
Molecular Analysis of a Congenital Myasthenic Syndrome Due to a Pathogenic Variant Affecting the C-Terminus of ColQ

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Clinical Case Description
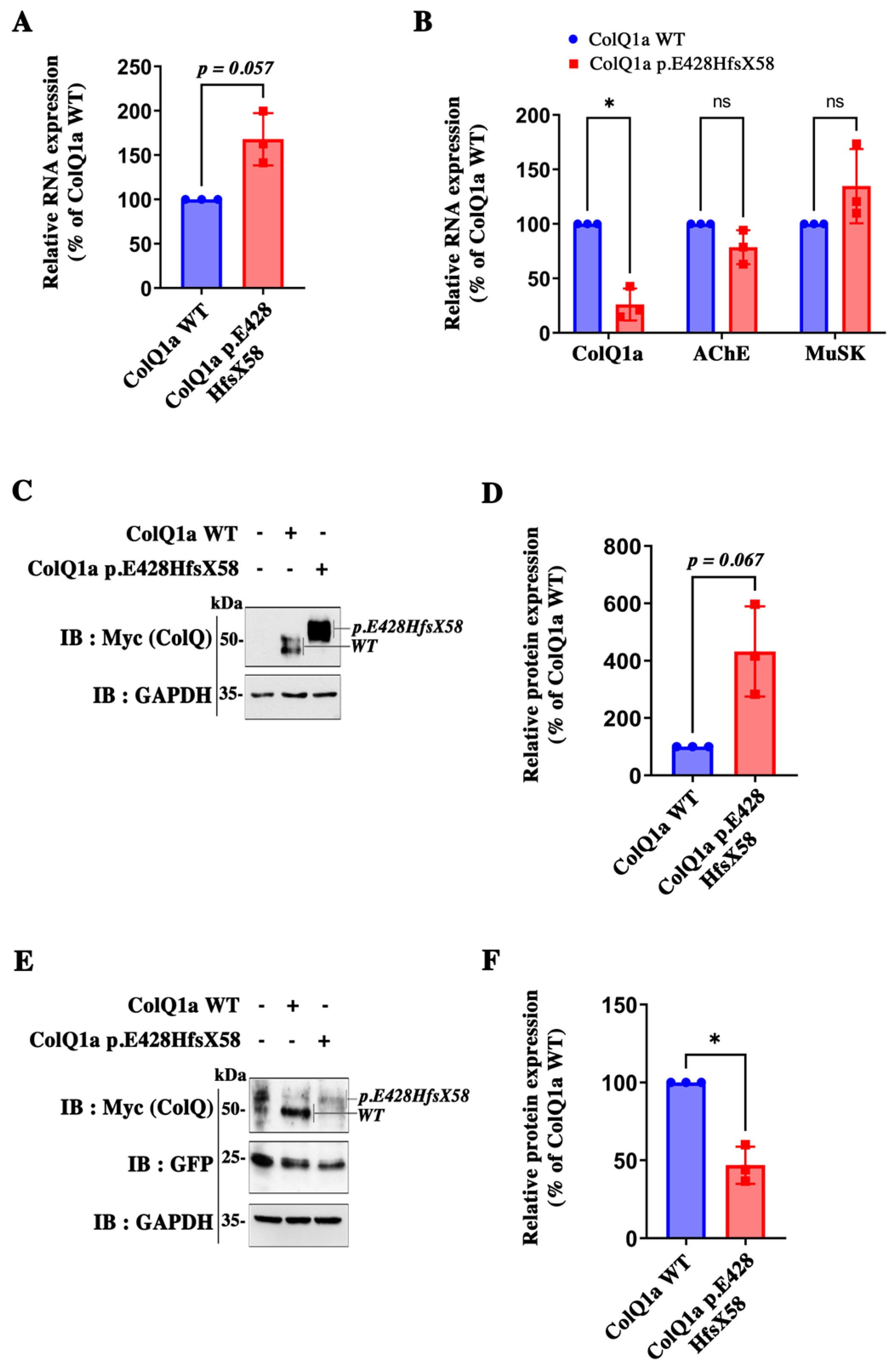
2.2. The Pathogenic Variant Neither Impairs Transcription nor Translation
