
Epidermal Growth Factor Receptors in Vascular Endothelial Cells Contribute to Functional Hyperemia in the Brain

Abstract
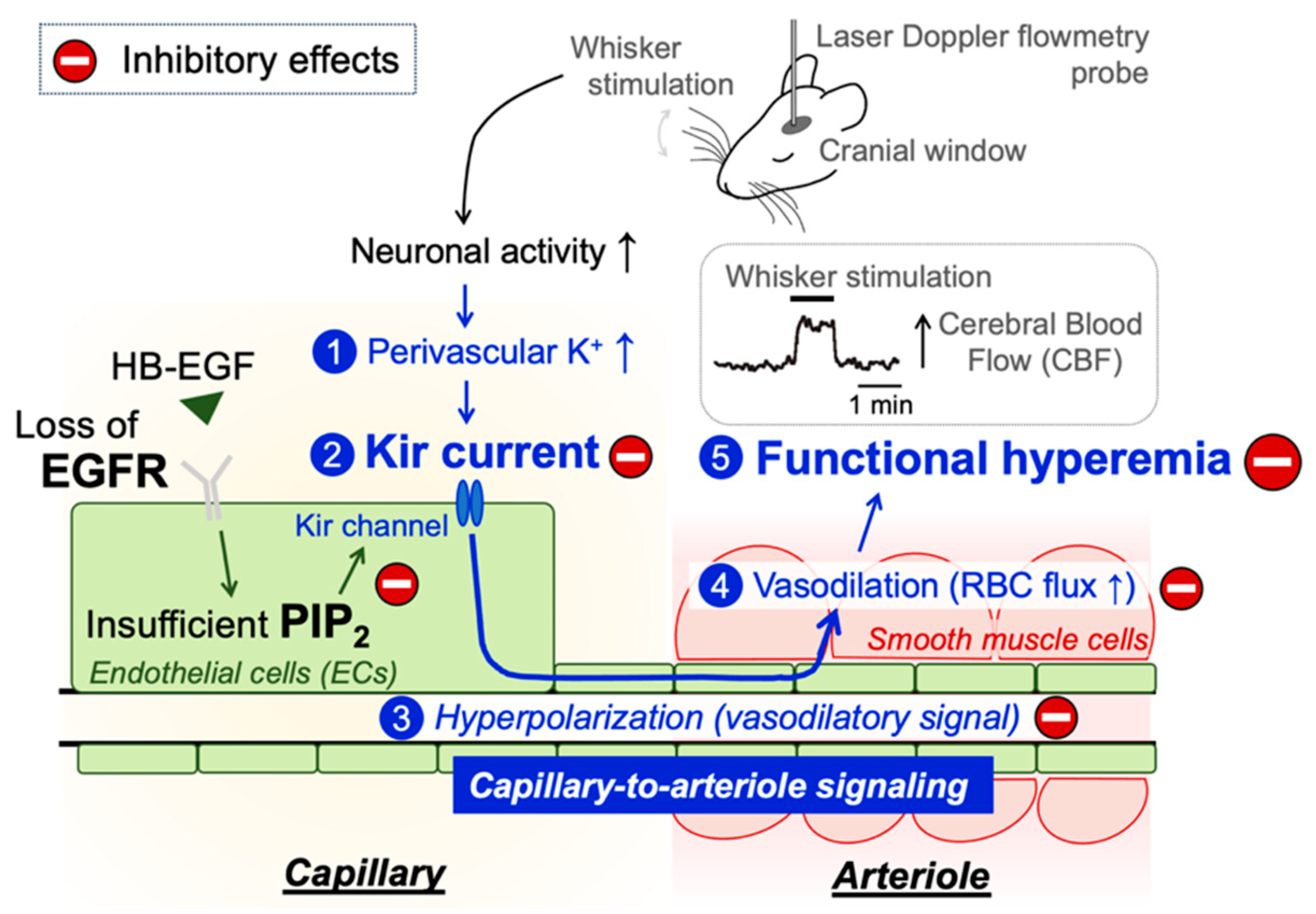
:1. Introduction
2. Results
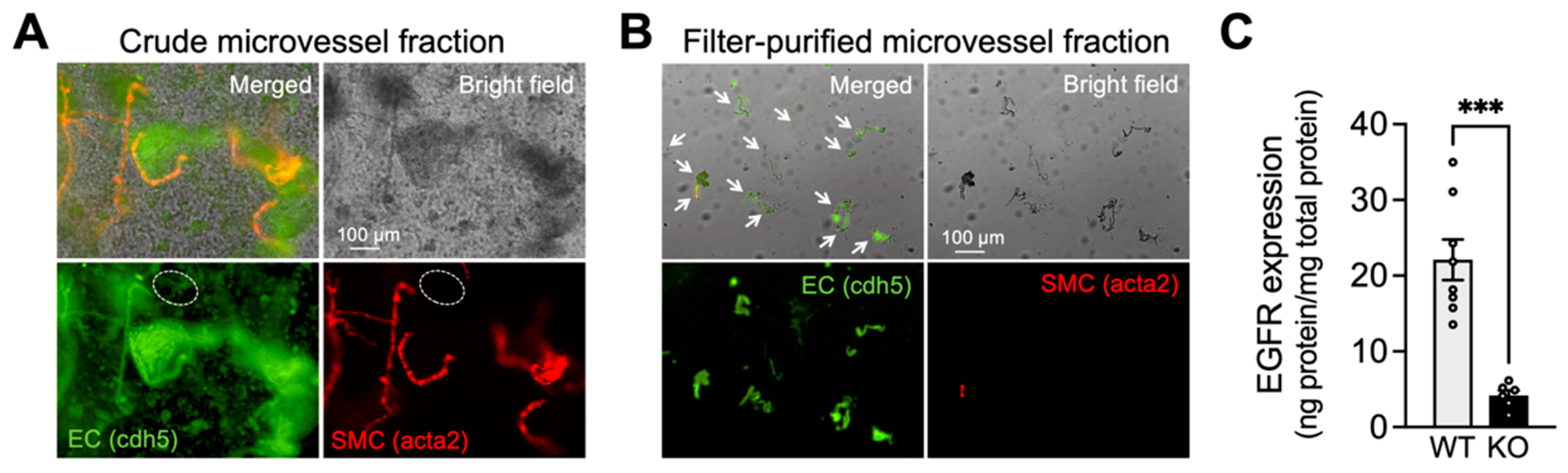
2.1. Characterization of Capillary ECs from EC-EGFR-KO Mice
2.2. Impaired Functional Hyperemia in EC-EGFR-KO Mice
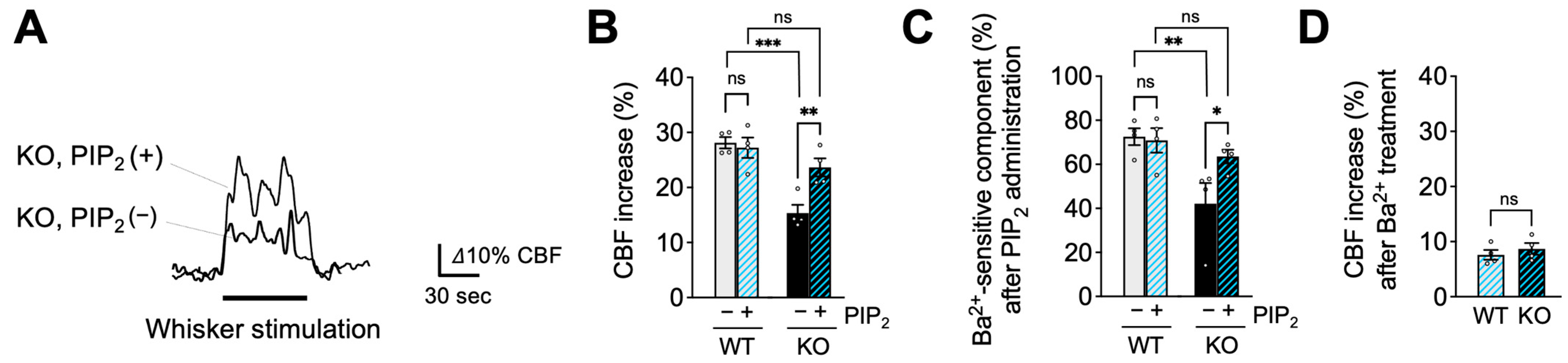
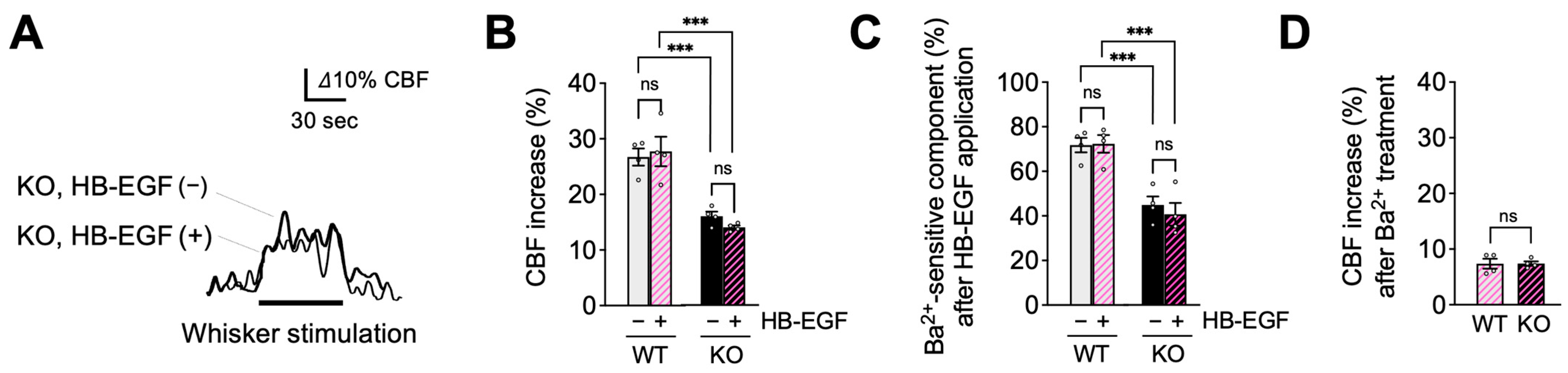
2.3. PIP2, but Not HB-EGF, Restores Functional Hyperemia in EC-EGFR-KO Mice
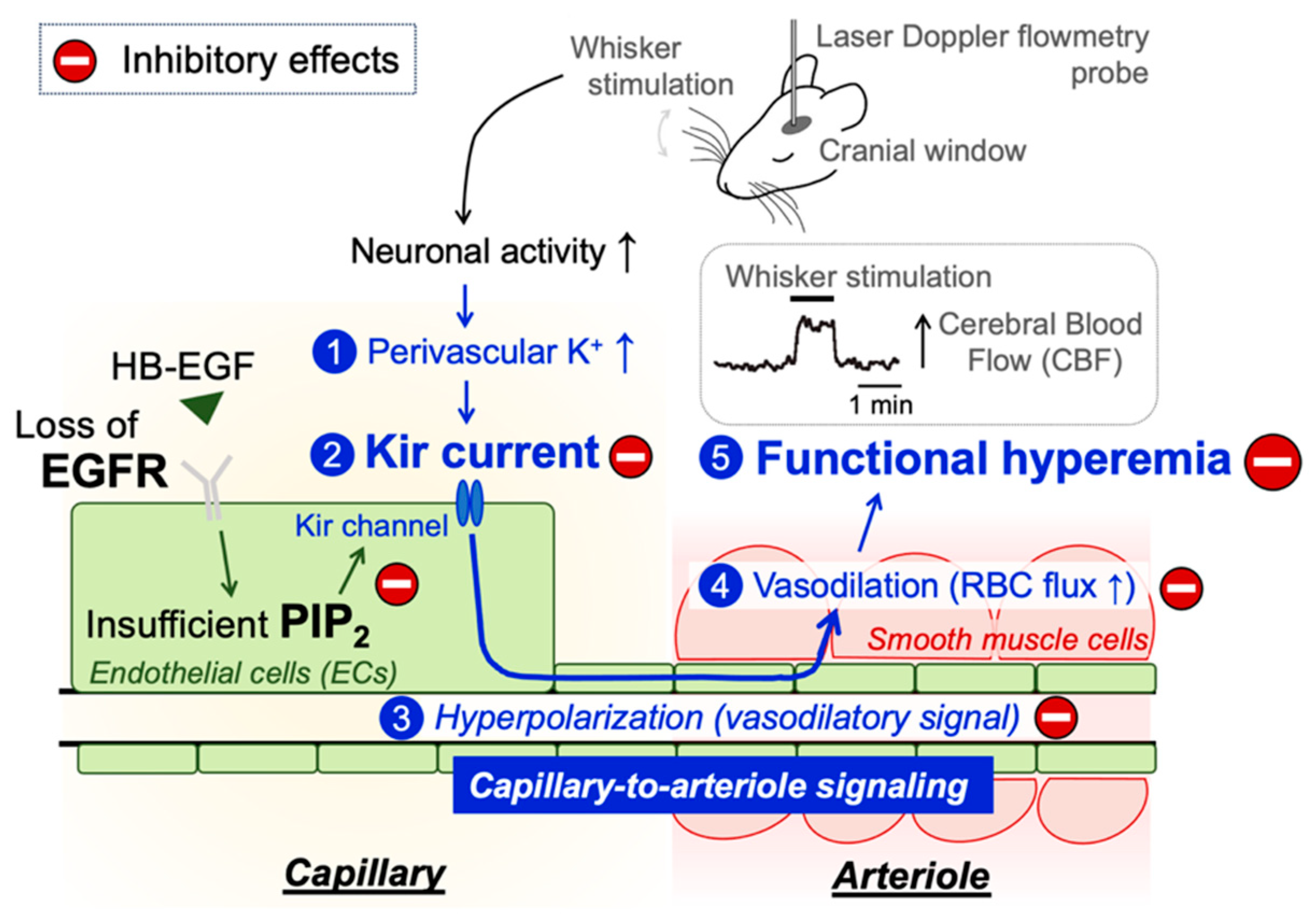
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Isolation of Brain Parenchymal Capillaries
4.3. Epi-Fluorescent Microscopy
4.4. Measurement of EGFR Expression
4.5. Laser Doppler Flowmetry
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Lecrux, C.; Hamel, E. The neurovascular unit in brain function and disease. Acta Physiol. 2011, 203, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.M.; Iadecola, C.; Carnevale, D. Hypertension, Neurovascular Dysfunction, and Cognitive Impairment. Hypertension 2023, 80, 22–34. [Google Scholar] [CrossRef]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef]
- Filosa, J.A.; Blanco, V.M. Neurovascular coupling in the mammalian brain. Exp. Physiol. 2007, 92, 641–646. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. Elife 2021, 10, e63040. [Google Scholar] [CrossRef]
- Bracko, O.; Harraz, O.F. Editorial: Small and mighty: Brain capillaries in health and disease. Front. Mol. Neurosci. 2022, 15, 1108978. [Google Scholar] [CrossRef]
- Jackson, W.F. Boosting the signal: Endothelial inward rectifier K+ channels. Microcirculation 2017, 24, e12319. