
Gut Microbiome–Estrobolome Profile in Reproductive-Age Women with Endometriosis

 , ,
, ,
Abstract
:1. Introduction
2. Results
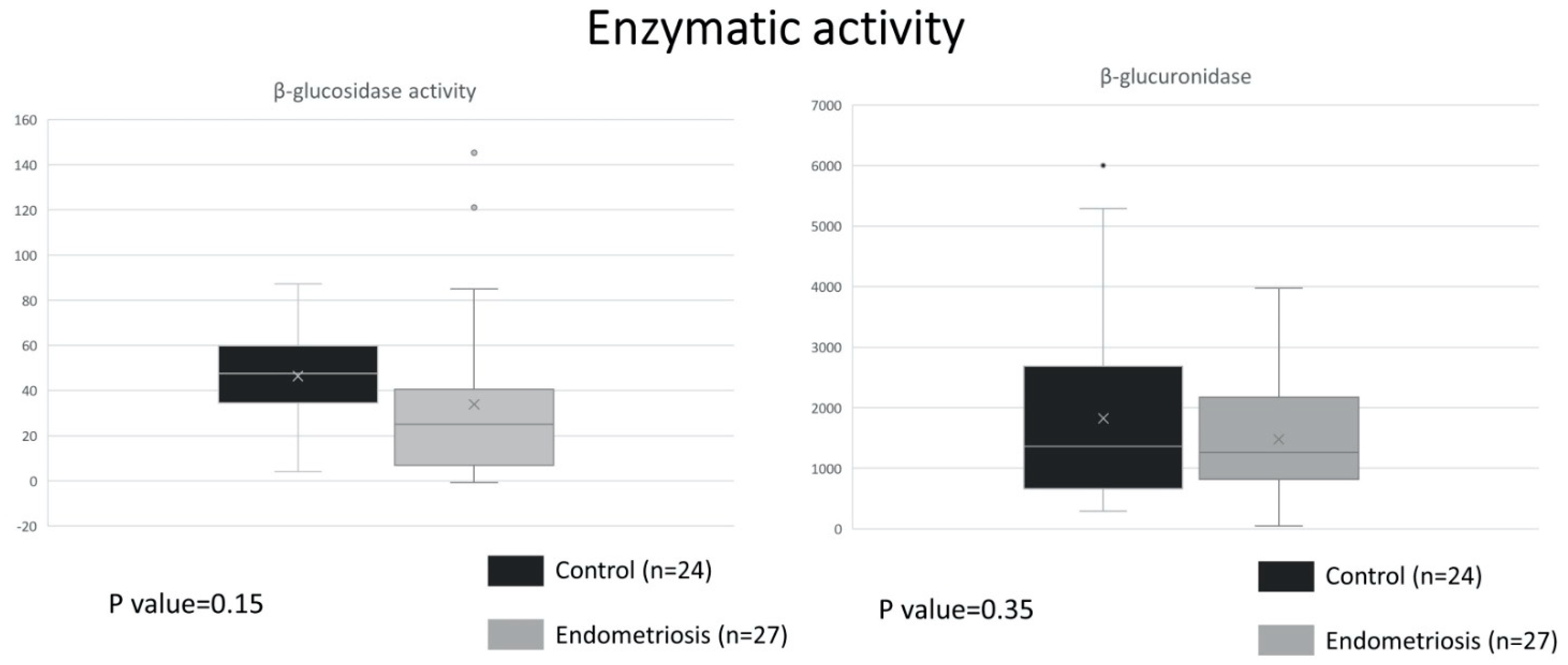
2.1. Similar ß-Glucuronidase Activity between Endometriosis and Control
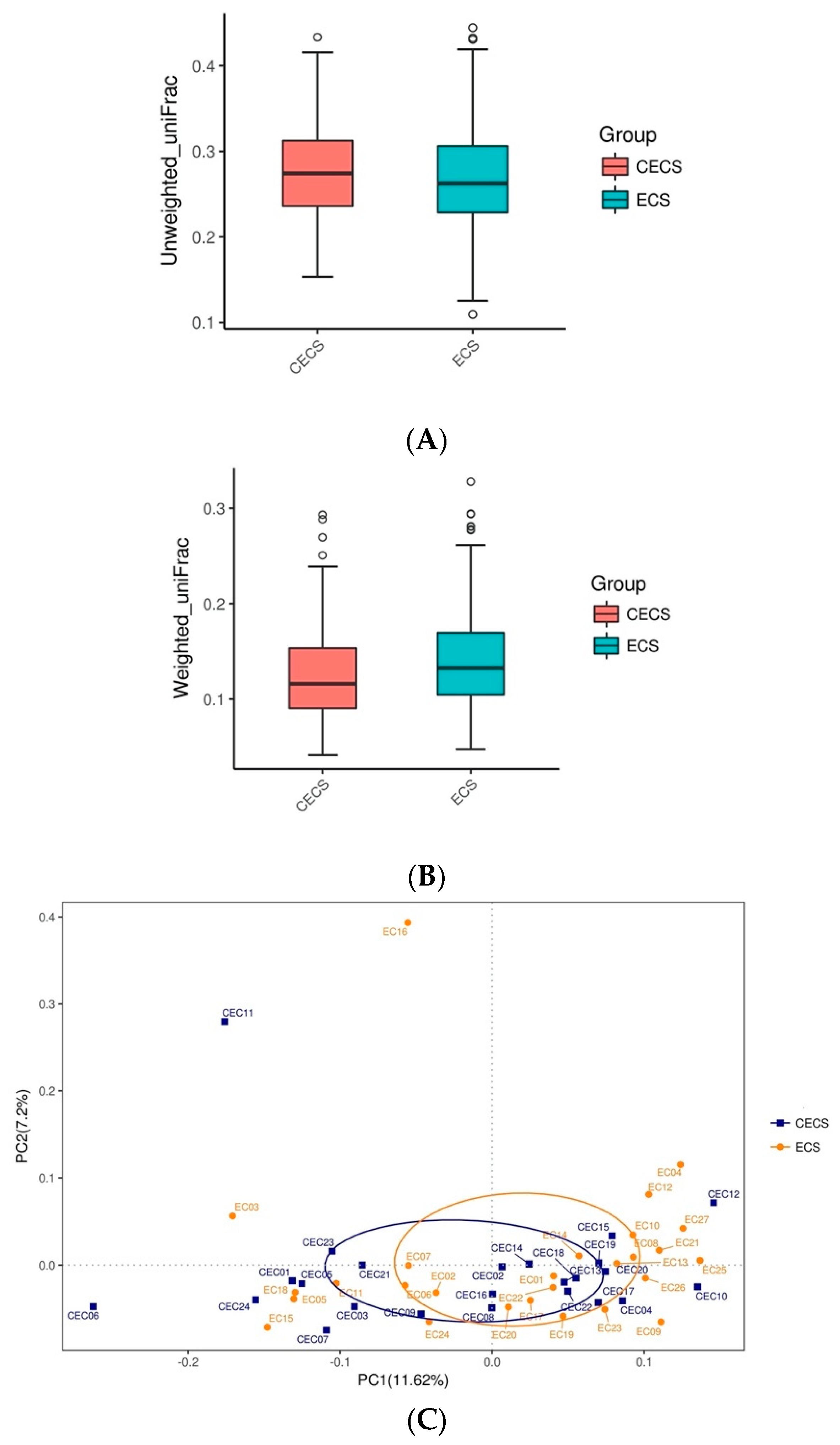
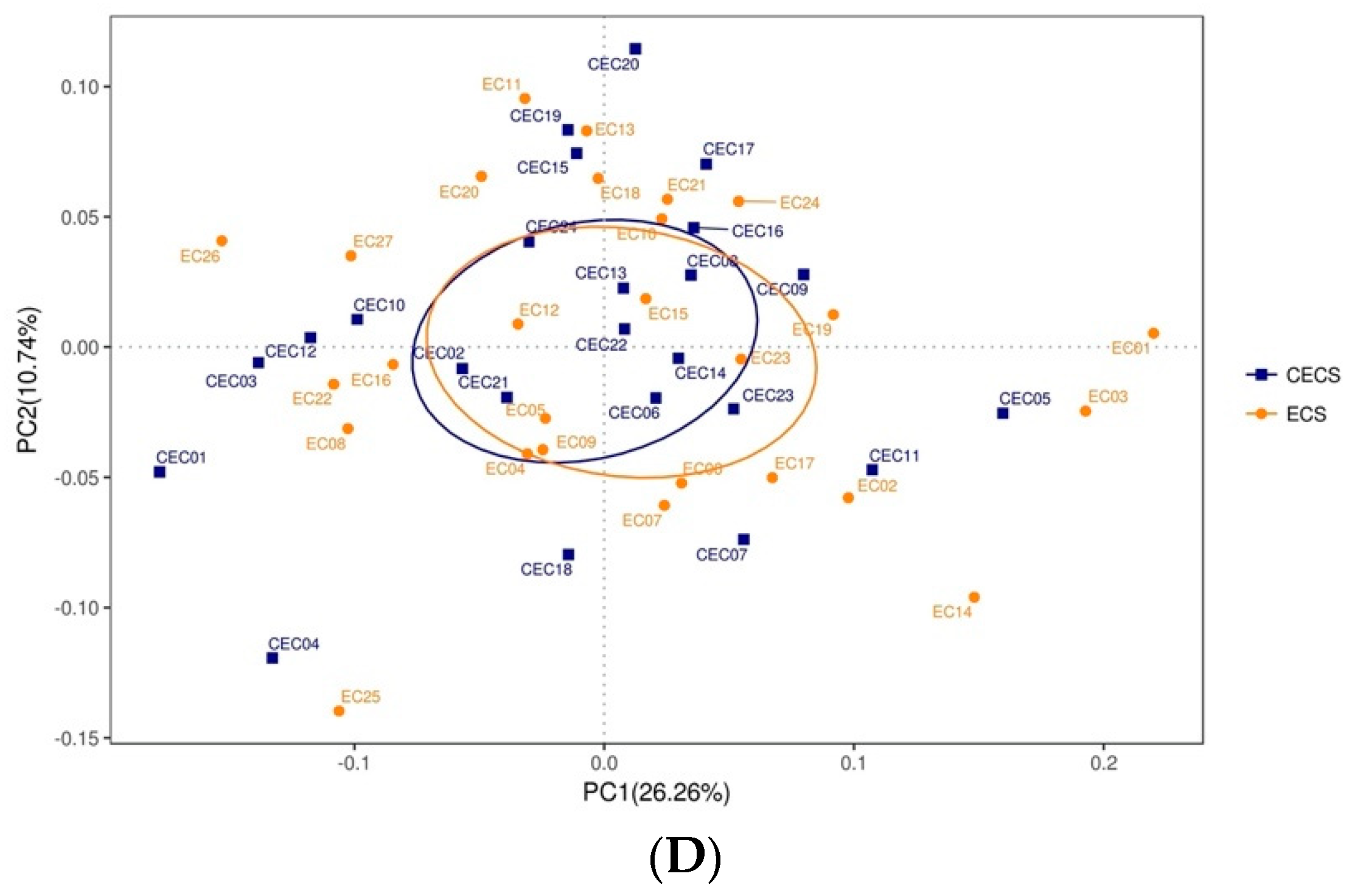
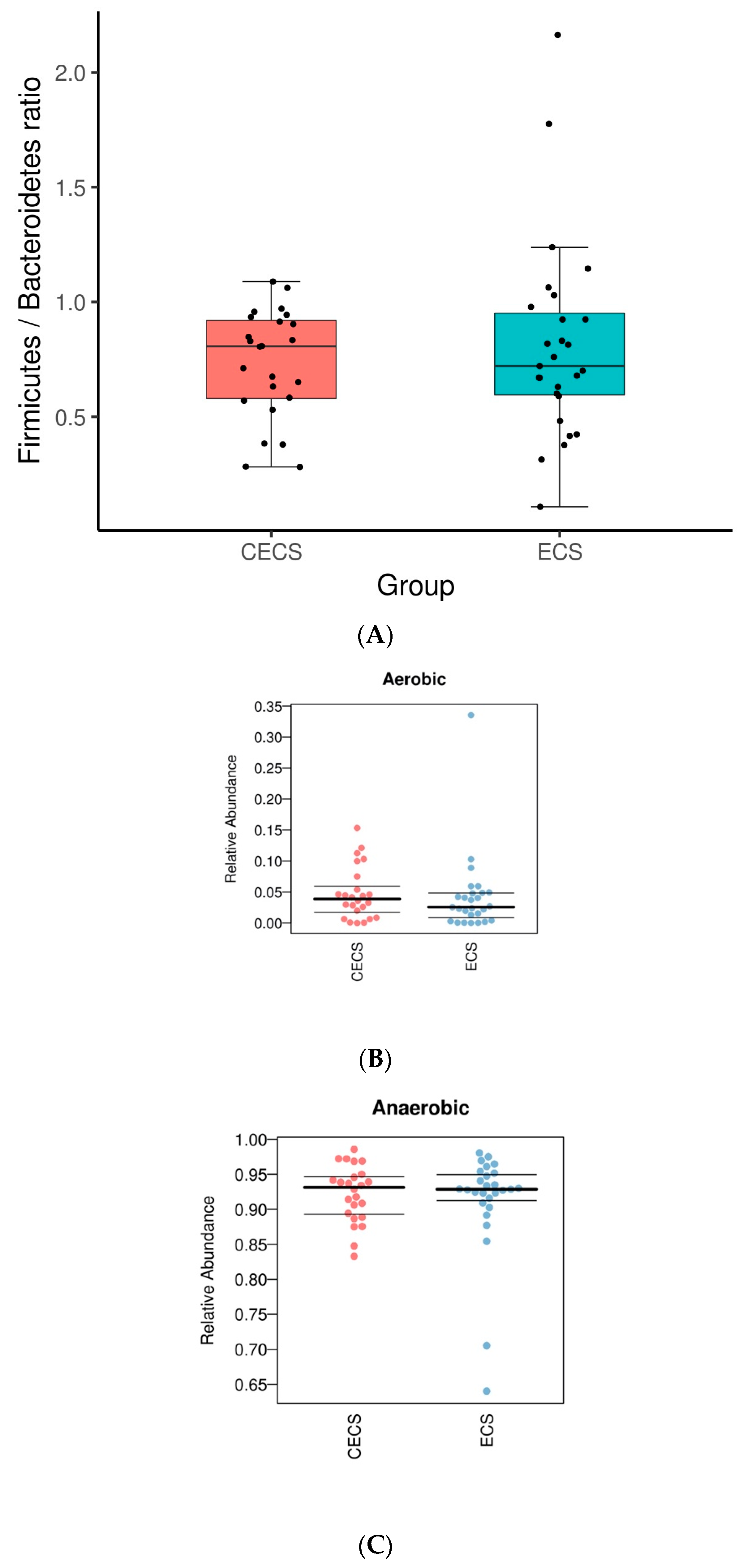
2.2. Patients with and without Endometriosis Shared Similar Bacterial Abundance, Diversity, Richness, and Evenness in the Gut
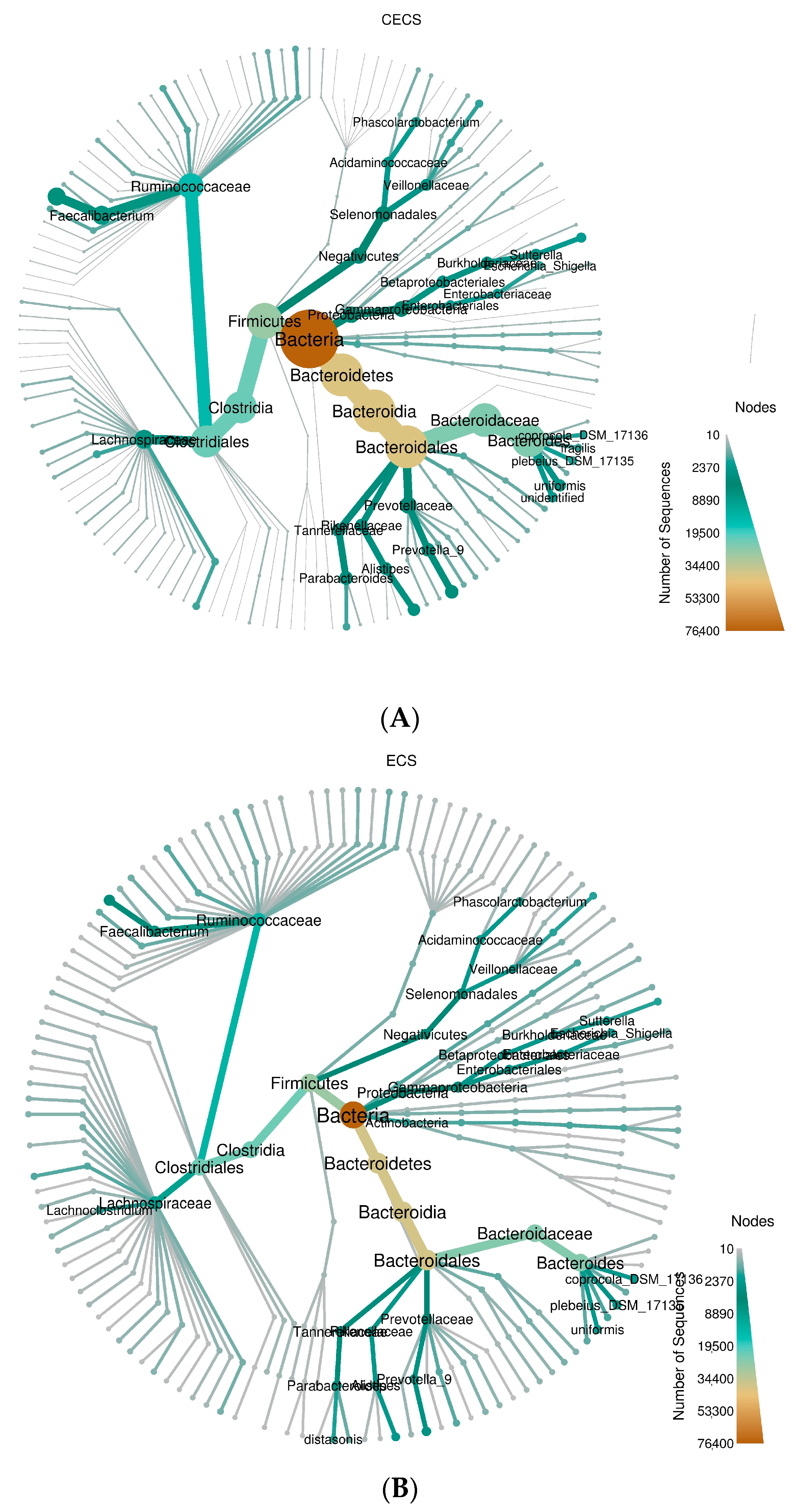
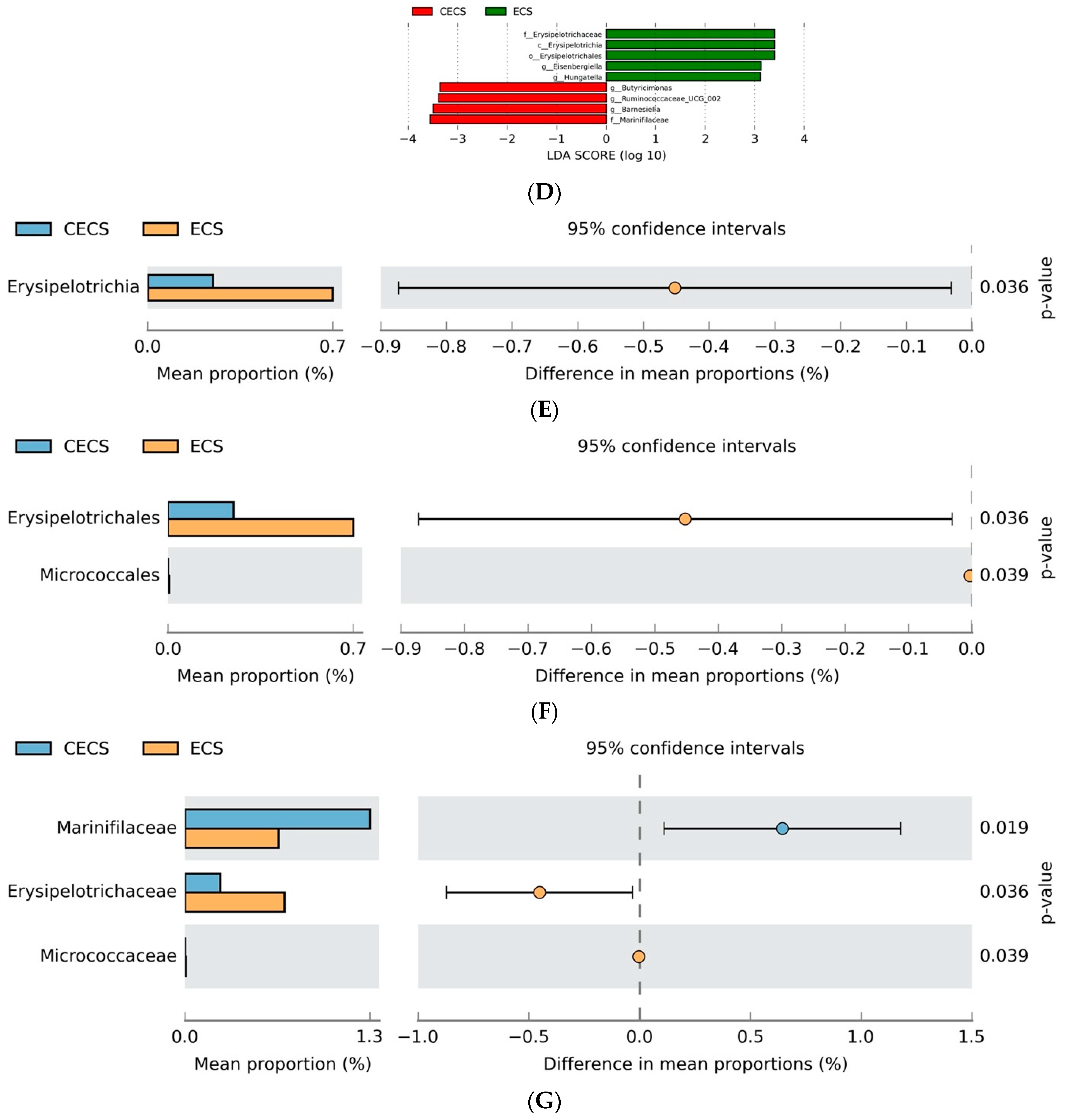
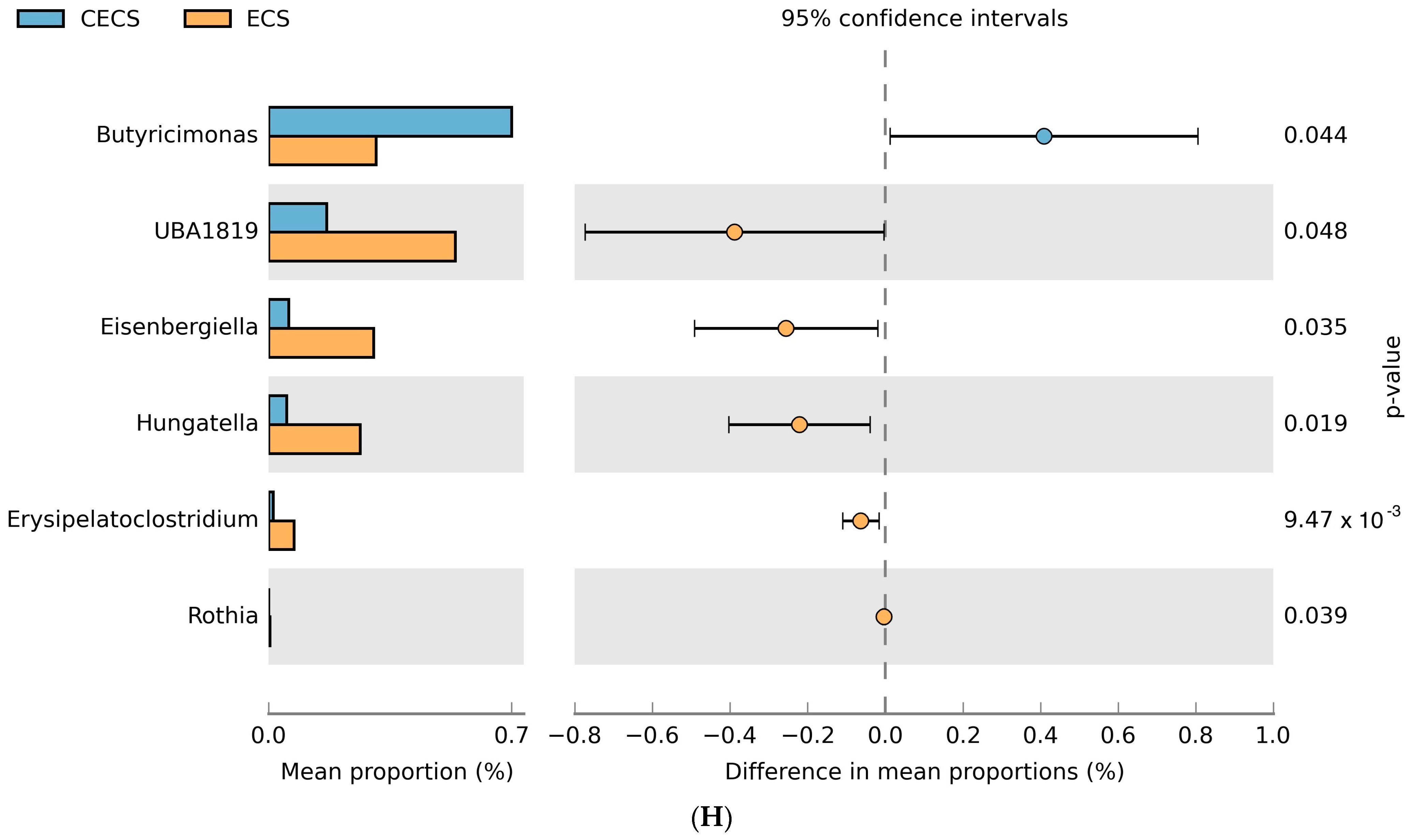
2.3. Although Dysbiosis Was Not Demonstrated in the Endometriosis Group, a Higher Composition of Certain Bacteria Was Detected
2.4. Differences in Estrogen Metabolites Were Seen in the Fecal but Not Urine Samples from Patients with Endometriosis
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Sample Collection
4.3. Protein Extraction and Enzyme Activity Assay
4.4. Genomic DNA Extraction and 16S rRNA Gene Sequencing
4.5. Liquid Chromatography and Tandem Mass Spectrometry
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, I.; Yong, P.J.; Allaire, C.; Bedaiwy, M.A. Intricate Connections between the Microbiota and Endometriosis. Int. J. Mol. Sci. 2021, 22, 5644. [Google Scholar] [CrossRef] [PubMed]
