L. Reducing the iron burden and improving survival in transfusion-dependent thalassemia patients: Current perspectives. J. Blood Med. 2016, 7, 159–169. [Google Scholar] [CrossRef]
- Brittenham, G.M. Iron-chelating therapy for transfusional iron overload. N. Engl. J. Med. 2011, 364, 146–156. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.; Florence, A.; Aouad, F.; Hider, R.; Jenner, P.; Crichton, R.R. Brain iron in the ferrocene-loaded rat: Its chelation and influence on dopamine metabolism. Biochem. Pharmacol. 1995, 49, 1821–1826. [Google Scholar] [CrossRef]
- Shachar, D.B.; Kahana, N.; Kampel, V.; Warshawsky, A.; Youdim, M.B. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lession in rats. Neuropharmacology 2004, 46, 254–263. [Google Scholar] [CrossRef]
- Hanson, L.R.; Fine, J.M.; Renner, D.B.; Svitak, A.L.; Burns, R.B.; Nguyen, T.M.; Tuttle, N.J.; Marti, D.L.; Panter, S.S.; Frey, W.H., 2nd. Intranasal delivery of deferoxamine reduces spatial memory loss in APP/PS1 mice. Drug Deliv. Transl. Res. 2012, 2, 160–168. [Google Scholar] [CrossRef]
- Zhu, D.; Liang, R.; Liu, Y.; Li, Z.; Cheng, L.; Ren, J.; Guo, Y.; Wang, M.; Chai, H.; Niu, Q.; et al. Deferoxamine ameliorated Al(mal)(3)-induced neuronal ferroptosis in adult rats by chelating brain iron to attenuate oxidative damage. Toxicol. Mech. Methods 2022, 32, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, Y.X.; Wang, T.; Zhong, M.L.; Yang, Z.H.; Hao, L.J.; Chai, R.; Zhang, S. Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1α pathway on the brain of APP/PS1 transgenic mice. Front. Aging Neurosci. 2015, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, P.; Zhong, M.L.; Wang, T.; Huang, X.S.; Li, J.Y.; Wang, Z.Y. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 2013, 62, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Poggiali, E.; Cassinerio, E.; Zanaboni, L.; Cappellini, M.D. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411–422. [Google Scholar]
- Maher, P.; Kontoghiorghes, G.J. Characterization of the neuroprotective potential of derivatives of the iron chelating drug deferiprone. Neurochem. Res. 2015, 40, 609–620. [Google Scholar] [CrossRef]
- Rao, S.S.; Portbury, S.D.; Lago, L.; Bush, A.I.; Adlard, P.A. The Iron Chelator Deferiprone Improves the Phenotype in a Mouse Model of Tauopathy. J. Alzheimers Dis. 2020, 77, 753–771. [Google Scholar] [CrossRef]
- Chand, K.; Rajeshwari; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Tacrine-deferiprone hybrids as multi-target-directed metal chelators against Alzheimer’s disease: A two-in-one drug. Metallomics 2018, 10, 1460–1475. [Google Scholar] [CrossRef]
- Cappellini, M.D. Long-term efficacy and safety of deferasirox. Blood Rev. 2008, 22 (Suppl. S2), S35–S41. [Google Scholar] [CrossRef]
- Banerjee, P.; Sahoo, A.; Anand, S.; Bir, A.; Chakrabarti, S. The Oral Iron Chelator, Deferasirox, Reverses the Age-Dependent Alterations in Iron and Amyloid-β Homeostasis in Rat Brain: Implications in the Therapy of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 49, 681–693. [Google Scholar] [CrossRef]
- Kwan, P.; Ho, A.; Baum, L. Effects of Deferasirox in Alzheimer’s Disease and Tauopathy Animal Models. Biomolecules 2022, 12, 365. [Google Scholar] [CrossRef]
- Feng, W.; Xiao, Y.; Zhao, C.; Zhang, Z.; Liu, W.; Ma, J.; Ganz, T.; Zhang, J.; Liu, S. New Deferric Amine Compounds Efficiently Chelate Excess Iron to Treat Iron Overload Disorders and to Prevent Ferroptosis. Adv. Sci. 2022, 9, e2202679. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J.; Kolnagou, A.; Kontoghiorghe, C.N.; Mourouzidis, L.; Timoshnikov, V.A.; Polyakov, N.E. Trying to Solve the Puzzle of the Interaction of Ascorbic Acid and Iron: Redox, Chelation and Therapeutic Implications. Medicines 2020, 7, 45. [Google Scholar] [CrossRef]