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.B.; Tao, X.; MacKinnon, R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 2011, 477, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.L.; Feng, S.; Hilgemann, D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 1998, 391, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Harraz, O.F.; Longden, T.A.; Dabertrand, F.; Hill-Eubanks, D.; Nelson, M.T. Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc. Natl. Acad. Sci. USA 2018, 115, E3569–E3577. [Google Scholar] [CrossRef] [PubMed]
- Dabertrand, F.; Harraz, O.F.; Koide, M.; Longden, T.A.; Rosehart, A.C.; Hill-Eubanks, D.C.; Joutel, A.; Nelson, M.T. PIP2 corrects cerebral blood flow deficits in small vessel disease by rescuing capillary Kir2.1 activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2025998118. [Google Scholar] [CrossRef]
- Joutel, A.; Corpechot, C.; Ducros, A.; Vahedi, K.; Chabriat, H.; Mouton, P.; Alamowitch, S.; Domenga, V.; Cécillion, M.; Maréchal, E.; et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 1996, 383, 707–710. [Google Scholar] [CrossRef]
- Joutel, A.; Vahedi, K.; Corpechot, C.; Troesch, A.; Chabriat, H.; Vayssière, C.; Cruaud, C.; Maciazek, J.; Weissenbach, J.; Bousser, M.-G.; et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 1997, 350, 1511–1515. [Google Scholar] [CrossRef]
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.G. Cadasil. Lancet Neurol. 2009, 8, 643–653. [Google Scholar] [CrossRef]
- Joutel, A. Prospects for Diminishing the Impact of Nonamyloid Small-Vessel Diseases of the Brain. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 437–456. [Google Scholar] [CrossRef]
- Chabriat, H.; Joutel, A.; Tournier-Lasserve, E.; Bousser, M.G. CADASIL: Yesterday, today, tomorrow. Eur. J. Neurol. 2020, 27, 1588–1595. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Vanlandewijck, M.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci. Data 2018, 5, 180160. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, T.; Ragno, M.; Paolinelli, F.; Castellucci, C.; Scarpelli, M.; Morroni, M. CADASIL: Ultrastructural insights into the morphology of granular osmiophilic material. Brain Behav. 2017, 7, e00624. [Google Scholar] [CrossRef]