- Seksik, P.; Landman, C. Understanding Microbiome Data: A Primer for Clinicians. Dig. Dis. 2015, 33 (Suppl. S1), 11–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Madak-Erdogan, Z. Estrogen and Microbiota Crosstalk: Should We Pay Attention? Trends Endocrinol. Metab. 2016, 27, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The Gut Microbiota and Inflammation: An Overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yun, C.; Pang, Y.; Qiao, J. The impact of the gut microbiota on the reproductive and metabolic endocrine system. Gut Microbes 2021, 13, 1894070. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef]
- Khan, A.A.; Shrivastava, A.; Khurshid, M. Normal to cancer microbiome transformation and its implication in cancer diagnosis. Biochim. Biophys. Acta 2012, 1826, 331–337. [Google Scholar] [CrossRef]
- Chase, D.; Goulder, A.; Zenhausern, F.; Monk, B.; Herbst-Kralovetz, M. The vaginal and gastrointestinal microbiomes in gynecologic cancers: A review of applications in etiology, symptoms and treatment. Gynecol. Oncol. 2015, 138, 190–200. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; Brummer, R.J.M.; Rastall, R.A.; Weersma, R.K.; Harmsen, H.J.M.; Faas, M.; Eggersdorfer, M. The role of the microbiome for human health: From basic science to clinical applications. Eur. J. Nutr. 2018, 57 (Suppl. S1), 1–14. [Google Scholar] [CrossRef]
- Laschke, M.W.; Menger, M.D. The gut microbiota: A puppet master in the pathogenesis of endometriosis? Am. J. Obstet. Gynecol. 2016, 215, 68.e1–68.e4. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, M.; Hicks, C.; El-Assaad, F.; El-Omar, E.; Condous, G. Endometriosis and the microbiome: A systematic review. BJOG 2020, 127, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Salliss, M.E.; Farland, L.V.; Mahnert, N.D.; Herbst-Kralovetz, M.M. The