
A Disintegrin and Metalloproteinase with Thrombospondin Motifs 4 Regulates Pulmonary Vascular Hyperpermeability through Destruction of Glycocalyx in Acute Respiratory Distress Syndrome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
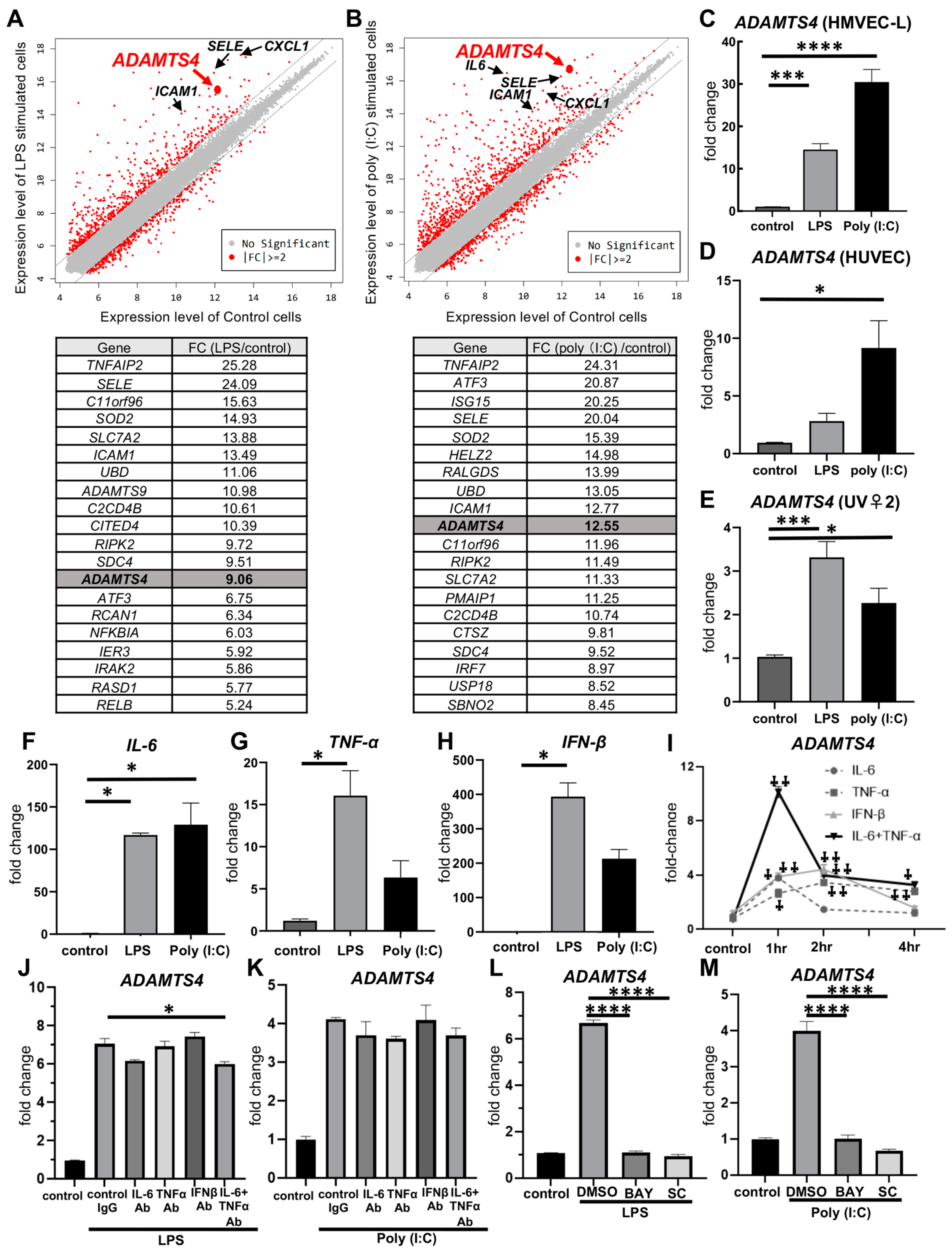
2.1. Stimulation with LPS or Poly (I:C) in Pulmonary VECs Increases ADAMTS4
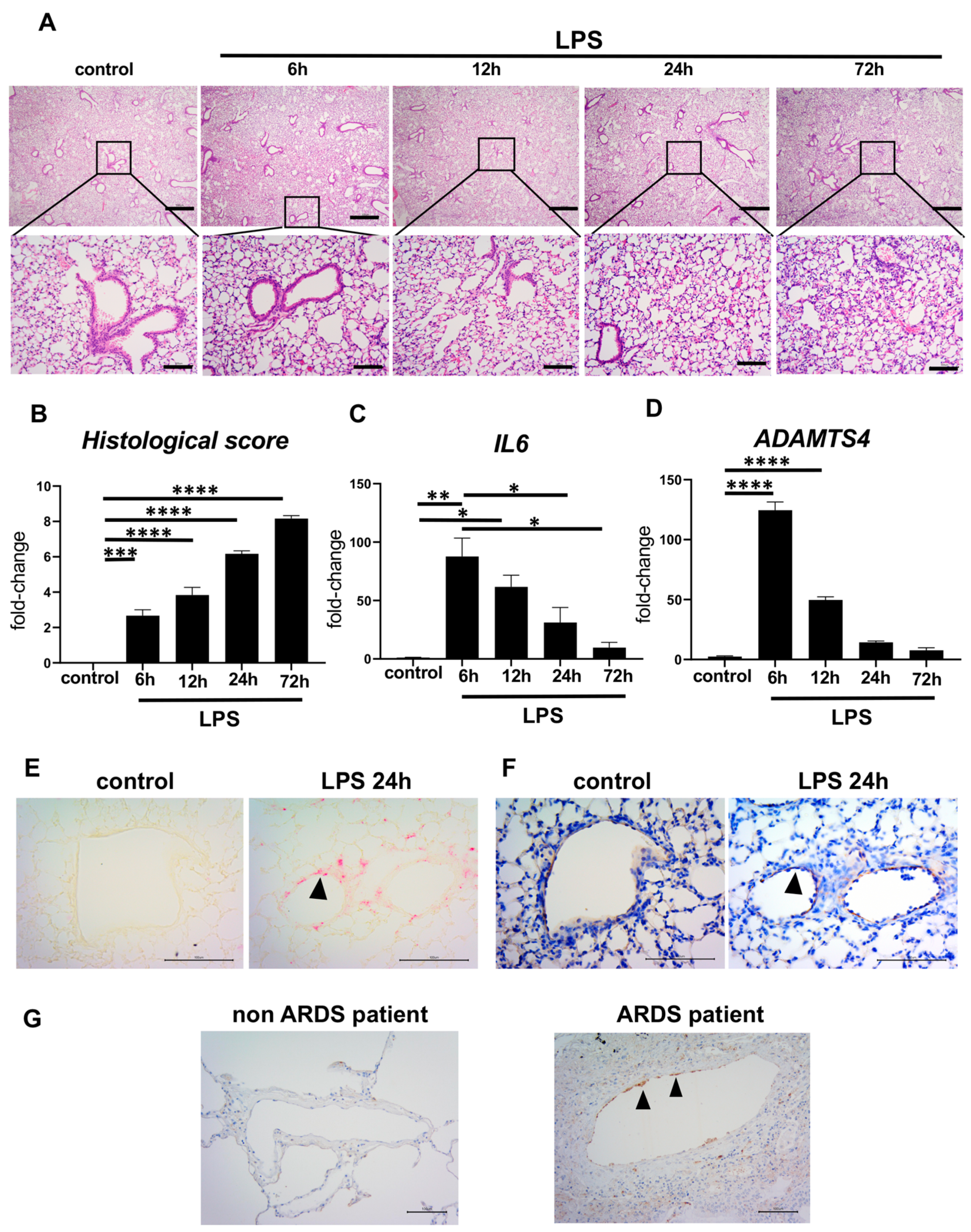
2.2. Elevated ADAMTS4 in Pulmonary VECs in the ARDS-Model Mice and Patients with ARDS
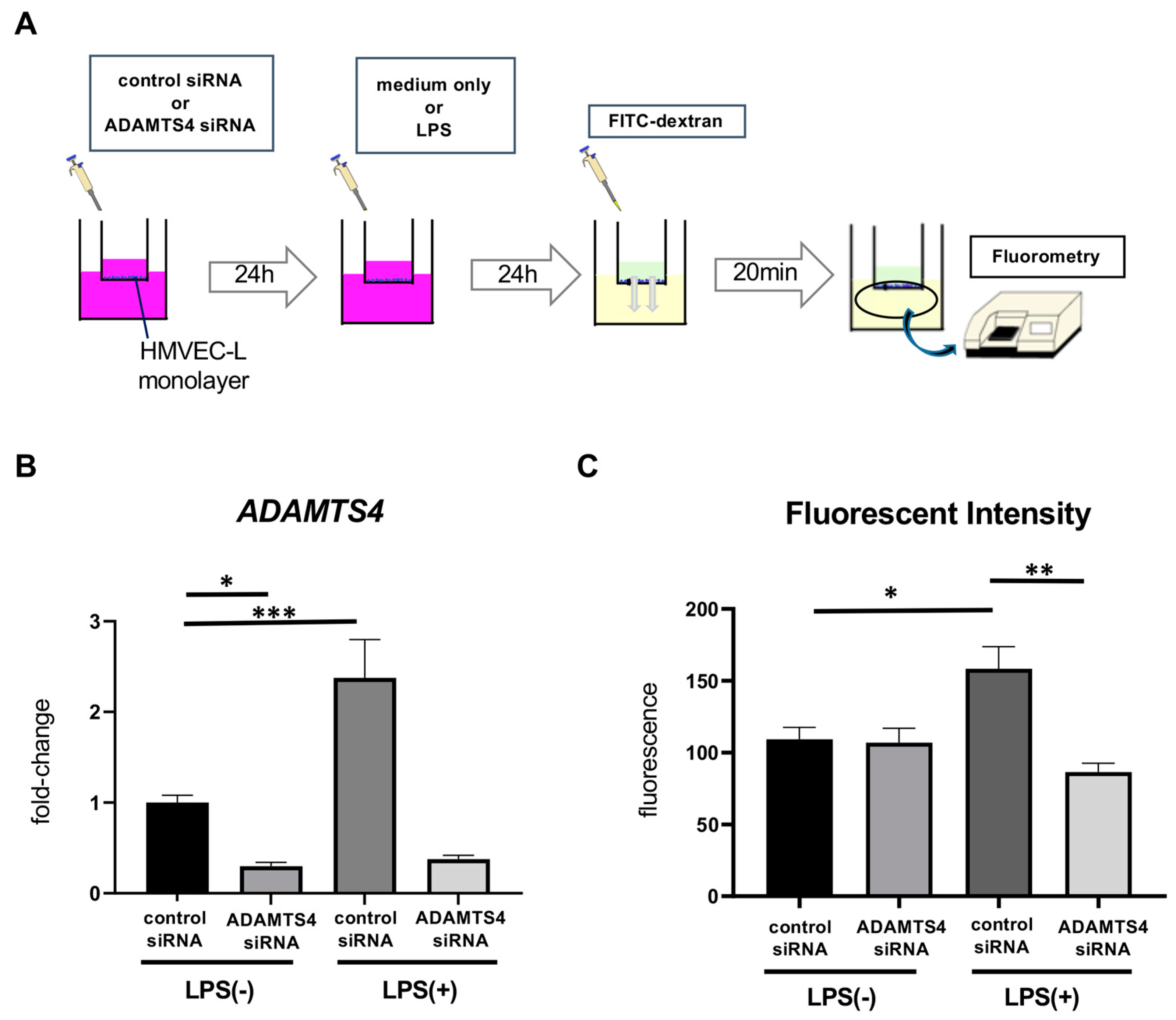
2.3. ADAMTS4 Is Involved in Increased Vascular Permeability
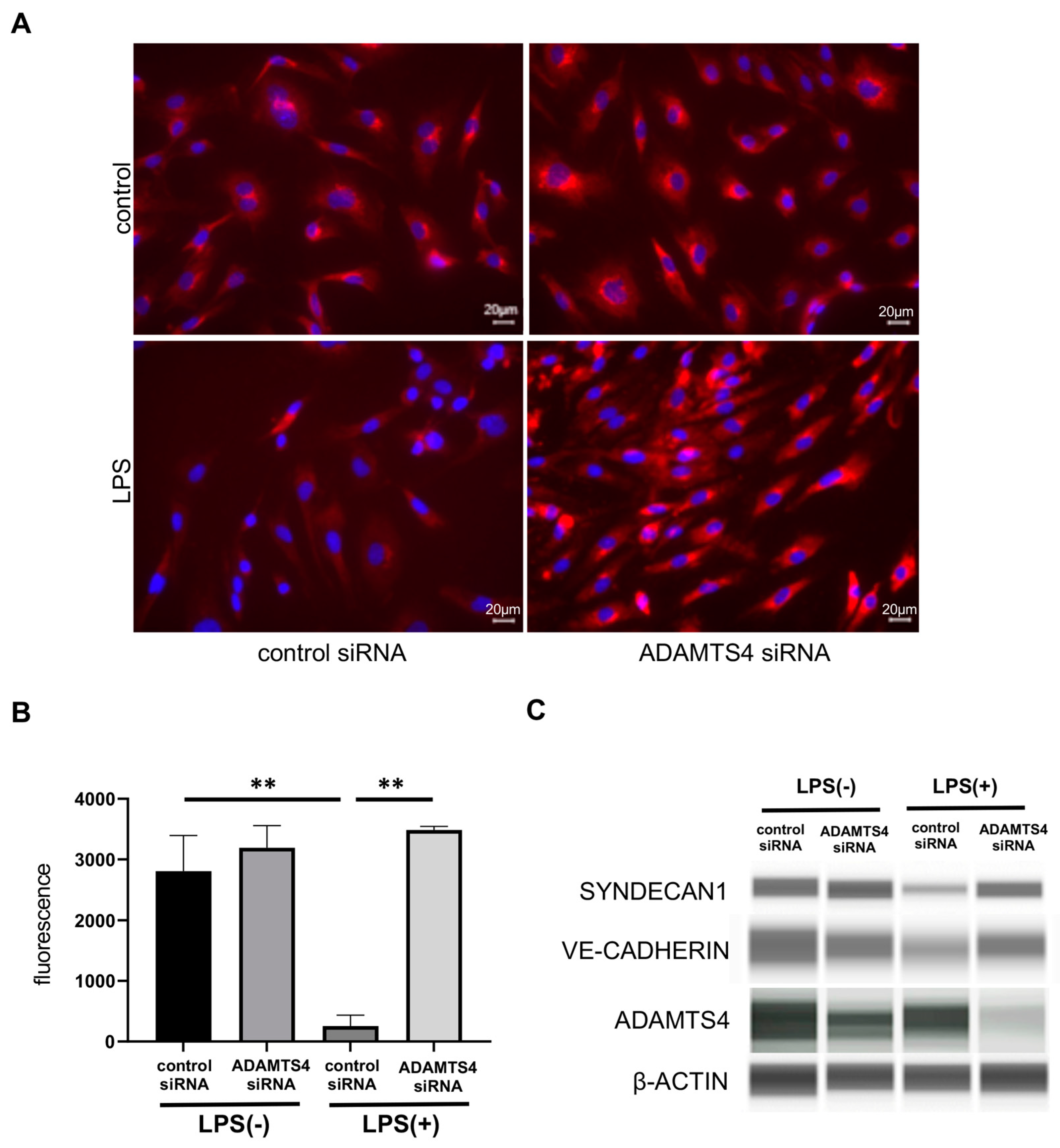
2.4. ADAMTS4 Contributes to the Destruction of Glycocalyx in HMVEC-Ls
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Human Lung Endothelial Cell Culture
4.3. ARDS Animal Model
4.4. Microarray Analysis
4.5. Real-Time Quantitative PCR
4.6. Histological Analysis
4.7. In Situ Hybridization
4.8. Immunohistochemical Staining
4.9. Transfection of siRNA
4.10. Permeability Assay
4.11. Immunofluorescent Staining
4.12. Simple Western Assays
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chambers, E.; Rounds, S.; Lu, Q. Pulmonary Endothelial Cell Apoptosis in Emphysema and Acute Lung Injury. Adv. Anat. Embryol. Cell Biol. 2018, 228, 63–86. [Google Scholar]
- Máca, J.; Jor, O.; Holub, M.; Sklienka, P.; Burša, F.; Burda, M.; Janout, V.; Ševčík, P. Past and Present ARDS Mortality Rates: A Systematic Review. Respir. Care 2017, 62, 113–122. [Google Scholar] [CrossRef]
