
Conformational States of the CXCR4 Inhibitor Peptide EPI-X4—A Theoretical Analysis


Abstract
:1. Introduction
2. Results
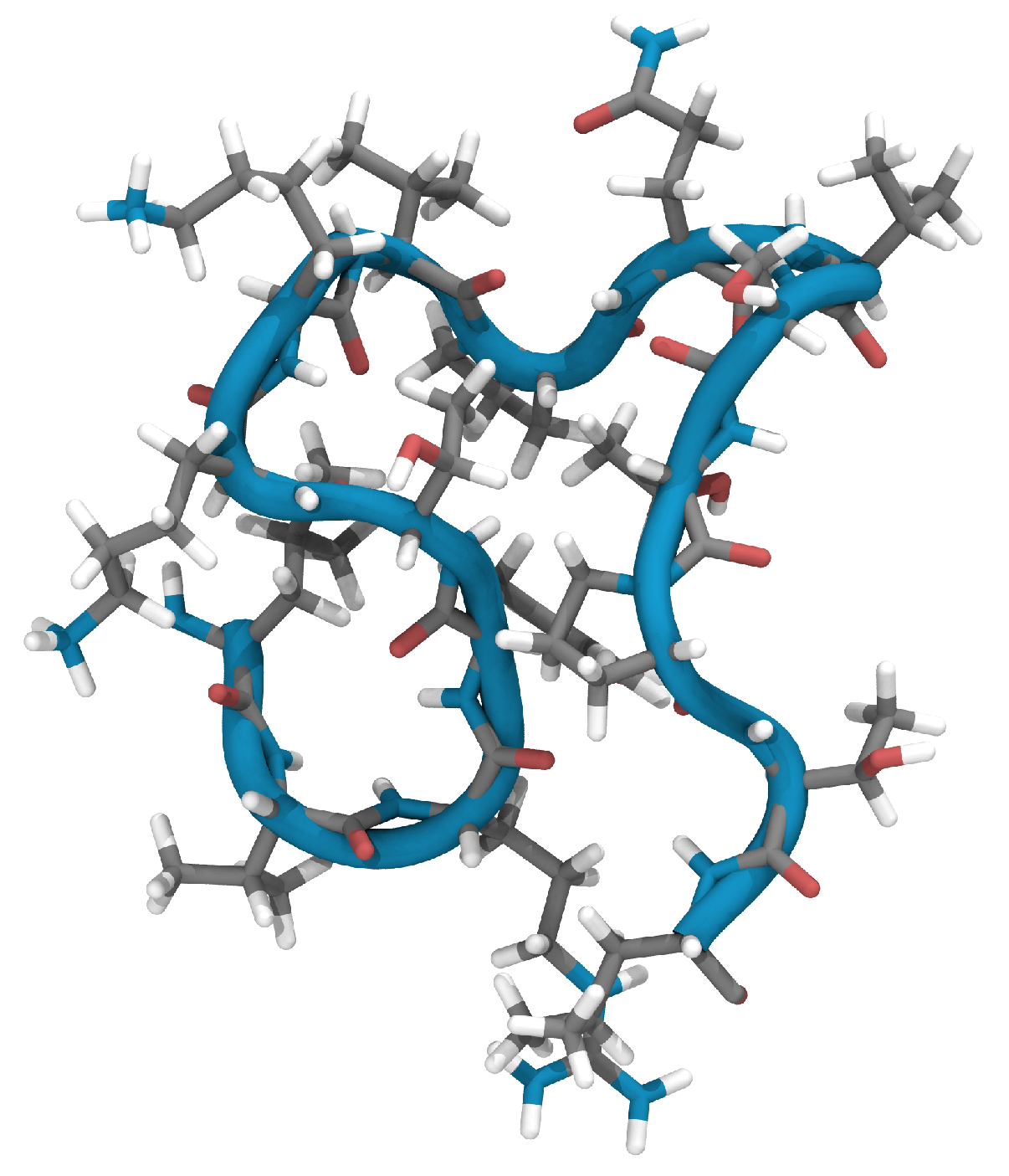
2.1. Characterization of the Experimentally Determined EPI-X4 Structure Using Reactive MD Simulations and DFT
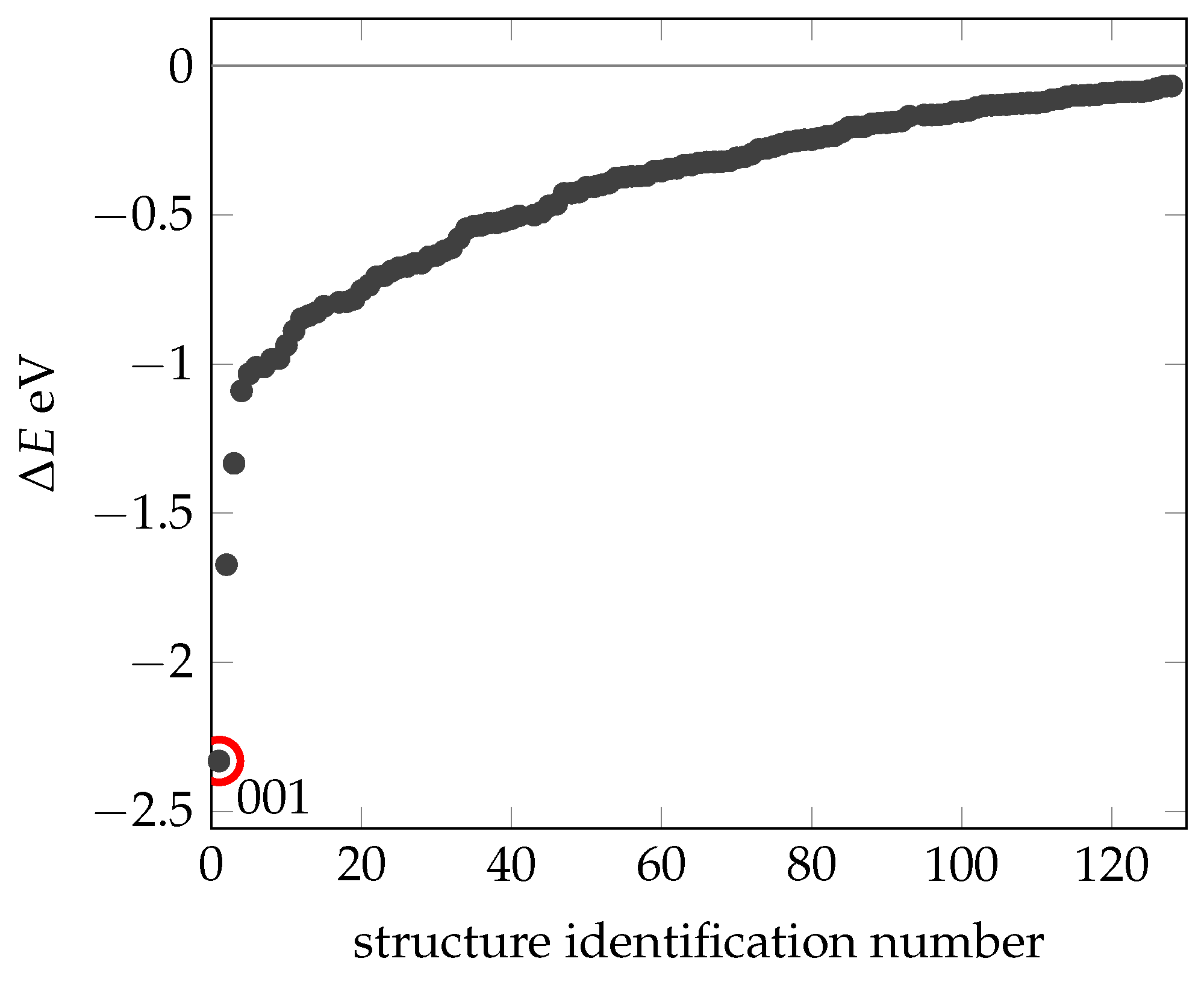
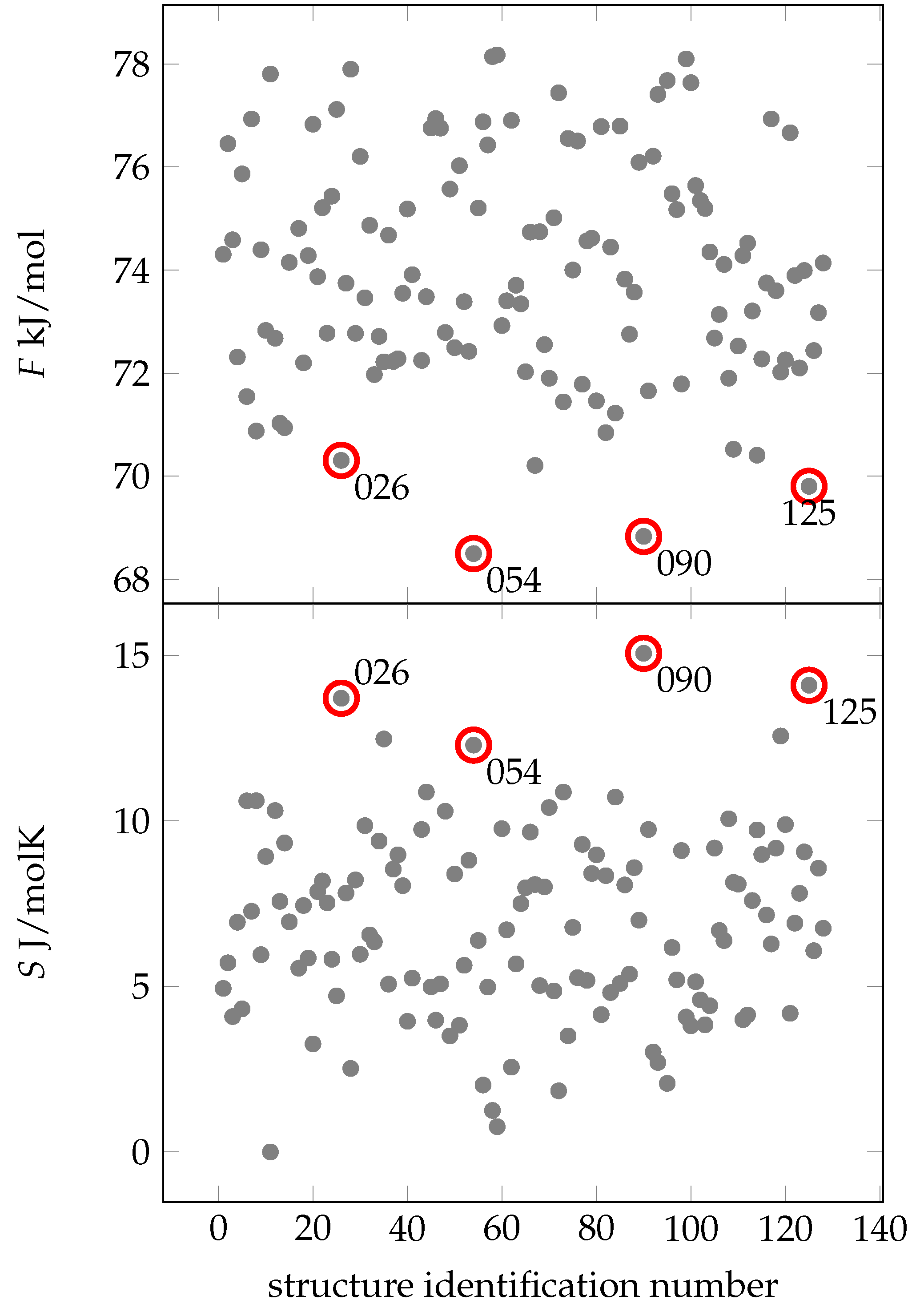
2.2. Creating and Analyzing Further Stable Conformations of EPI-X4
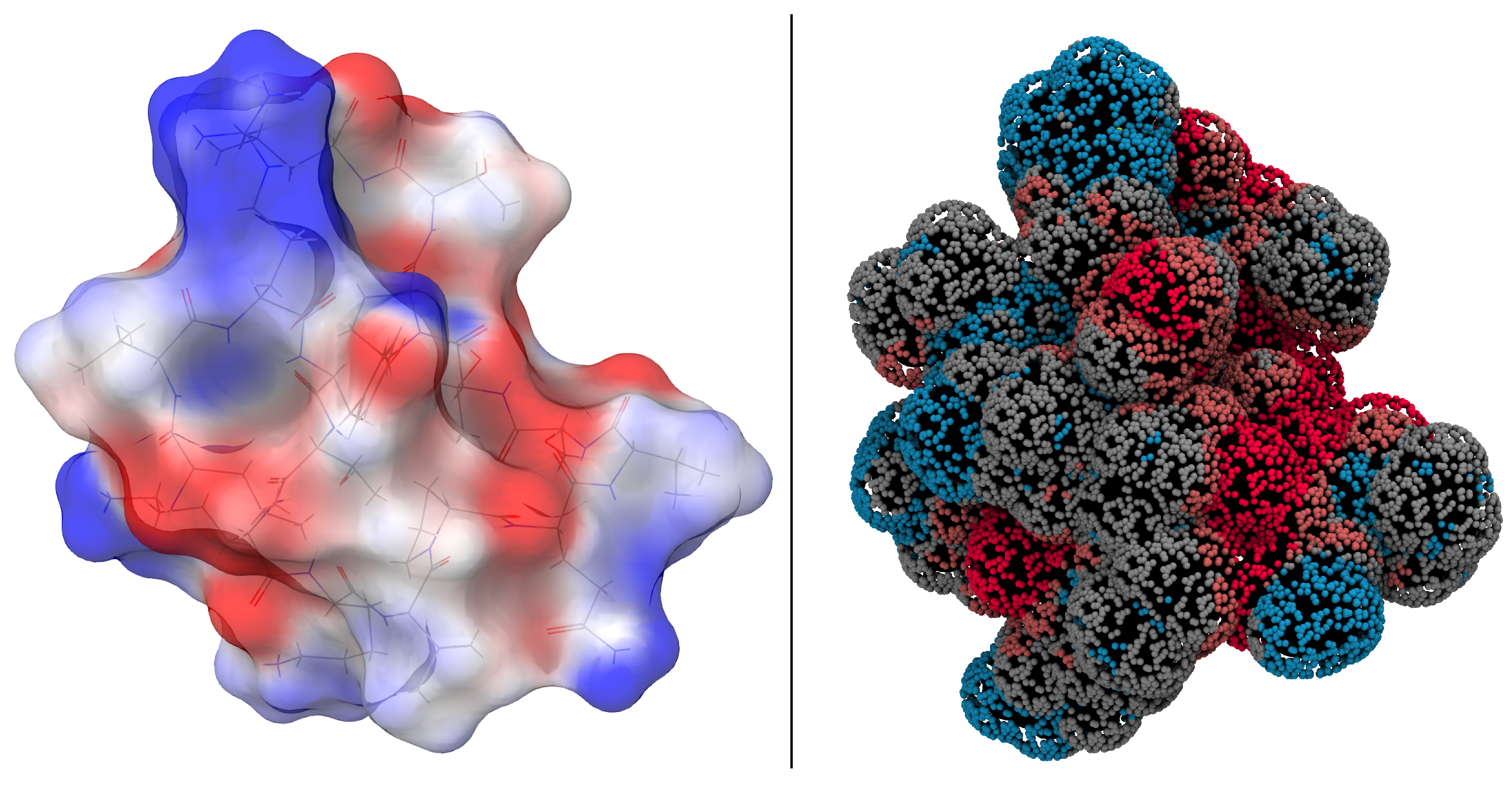
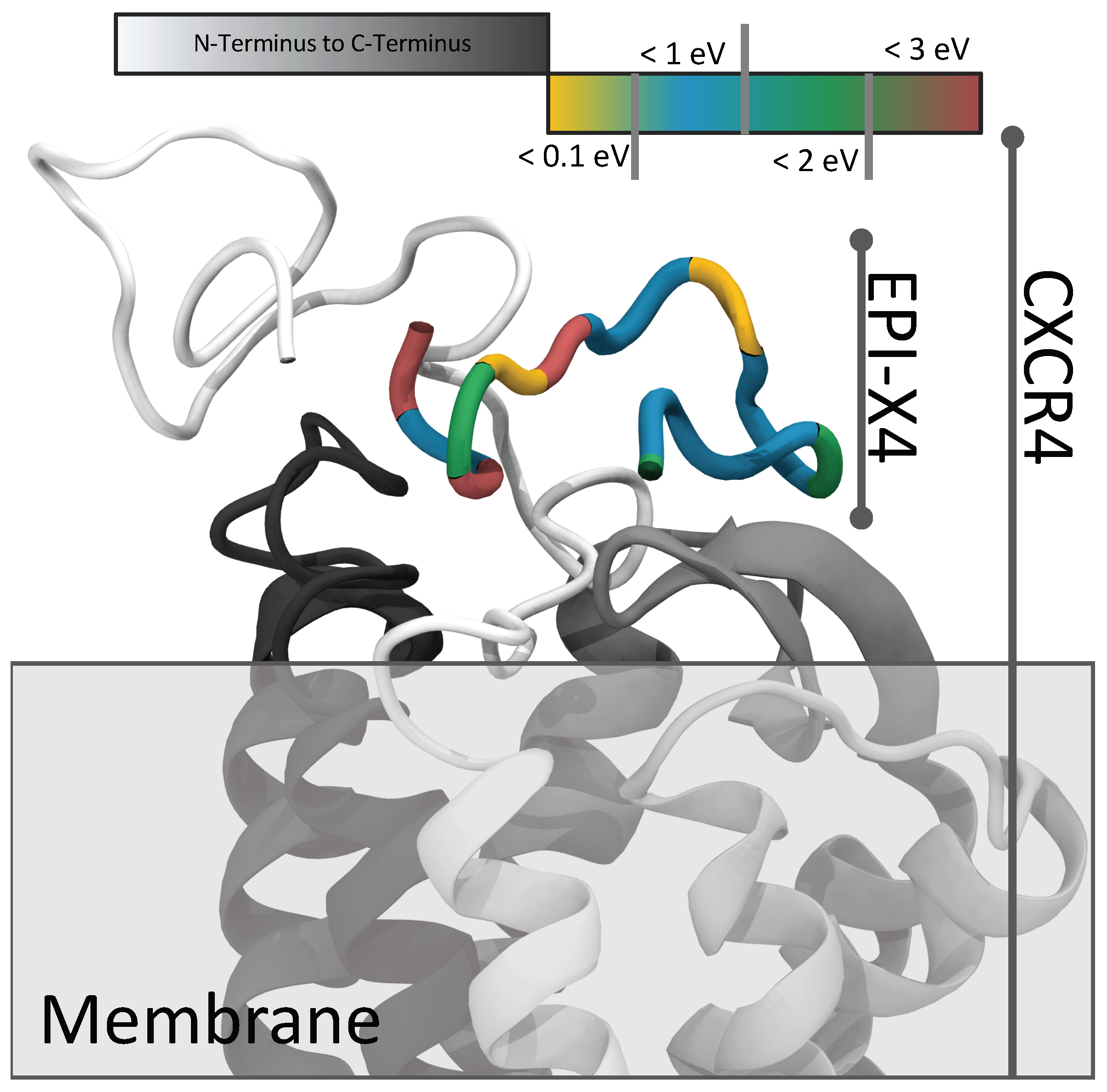
2.3. Docking of EPI-X4 to CXCR4
3. Conclusions
4. Theoretical Methods
4.1. DFT
4.2. ReaxFF
4.3. 2PT Approach
4.4. Interaction Potential of Structures
4.5. Exploration of Binding Sites through Grid-Based Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qin, L.; Kufareva, I.; Holden, L.G.; Wang, C.; Zheng, Y.; Zhao, C.; Fenalti, G.; Wu, H.; Han, G.W.; Cherezov, V.; et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015, 347, 1117–1122. [Google Scholar] [CrossRef]