- Gravesteijn, G.; Munting, L.P.; Overzier, M.; Mulder, A.A.; Hegeman, I.; Derieppe, M.; Koster, A.J.; van Duinen, S.G.; Meijer, O.C.; Aartsma-Rus, A.; et al. Progression and Classification of Granular Osmiophilic Material (GOM) Deposits in Functionally Characterized Human NOTCH3 Transgenic Mice. Transl. Stroke Res. 2020, 11, 517–527. [Google Scholar] [CrossRef]
- Monet-Leprêtre, M.; Haddad, I.; Baron-Menguy, C.; Fouillot-Panchal, M.; Riani, M.; Domenga-Denier, V.; Dussaule, C.; Cognat, E.; Vinh, J.; Joutel, A. Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: A new pathomechanism in, CADASIL. Brain 2013, 136, 1830–1845. [Google Scholar] [CrossRef]
- Baron-Menguy, C.; Domenga-Denier, V.; Ghezali, L.; Faraci, F.M.; Joutel, A. Increased Notch3 Activity Mediates Pathological Changes in Structure of Cerebral Arteries. Hypertension 2017, 69, 60–70. [Google Scholar] [CrossRef]
- Schoemaker, D.; Arboleda-Velasquez, J.F. Notch3 Signaling and Aggregation as Targets for the Treatment of CADASIL and Other NOTCH3-Associated Small-Vessel Diseases. Rev. Am. J. Pathol. 2021, 191, 1856–1870. [Google Scholar] [CrossRef]
- Capone, C.; Dabertrand, F.; Baron-Menguy, C.; Chalaris, A.; Ghezali, L.; Domenga-Denier, V.; Schmidt, S.; Huneau, C.; Rose-John, S.; Nelson, M.T.; et al. Mechanistic insights into a TIMP3-sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics. eLife 2016, 5, e17536. [Google Scholar] [CrossRef]
- Dabertrand, F.; Krøigaard, C.; Bonev, A.D.; Cognat, E.; Dalsgaard, T.; Domenga-Denier, V.; Hill-Eubanks, D.C.; Brayden, J.E.; Joutel, A.; Nelson, M.T. Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease. Proc. Natl. Acad. Sci. USA 2015, 112, E796–E805. [Google Scholar] [CrossRef]