- Optiz, B.; van Laak, V. Innate immune recognition in infectious and noninfectious diseases of the lung. Am. J. Respir. Crit. Care Med. 2010, 181, 1294–1309. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Jang, Y.H.; Lee, G.Y.; Han, G.M.; Jeong, H.J.; Kim, J.W.; Lee, J.-O. TLR3 forms a highly organized cluster when bound to a poly(I:C) RNA ligand. Nat. Commun. 2022, 12, 136876. [Google Scholar] [CrossRef] [PubMed]
- Płóciennikowska, A.; Hromada-Judycka, A.; Borzęcka, K.; Kwiatkowska, K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2015, 72, 557–581. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K. Identification of oxidative stress and Toll-like receptor 4 signaling as key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Mariotti, M. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 9815–9820. [Google Scholar] [CrossRef]
- Li, K.; Wang, Z.-W.; Hu, Z.; Ren, Z.; Hu, X.; Li, L.; Wu, Z.; Wu, H.; Li, B.; Huang, J.; et al. Assessing serum levels of ADAMTS1 and ADAMTS4 as new biomarkers for patients with type A acute aortic dissection. Med. Sci. 2017, 23, 3913–3922. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Scislowska-Czarnecka, A.; Chadzinska, M.; Plytycz, B.; van Rooijen, N.; Opdenakker, G.; Arnold, B. Enhanced early vascular permeability in gelatinase B (MMP-9)- deficient mice: Putative contribution of COX-1-derived PGE2 of macrophage origin. J. Leuloc. Biol. 2006, 80, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, S.; Kitabatake, M.; Hara, A.; Konda, M.; Ouji-Sageshima, N.; Terada-Ikeda, C.; Furukawa, R.; Imakita, N.; Oda, A.; Takeda, M.; et al. The critical role of the histone modification enzyme Setdb2 in the pathogenesis of acute respiratory distress syndrome. Shock 2023, 1, 137–145. [Google Scholar] [CrossRef]