- Choi, W.T.; Duggineni, S.; Xu, Y.; Huang, Z.; An, J. Drug Discovery Research Targeting the CXC Chemokine Receptor 4 (CXCR4). J. Med. Chem. 2012, 55, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Stevens, R.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 Entry Cofactor: Functional cDNA Cloning of a Seven-Transmembrane, G Protein-Coupled Receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef]
- Moore, J.P.; Kitchen, S.G.; Pugach, P.; Zack, J.A. The CCR5 and CXCR4 Coreceptors—Central to Understanding the Transmission and Pathogenesis of Human Immunodeficiency Virus Type 1 Infection. AIDS Res. Hum. Retroviruses 2004, 20, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Zirafi, O.; Kim, K.A.; Ständker, L.; Mohr, K.B.; Sauter, D.; Heigele, A.; Kluge, S.F.; Wiercinska, E.; Chudziak, D.; Richter, R.; et al. Discovery and Characterization of an Endogenous CXCR4 Antagonist. Cell Rep. 2015, 11, 737–747. [Google Scholar] [CrossRef]
- Gilg, A.; Harms, M.; Olari, L.R.; Urbanowitz, A.K.; Bonig, H.; Münch, J. Absence of the CXCR4 antagonist EPI-X4 from pharmaceutical human serum albumin preparations. J. Transl. Med. 2021, 19, 190. [Google Scholar] [CrossRef] [PubMed]