role of gut and genital microbiota and the estrobolome in endometriosis, infertility and chronic pelvic pain. Hum. Reprod. Update 2021, 28, 92–131. [Google Scholar] [CrossRef] [PubMed]
- Qin, R.; Tian, G.; Liu, J.; Cao, L. The gut microbiota and endometriosis: From pathogenesis to diagnosis and treatment. Front. Cell. Infect. Microbiol. 2022, 12, 1069557. [Google Scholar] [CrossRef]
- Chen, L.H.; Lo, W.C.; Huang, H.Y.; Wu, H.M. A Lifelong Impact on Endometriosis: Pathophysiology and Pharmacological Treatment. Int. J. Mol. Sci. 2023, 24, 7503. [Google Scholar] [CrossRef]
- Talwar, C.; Singh, V.; Kommagani, R. The gut microbiota: A double-edged sword in endometriosisdagger. Biol. Reprod. 2022, 107, 881–901. [Google Scholar] [CrossRef]
- Ellis, K.; Munro, D.; Clarke, J. Endometriosis Is Undervalued: A Call to Action. Front. Glob. Womens Health 2022, 3, 902371. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar]
- Dabek, M.; McCrae, S.I.; Stevens, V.J.; Duncan, S.H.; Louis, P. Distribution of beta-glucosidase and beta-glucuronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol. Ecol. 2008, 66, 487–495. [Google Scholar] [CrossRef]
- Parida, S.; Sharma, D. The Microbiome-Estrogen Connection and Breast Cancer Risk. Cells 2019, 8, 1642. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ding, Q.; Zhang, W.; Kang, M.; Ma, J.; Zhao, L. Gut microbial beta-glucuronidase: A vital regulator in female estrogen metabolism. Gut Microbes 2023, 15, 2236749. [Google Scholar] [CrossRef]