- Vistnes, M.; Aronsen, J.M.; Lunde, I.G.; Sjaastad, I.; Carlson, C.R.; Christensen, G. Pentosan polysulfate decreases myocardial expression of the extracellular matrix enzyme ADAMTS4 and improves cardiac function in vivo in rats subjected to pressure overload by aortic banding. PLoS ONE 2014, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Rienks, M.; Barallobre-Barreiro, J.; Mayr, M. Te emerging role of the ADAMTS family in vascular diseases. Circ. Res. 2018, 123, 1279–1281. [Google Scholar] [CrossRef] [PubMed]
- Mead, T.J.; Apte, S.S. Apte. ADAMTS proteins in human disorders. Matrix Biol. 2018, 71–72, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Cilek, M.Z.; de Vega, S.; Shiozawa, J.; Yoshinaga, C.; Miyamae, Y.; Chijiiwa, M.; Mochizuki, S.; Ito, M.; Kaneko, H.; Kaneko, K.; et al. Synergistic upregulation of ADAMTS4 (aggrecanase-a) by cytokines and its suppression in knee osteoarthritic synovial fibroblasts. Lab. Invest. 2022, 102, 102–111. [Google Scholar] [CrossRef]
- Salter, R.C.; Arnaoutakis, K. The expression of a disintegrin and metalloproteinase with thrombospondin motifs 4 in human macrophages is inhibited by the anti-atherogenic cytokine transforming growth factor-b and requires Smads, p38 mitogen-activated protein kinase and c-Jun. Int. J. Biochem. Cell Biol. 2011, 43, 805–811. [Google Scholar] [CrossRef]
- Ashlin, T.G.; Kwan, A.P.; Ramji, D.P. Regulation of ADAMTS-1, -4 and -5 expression in human macrophages: Differential regulation by key cytokines implicated in atherosclerosis and novel synergism between TL1A and IL-17. Cytokine 2013, 64, 234–242. [Google Scholar] [CrossRef]