- Elalfy, M.S.; Saber, M.M.; Adly, A.A.; Ismail, E.A.; Tarif, M.; Ibrahim, F.; Elalfy, O.M. Role of vitamin C as an adjuvant therapy to different iron chelators in young β-thalassemia major patients: Efficacy and safety in relation to tissue iron overload. Eur. J. Haematol. 2016, 96, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Bostanci, M.O.; Bas, O.; Bagirici, F. Alpha-tocopherol decreases iron-induced hippocampal and nigral neuron loss. Cell Mol. Neurobiol. 2010, 30, 389–394. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y.; Guo, S.Q.; Wang, S.; Guo, T.; Wang, Z.Y.; et al. α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Gaasch, J.A.; Geldenhuys, W.J.; Lockman, P.R.; Allen, D.D.; Van der Schyf, C.J. Voltage-gated calcium channels provide an alternate route for iron uptake in neuronal cell cultures. Neurochem. Res. 2007, 32, 1686–1693. [Google Scholar] [CrossRef]
- Bostanci, M.; Bagirici, F. Blocking of L-type calcium channels protects hippocampal and nigral neurons against iron neurotoxicity. The role of L-type calcium channels in iron-induced neurotoxicity. Int. J. Neurosci. 2013, 123, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Grossi, C.; Francese, S.; Casini, A.; Rosi, M.C.; Luccarini, I.; Fiorentini, A.; Gabbiani, C.; Messori, L.; Moneti, G.; Casamenti, F. Clioquinol decreases amyloid-beta burden and reduces working memory impairment in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 423–440. [Google Scholar] [CrossRef]
- Biswas, S.; Torchilin, V.P. Nanopreparations for organelle-specific delivery in cancer. Adv. Drug Deliv. Rev. 2014, 66, 26–41. [Google Scholar] [CrossRef]
- Liu, G.; Men, P.; Kudo, W.; Perry, G.; Smith, M.A. Nanoparticle-chelator conjugates as inhibitors of amyloid-beta aggregation and neurotoxicity: A novel therapeutic approach for Alzheimer disease. Neurosci. Lett. 2009, 455, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Liu, G.; Men, P.; Perry, G.; Smith, M.A.; Zhu, X. Nanoparticle delivery of transition-metal chelators to the brain: Oxidative stress will never see it coming! CNS Neurol. Disord. Drug Targets 2012, 11, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Men, P.; Perry, G.; Smith, M.A. Nanoparticle and iron chelators as a potential novel Alzheimer therapy. Methods Mol. Biol. 2010, 610, 123–144. [Google Scholar] [PubMed]
- Krol, S. Challenges in drug delivery to the brain: Nature is against us. J. Control. Release 2012, 164, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, S.; Akiyama, H.; Saito, H.; McGeer, P.L. Ultrastructural localization of complement membrane attack complex (MAC)-like immunoreactivity in brains of patients with Alzheimer’s disease. Brain Res. 1994, 645, 78–84. [Google Scholar] [CrossRef]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am. J. Pathol. 1999, 154, 927–936. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Fu, J.; Zhao, Y.; Liu, Q.; Yan, X.; Su, J. Iron and Targeted Iron Therapy in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 16353. https://doi.org/10.3390/ijms242216353
Wang J, Fu J, Zhao Y, Liu Q, Yan X, Su J. Iron and Targeted Iron Therapy in Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(22):16353. https://doi.org/10.3390/ijms242216353
Chicago/Turabian StyleWang, Jian, Jiaying Fu, Yuanxin Zhao, Qingqing Liu, Xiaoyu Yan, and Jing Su. 2023. "Iron and Targeted Iron Therapy in Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 22: 16353. https://doi.org/10.3390/ijms242216353
APA StyleWang, J., Fu, J., Zhao, Y., Liu, Q., Yan, X., & Su, J. (2023). Iron and Targeted Iron Therapy in Alzheimer’s Disease. International Journal of Molecular Sciences, 24(22), 16353. https://doi.org/10.3390/ijms242216353