- Wei, Y.; Tan, H.; Yang, R.; Yang, F.; Liu, D.; Huang, B.; OuYang, L.; Lei, S.; Wang, Z.; Jiang, S.; et al. Gut dysbiosis-derived beta-glucuronidase promotes the development of endometriosis. Fertil. Steril. 2023, 120, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Li, D.; Zhang, Z.; Sun, H.; An, M.; Wang, G. Endometriosis induces gut microbiota alterations in mice. Hum. Reprod. 2018, 33, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Sun, S.; Bi, Y.; Ding, J.; Cheng, W.; Yu, J.; Zhou, L.; Li, M.; Yu, C. Correlation of fecal metabolomics and gut microbiota in mice with endometriosis. Am. J. Reprod. Immunol. 2020, 84, e13307. [Google Scholar] [CrossRef] [PubMed]
- Chadchan, S.B.; Cheng, M.; Parnell, L.A.; Yin, Y.; Schriefer, A.; Mysorekar, I.U.; Kommagani, R. Antibiotic therapy with metronidazole reduces endometriosis disease progression in mice: A potential role for gut microbiota. Hum. Reprod. 2019, 34, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, J.; Weis, S.; Schafer, K.H.; Menger, M.D.; Kohl, M.; Egert, M.; Laschke, M.W. Effect of endometriosis on the fecal bacteriota composition of mice during the acute phase of lesion formation. PLoS ONE 2019, 14, e0226835. [Google Scholar] [CrossRef]
- Wessels, J.M.; Dominguez, M.A.; Leyland, N.A.; Agarwal, S.K.; Foster, W.G. Endometrial microbiota is more diverse in people with endometriosis than symptomatic controls. Sci. Rep. 2021, 11, 18877. [Google Scholar] [CrossRef]