- Kelwick, R.; Desanlis, I. The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef]
- Ashlin, T.G.; Buckley, M.L. The anti-atherogenic cytokine interleukin-33 inhibits the expression of a disintegrin and metalloproteinase with thrombospondin motifs-a,-4 and -5 in human macrophages; requirement of extracellular signal-regulated kinase, c-Jun N-terminal kinase and phosphoinositide 3-kinase signaling pathways. Int. J. Biochem. Cell Biol. 2014, 46, 113–123. [Google Scholar]
- Boyd, D.F.; PALISI Pediatric Intensive Care Influenza (PICFLU) Investigators; Allen, E.K.; Randolph, A.G.; Guo, X.-Z.J.; Weng, Y.; Sanders, C.J.; Bajracharya, R.; Lee, N.K.; Guy, C.S.; et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature 2020, 587, 466–471. [Google Scholar] [CrossRef]
- Chen, H.; Bai, C.; Wang, X. The value of the lipopolysaccharide-induced acute lung injury model in respiratory medicine. Expert Rev. Respir. Med. 2010, 4, 773–783. [Google Scholar] [CrossRef]
- Xu, Y.; Ito, T.; Fushimi, S. Spred-2 deficiency exacerbates lipopolysaccharide induced acute lung inflammation in mice. PLoS ONE 2014, 9, e108914. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, N.N.; Sivak, K.V. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Vink, H.; Duling, B.R. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ. Res. 1996, 79, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Megens, R.T.A.; Reitsma, S. Two-photon microscopy of vital murine elastic and muscular arteries. Combined structural and functional imaging with subcellular resolution. J. Vasc. Res. 2007, 44, 87–98. [Google Scholar] [CrossRef]