- Zirafi, O.; Hermann, P.C.; Münch, J. Proteolytic processing of human serum albumin generates EPI-X4, an endogenous antagonist of CXCR4. J. Leukoc. Biol. 2016, 99, 863–868. [Google Scholar] [CrossRef]
- Sokkar, P.; Harms, M.; Stürzel, C.; Gilg, A.; Kizilsavas, G.; Raasholm, M.; Preising, N.; Wagner, M.; Kirchhoff, F.; Ständker, L.; et al. Computational modeling and experimental validation of the EPI-X4/CXCR4 complex allows rational design of small peptide antagonists. Commun. Biol. 2021, 4, 1113. [Google Scholar] [CrossRef]
- Jorgensen, W.L. The Many Roles of Computation in Drug Discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Harvey, M.J.; De Fabritiis, G. High-throughput molecular dynamics: The powerful new tool for drug discovery. Drug Discov. Today 2012, 17, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Alfonso, A.; Heck, A.; Ruiz-Blanco, Y.B.; Gilg, A.; Ständker, L.; Kuan, S.L.; Weil, T.; Sanchez-Garcia, E.; Wiese, S.; Münch, J.; et al. Advanced EPI-X4 Derivatives Covalently Bind Human Serum Albumin Resulting in Prolonged Plasma Stability. Int. J. Mol. Sci. 2022, 23, 15029. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4033–4414. [Google Scholar] [CrossRef]