- Joutel, A.; Monet-Leprêtre, M.; Gosele, C.; Baron-Menguy, C.; Hammes, A.; Schmidt, S.; Lemaire-Carrette, B.; Domenga, V.; Schedl, A.; Lacombe, P.; et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J. Clin. Investig. 2010, 120, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Uchida, H.; Smith, H.; Ito, A.; Sanchez, T. The isolation and molecular characterization of cerebral microvessels. Nat. Protoc. 2019, 14, 3059–3081. [Google Scholar] [CrossRef] [PubMed]
- Schreier, B.; Stern, C.; Dubourg, V.; Nolze, A.; Rabe, S.; Mildenberger, S.; Wickenhauser, C.; Gekle, M. Endothelial epidermal growth factor receptor is of minor importance for vascular and renal function and obesity-induced dysfunction in mice. Sci. Rep. 2021, 11, 7269. [Google Scholar] [CrossRef]
- Koide, M.; Harraz, O.F.; Dabertrand, F.; Longden, T.A.; Ferris, H.R.; Wellman, G.C.; Hill-Eubanks, D.C.; Greenstein, A.S.; Nelson, M.T. Differential restoration of functional hyperemia by antihypertensive drug classes in hypertension-related cerebral small vessel disease. J. Clin. Investig. 2021, 131, e149029. [Google Scholar] [CrossRef]
- Merlino, G.T.; Xu, Y.H.; Ishii, S.; Clark, A.J.; Semba, K.; Toyoshima, K.; Yamamoto, T.; Pastan, I. Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. Science 1984, 224, 417–419. [Google Scholar] [CrossRef]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Schreier, B.; Gekle, M.; Grossmann, C. Role of epidermal growth factor receptor in vascular structure and function. Curr. Opin. Nephrol. Hypertens. 2014, 23, 113–121. [Google Scholar] [CrossRef]
- Roberts, R.B.; Arteaga, C.L.; Threadgill, D.W. Modeling the cancer patient with genetically engineered mice: Prediction of toxicity from molecule-targeted therapies. Cancer Cell. 2004, 5, 115–120. [Google Scholar] [CrossRef]