- Weinbaum, S.; Tarbell, J.M.; Damiano, E.R. The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Takemura, G. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Crit. Care 2017, 21, 261. [Google Scholar] [CrossRef] [PubMed]
- Broekhuizen, L.N.; Lemkes, B.A. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 2010, 53, 2646–2655. [Google Scholar] [CrossRef]
- Molecular functions of syndecan-1 in disease. Mtrix Biol. 2012, 31, 3–16. [CrossRef]
- Bernfield, M.; Kokenyesi, R. Biology of the syndecans: A family of transmembrane heparan sulfate proteoglycans. Annu. Rev. Cell Biol. 1992, 8, 365–393. [Google Scholar] [CrossRef]
- Luft, J.H. Fine structures of capillary and endocapillary layer as revealed by ruthenim red. Fed. Proc. 1966, 25, 1773–1783. [Google Scholar]
- Sullivan, R.C.; Rockstrom, M.D.; Schmidt, E.P.; Hippensteel, J.A. Endothelial glycocalyx degradation during sepsis: Causes and consequences. Matrix Biology Plus. 2021, 12, 100094. [Google Scholar] [CrossRef] [PubMed]
- Torres Filho, I.P.; Torres, L.N.; Salgado, C.; Dubick, M.A. Plasma sundecan-1 and heparin sulfate correlate with microvascular glycocalyx degradation in hemorrhaged rats after different resuscitation fluids. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, 1468–1478. [Google Scholar] [CrossRef]
- Gao, G.; Plass, A. ADAMTS4 (aggrecanase-1) activation on the cell surface involves C-terminal cleavage by glycosylphosphatidyl inositol-anchored membrane type 4—matrix metalloproteinase and binding of the activated proteinase to chondroitin sulfate and heparin sulfate on syndecan—1. J. Biol. Chem. 2004, 279, 10042–10051. [Google Scholar] [PubMed]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Derangement of the endothelial glycocalyx in sepsis. J. Thromb. Haemost. 2019, 17, 283–2943. [Google Scholar] [CrossRef]