- Chass, G.; Lovas, S.; Murphy, R.; Csizmadia, I. The role of enhanced aromatic π-electron donating aptitude of the tyrosyl sidechain with respect to that of phenylalanyl in intramolecular interactions. Eur. Phys. J. D 2002, 20, 481–497. [Google Scholar] [CrossRef]
- Jensen, F. Introduction to Computational Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2007; Volume 2, p. 599. [Google Scholar]
- Mo, P.; Li, C.; Zhao, D.; Zhang, Y.; Shi, M.; Li, J.; Liu, J. Accurate and efficient molecular dynamics based on machine learning and non von Neumann architecture. NPJ Comput. Mater. 2022, 8, 107. [Google Scholar] [CrossRef]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Monti, S.; Corozzi, A.; Fristrup, P.; Joshi, K.L.; Shin, Y.K.; Oelschlaeger, P.; van Duin, A.C.T.; Barone, V. Exploring the conformational and reactive dynamics of biomolecules in solution using an extended version of the glycine reactive force field. Phys. Chem. Chem. Phys. 2013, 15, 15062–15077. [Google Scholar] [CrossRef]
- Strange, P.G. Agonist binding, agonist affinity and agonist efficacy at G protein-coupled receptors. Br. J. Pharmacol. 2008, 153, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.K.; Braunwarth, L.; Sinyavskiy, A.; Jacob, T. Thermodynamic Description of Interfaces Applying the 2PT Method on ReaxFF Molecular Dynamics Simulations. J. Phys. Chem. C 2021, 125, 24663–24670. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-3: Jaguar; Schrödinger, LLC: New York, NY, USA, 2018.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Slater, J.C. The Self-Consistent Field for Molecules and Solids. Quantum Theory Mol. Solids 1974, 27, 49–50. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Chenoweth, K.; van Duin, A.C.T.; Goddard, W.A. ReaxFF Reactive Force Field for Molecular Dynamics Simulations of Hydrocarbon Oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef]
- Rahaman, O.; Van Duin, A.C.T.; Goddard, W.A.; Doren, D.J. Development of a ReaxFF reactive force field for glycine and application to solvent effect and tautomerization. J. Phys. Chem. B 2011, 115, 249–261. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Postma, J.P.