- Sancho, M.; Fabris, S.; Hald, B.O.; Brett, S.E.; Sandow, S.L.; Poepping, T.L.; Welsh, D.G. Membrane Lipid-KIR2.x Channel Interactions Enable Hemodynamic Sensing in Cerebral Arteries. Arter. Thromb. Vasc. Biol. 2019, 39, 1072–1087. [Google Scholar] [CrossRef]
- Fujimoto, M.; Shiba, M.; Kawakita, F.; Liu, L.; Nakasaki, A.; Shimojo, N.; Imanaka-Yoshida, K.; Yoshida, T.; Suzuki, H. Epidermal growth factor-like repeats of tenascin-C-induced constriction of cerebral arteries via activation of epidermal growth factor receptors in rats. Brain Res. 2016, 1642, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Penar, P.L.; Tranmer, B.I.; Wellman, G.C. Heparin-binding EGF-like growth factor mediates oxyhemoglobin-induced suppression of voltage-dependent potassium channels in rabbit cerebral artery myocytes. Am. J. Physiol. Circ. Physiol. 2007, 293, H1750-9. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Du, M.; Lopez-Campistrous, A.; Fernandez-Patron, C. Agonist-induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ. Res. 2004, 94, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Chansel, D.; Ciroldi, M.; Vandermeersch, S.; Jackson, L.F.; Gomez, A.M.; Henrion, D.; Lee, D.C.; Coffman, T.M.; Richard, S.; Dussaule, J.; et al. Heparin binding EGF is necessary for vasospastic response to endothelin. Faseb J. 2006, 20, 1936–1938. [Google Scholar] [CrossRef]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Lüthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483–486. [Google Scholar] [CrossRef]
- Lee, T.C.; Threadgill, D.W. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 2009, 47, 85–92. [Google Scholar] [CrossRef]
- Lee, F.K.; Lee, J.C.; Shui, B.; Reining, S.; Jibilian, M.; Small, D.M.; Jones, J.S.; Allan-Rahill, N.H.; Lamont, M.R.; Rizzo, M.A. Genetically engineered mice for combinatorial cardiovascular optobiology. Elife 2021, 10, e67858. [Google Scholar] [CrossRef]