- Wheeler-Jones, C.P.D.; E Farrar, C.; A Pitsillides, A. Targeting hyaluronan of the endothelial glycocalyx for therapeutic intervention. Curr. Opin Invest. Drugs 2010, 11, 997–1006. [Google Scholar]
- Chan, Y.H.; Harith, H.H.; Israf, D.A.; Tham, C.L. Differential Regulation of LPS- Mediated VE-Cadherin Disruption in Human Endothelial Cells and the Underlying Signaling Pathways: A Mini Review. Front. Cell Dev. Biol. 2020, 7, 280. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, S.C.; Poth, J.M.; Fini, M.A.; Olschewski, A.; El Kasmi, K.C.; Stenmark, K.R. The role of inflammation in hypoxic pulmonary hypertension: From cellular mechanisms to clinical phenotypes. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, 229–252. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Li, L.; Sun, M.; Zhang, Y.; Chen, L.; Rong, Y.; Li, Y. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res. Ther. 2015, 6, 103. [Google Scholar] [CrossRef]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAM10 regulates endothelial peremeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef]
- Schliehe, C.; Flynn, E.K.; Vilagos, B.; Richson, U.; Swaminathan, S.; Bosnjak, B.; Bauer, L.; Kandasamy, R.K.; Griesshammer, I.M.; Kosack, L.; et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat. Immunol. 2015, 16, 67–74. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konda, M.; Kitabatake, M.; Ouji-Sageshima, N.; Tonomura, R.; Furukawa, R.; Sonobe, S.; Terada-Ikeda, C.; Takeda, M.; Kawaguchi, M.; Ito, T. A Disintegrin and Metalloproteinase with Thrombospondin Motifs 4 Regulates Pulmonary Vascular Hyperpermeability through Destruction of Glycocalyx in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2023, 24, 16230. https://doi.org/10.3390/ijms242216230
Konda M, Kitabatake M, Ouji-Sageshima N, Tonomura R, Furukawa R, Sonobe S, Terada-Ikeda C, Takeda M, Kawaguchi M, Ito T. A Disintegrin and Metalloproteinase with Thrombospondin Motifs 4 Regulates Pulmonary Vascular Hyperpermeability through Destruction of Glycocalyx in Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences. 2023; 24(22):16230. https://doi.org/10.3390/ijms242216230
Chicago/Turabian StyleKonda, Makiko, Masahiro Kitabatake, Noriko Ouji-Sageshima, Rei Tonomura, Ryutaro Furukawa, Shota Sonobe, Chiyoko Terada-Ikeda, Maiko Takeda, Masahiko Kawaguchi, and Toshihiro Ito. 2023. "A Disintegrin and Metalloproteinase with Thrombospondin Motifs 4 Regulates Pulmonary Vascular Hyperpermeability through Destruction of Glycocalyx in Acute Respiratory Distress Syndrome" International Journal of Molecular Sciences 24, no. 22: 16230. https://doi.org/10.3390/ijms242216230
APA StyleKonda, M., Kitabatake, M., Ouji-Sageshima, N., Tonomura, R., Furukawa, R., Sonobe, S., Terada-Ikeda, C., Takeda, M., Kawaguchi, M., & Ito, T. (2023). A Disintegrin and Metalloproteinase with Thrombospondin Motifs 4 Regulates Pulmonary Vascular Hyperpermeability through Destruction of Glycocalyx in Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences, 24(22), 16230. https://doi.org/10.3390/ijms242216230