- Ata, B.; Yildiz, S.; Turkgeldi, E.; Brocal, V.P.; Dinleyici, E.C.; Moya, A.; Urman, B. The Endobiota Study: Comparison of Vaginal, Cervical and Gut Microbiota Between Women with Stage 3/4 Endometriosis and Healthy Controls. Sci. Rep. 2019, 9, 2204. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, A.R.; Borrelli, G.M.; Martins, C.O.; Kallas, E.G.; Sanabani, S.S.; Griffith, L.G.; Alm, E.J.; Abrao, M.S. The Vaginal Microbiome as a Tool to Predict rASRM Stage of Disease in Endometriosis: A Pilot Study. Reprod. Sci. 2020, 27, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Beaud, D.; Tailliez, P.; Anba-Mondoloni, J. Genetic characterization of the beta-glucuronidase enzyme from a human intestinal bacterium, Ruminococcus gnavus. Microbiology 2005, 151 Pt 7, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2015, 3, 207–215. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef]
- Rajilic-Stojanovic, M.; Biagi, E.; Heilig, H.G.; Kajander, K.; Kekkonen, R.A.; Tims, S.; de Vos, W.M. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 2011, 141, 1792–1801. [Google Scholar] [CrossRef]
- Greiner, T.; Backhed, F. Effects of the gut microbiota on obesity and glucose homeostasis. Trends Endocrinol. Metab. 2011, 22, 117–123. [Google Scholar] [CrossRef]
- Tiffany, C.R.; Lee, J.Y.; Rogers, A.W.L.; Olsan, E.E.; Morales, P.; Faber, F.; Baumler, A.J. The metabolic footprint of Clostridia and Erysipelotrichia reveals their role in depleting sugar alcohols in the cecum. Microbiome 2021, 9, 174. [Google Scholar] [CrossRef]
- Pettersen, V.K.; Steinsland, H.; Wiker, H.G. Distinct Metabolic Features of Pathogenic Escherichia coli and Shigella spp. Determined by Label-Free Quantitative Proteomics. Proteomics 2021, 21, e2000072. [Google Scholar] [CrossRef]