- Bethge, P.; Carta, S.; Lorenzo, D.A.; Egolf, L.; Goniotaki, D.; Madisen, L.; Voigt, F.F.; Chen, J.L.; Schneider, B.; Ohkura, M.; et al. An R-CaMP1.07 reporter mouse for cell-type-specific expression of a sensitive red fluorescent calcium indicator. PLoS ONE 2017, 12, e0179460. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT (n = 8) | EC-EGFR-KO (n = 8) | |
|---|---|---|
| Age (weeks old) | 20.2 ± 2.2 | 20.8 ± 1.3 |
| Body weight (g) | 26.8 ± 1.2 | 27.3 ± 1.4 |
| Blood pressure (mmHg) | 91.3 ± 2.9 | 91.8 ± 1.1 |
| Heart rate (bpm) | 498 ± 8 | 491 ± 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferris, H.R.; Stine, N.C.; Hill-Eubanks, D.C.; Nelson, M.T.; Wellman, G.C.; Koide, M. Epidermal Growth Factor Receptors in Vascular Endothelial Cells Contribute to Functional Hyperemia in the Brain. Int. J. Mol. Sci. 2023, 24, 16284. https://doi.org/10.3390/ijms242216284
Ferris HR, Stine NC, Hill-Eubanks DC, Nelson MT, Wellman GC, Koide M. Epidermal Growth Factor Receptors in Vascular Endothelial Cells Contribute to Functional Hyperemia in the Brain. International Journal of Molecular Sciences. 2023; 24(22):16284. https://doi.org/10.3390/ijms242216284
Chicago/Turabian StyleFerris, Hannah R., Nathan C. Stine, David C. Hill-Eubanks, Mark T. Nelson, George C. Wellman, and Masayo Koide. 2023. "Epidermal Growth Factor Receptors in Vascular Endothelial Cells Contribute to Functional Hyperemia in the Brain" International Journal of Molecular Sciences 24, no. 22: 16284. https://doi.org/10.3390/ijms242216284
APA StyleFerris, H. R., Stine, N. C., Hill-Eubanks, D. C., Nelson, M. T., Wellman, G. C., & Koide, M. (2023). Epidermal Growth Factor Receptors in Vascular Endothelial Cells Contribute to Functional Hyperemia in the Brain. International Journal of Molecular Sciences, 24(22), 16284. https://doi.org/10.3390/ijms242216284