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Guthrie, J.P. Use of DFT methods for the calculation of the entropy of gas phase organic molecules: An examination of the quality of results from a simple approach. J. Phys. Chem. A 2001, 105, 8495–8499. [Google Scholar] [CrossRef]
- Lin, S.T.; Maiti, P.K.; Goddard, W.A. Two-Phase Thermodynamic Model for Efficient and Accurate Absolute Entropy of Water from Molecular Dynamics Simulations. J. Phys. Chem. B 2010, 114, 8191–8198. [Google Scholar] [CrossRef]
- Lin, S.T.; Blanco, M.; Goddard, W.A. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: Validation for the phase diagram of Lennard-Jones fluids. J. Chem. Phys. 2003, 119, 11792–11805. [Google Scholar] [CrossRef]
- Pascal, T.A.; Schärf, D.; Jung, Y.; Kühne, T.D. On the absolute thermodynamics of water from computer simulations: A comparison of first-principles molecular dynamics, reactive and empirical force fields. J. Chem. Phys. 2012, 137, 244507. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C001 | C026 | C054 | C090 | C125 | N1 | |
|---|---|---|---|---|---|---|
| Surface [Å] | 1986 | 1868 | 1980 | 1913 | 1916 | 1969 |
| Hydrophil [Å] | 601 | 615 | 600 | 749 | 635 | 607 |
| Else [Å] | 448 | 435 | 351 | 541 | 451 | 178 |
| Hydrophob [Å] | 935 | 817 | 1027 | 621 | 829 | 1183 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, C.K.; Münch, J.; Jacob, T. Conformational States of the CXCR4 Inhibitor Peptide EPI-X4—A Theoretical Analysis. Int. J. Mol. Sci. 2023, 24, 16229. https://doi.org/10.3390/ijms242216229
Jung CK, Münch J, Jacob T. Conformational States of the CXCR4 Inhibitor Peptide EPI-X4—A Theoretical Analysis. International Journal of Molecular Sciences. 2023; 24(22):16229. https://doi.org/10.3390/ijms242216229
Chicago/Turabian StyleJung, Christoph Karsten, Jan Münch, and Timo Jacob. 2023. "Conformational States of the CXCR4 Inhibitor Peptide EPI-X4—A Theoretical Analysis" International Journal of Molecular Sciences 24, no. 22: 16229. https://doi.org/10.3390/ijms242216229
APA StyleJung, C. K., Münch, J., & Jacob, T. (2023). Conformational States of the CXCR4 Inhibitor Peptide EPI-X4—A Theoretical Analysis. International Journal of Molecular Sciences, 24(22), 16229. https://doi.org/10.3390/ijms242216229