- Sanchez-Garrido, J.; Ruano-Gallego, D.; Choudhary, J.S.; Frankel, G. The type III secretion system effector network hypothesis. Trends Microbiol. 2022, 30, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolite: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Greenwood-Van Meerveld, B.; Johnson, A.C.; Travagli, R.A. Role of estrogen and stress on the brain-gut axis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G203–G209. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Pfeiffer, R.M.; Byrd, D.A.; Wan, Y.; Ansong, D.; Clegg-Lamptey, J.N.; Wiafe-Addai, B.; Edusei, L.; Adjei, E.; Titiloye, N.; et al. Associations of Circulating Estrogens and Estrogen Metabolites with Fecal and Oral Microbiome in Postmenopausal Women in the Ghana Breast Health Study. Microbiol. Spectr. 2023, 11, e0157223. [Google Scholar] [CrossRef] [PubMed]
- Coburn, S.B.; Stanczyk, F.Z.; Falk, R.T.; McGlynn, K.A.; Brinton, L.A.; Sampson, J.; Bradwin, G.; Xu, X.; Trabert, B. Comparability of serum, plasma, and urinary estrogen and estrogen metabolite measurements by sex and menopausal status. Cancer Causes Control 2019, 30, 75–86. [Google Scholar] [CrossRef]
- Liu, B.N.; Liu, X.T.; Liang, Z.H.; Wang, J.H. Gut microbiota in obesity. World J. Gastroenterol. 2021, 27, 3837–3850. [Google Scholar] [CrossRef]
- Van Hul, M.; Cani, P.D. The gut microbiota in obesity and weight management: Microbes as friends or foe? Nat. Rev. Endocrinol. 2023, 19, 258–271. [Google Scholar] [CrossRef]
- Muga, M.A.; Owili, P.O.; Hsu, C.Y.; Rau, H.H.; Chao, J.C. Dietary patterns, gender, and weight status among middle-aged and older adults in Taiwan: A cross-sectional study. BMC Geriatr. 2017, 17, 268. [Google Scholar] [CrossRef]
- Shafrir, A.L.; Farland, L.V.; Shah, D.K.; Harris, H.R.; Kvaskoff, M.; Zondervan, K.; Missmer, S.A. Risk for and consequences of endometriosis: A critical epidemiologic review. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 51, 1–15. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Control (n = 24) | Endometriosis (n = 27) | p-Value |
|---|---|---|---|
| Age, yr (mean ± SEM) | 37.7 ± 1.3 | 38.1 ± 1.0 | 0.770 |
| BMI, kg/m2 (mean ± SEM) | 24.04 ± 0.87 | 21.77 ± 0.73 | 0.051 |
| Gravida (median, IQR) | 1.0 (0.0–2.0) | 1.0 (0.0–2.0) | 0.825 |
| Parity (median, IQR) | 0.0 (0.0–1.75) | 0.0 (0.0–1.0) | 0.975 |
| CA-125, U/mL (mean ± SEM) | 21.54 ± 3.12 | 70.07 ± 9.07 | <0.001 * |
| rAFS score (mean ± SEM) | NA | 31.1 ± 6.4 | NA |
| Endometriosis stage (%) I | NA | 29.6 (8/27) | NA |
| II | NA | 14.8 (4/27) | NA |
| III | NA | 25.9 (7/27) | NA |
| IV | NA | 29.6 (8/27) | NA |
| Stool | Urine | |||
|---|---|---|---|---|
| Fold (Endometriosis/Control) | p-Value | Fold (Endometriosis/Control) | p-Value | |
| Estradiol | 2.093 | 0.121 | 1.218 | 0.224 |
| Estriol | 2.002 | 0.011 * | N.D. | N.D. |
| Estrone | 1.220 | 0.059 | 0.756 | 0.902 |
| 16-epiestriol | 1.681 | 0.018 * | 1.292 | 0.183 |
| 16α-hydroxyestrone | 2.991 | 0.016 * | 2.894 | 0.404 |
| 17-epiestriol | 0.834 | 0.21 | 1.189 | 0.328 |
| 2-hydroxyestradiol | 0.851 | 0.829 | 0.968 | 0.546 |
| 2-hydroxyestrone | 1.049 | 0.282 | 0.890 | 0.794 |
| 4-hydroxyestrone | 0.669 | 0.468 | 0.941 | 0.962 |
| 2-methoxyestradiol | 1.799 | 0.035 * | N.D. | N.D. |
| 4-methoxyestradiol | 1.578 | 0.196 | N.D. | N.D. |
| 2-methoxyestrone | 0.389 | 0.197 | 1.566 | 0.374 |
| 4-methoxyestrone | N.D. | N.D. | 1.712 | 0.595 |
| 2-hydroxyestrone-3 methyl ether | 0.558 | 0.84 | N.D. | N.D. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pai, A.H.-Y.; Wang, Y.-W.; Lu, P.-C.; Wu, H.-M.; Xu, J.-L.; Huang, H.-Y. Gut Microbiome–Estrobolome Profile in Reproductive-Age Women with Endometriosis. Int. J. Mol. Sci. 2023, 24, 16301. https://doi.org/10.3390/ijms242216301
Pai AH-Y, Wang Y-W, Lu P-C, Wu H-M, Xu J-L, Huang H-Y. Gut Microbiome–Estrobolome Profile in Reproductive-Age Women with Endometriosis. International Journal of Molecular Sciences. 2023; 24(22):16301. https://doi.org/10.3390/ijms242216301
Chicago/Turabian StylePai, Angel Hsin-Yu, Yi-Wen Wang, Pei-Chen Lu, Hsien-Ming Wu, Jia-Ling Xu, and Hong-Yuan Huang. 2023. "Gut Microbiome–Estrobolome Profile in Reproductive-Age Women with Endometriosis" International Journal of Molecular Sciences 24, no. 22: 16301. https://doi.org/10.3390/ijms242216301
APA StylePai, A. H.-Y., Wang, Y.-W., Lu, P.-C., Wu, H.-M., Xu, J.-L., & Huang, H.-Y. (2023). Gut Microbiome–Estrobolome Profile in Reproductive-Age Women with Endometriosis. International Journal of Molecular Sciences, 24(22), 16301. https://doi.org/10.3390/ijms242216301