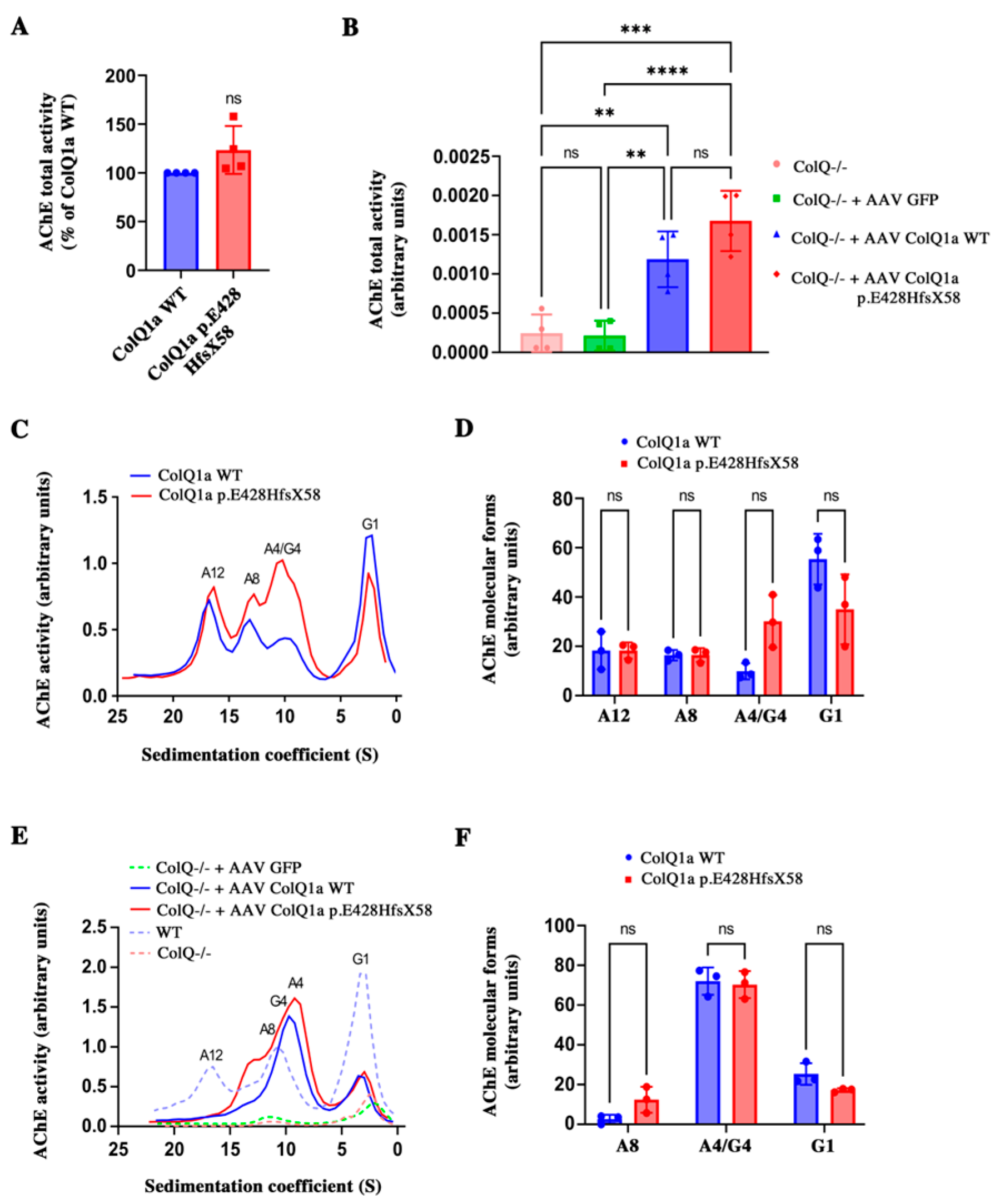
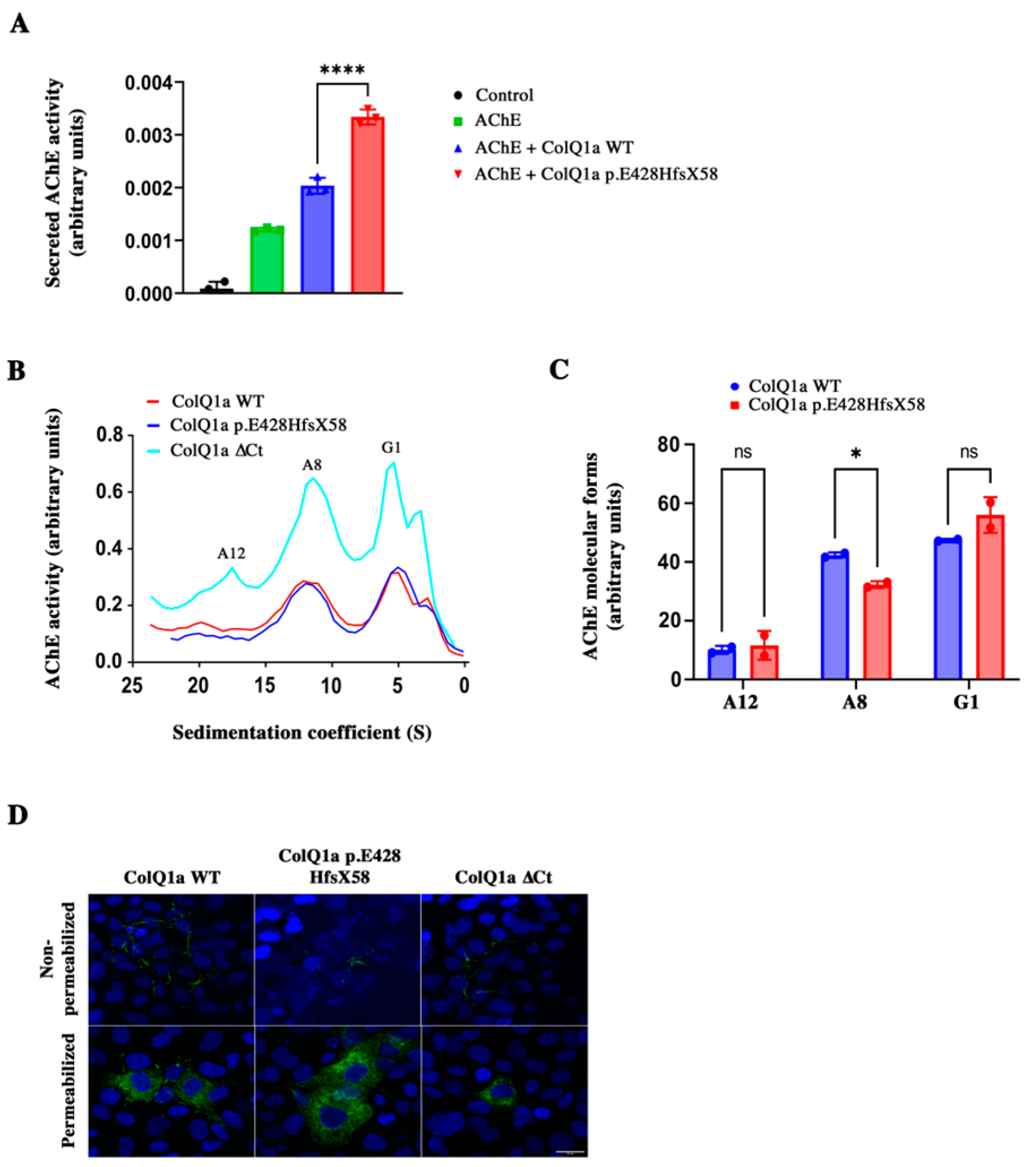
2.3. The Variant Protein: The Interaction between the ColQ Variant and LRP4 Is Reduced Is Secreted
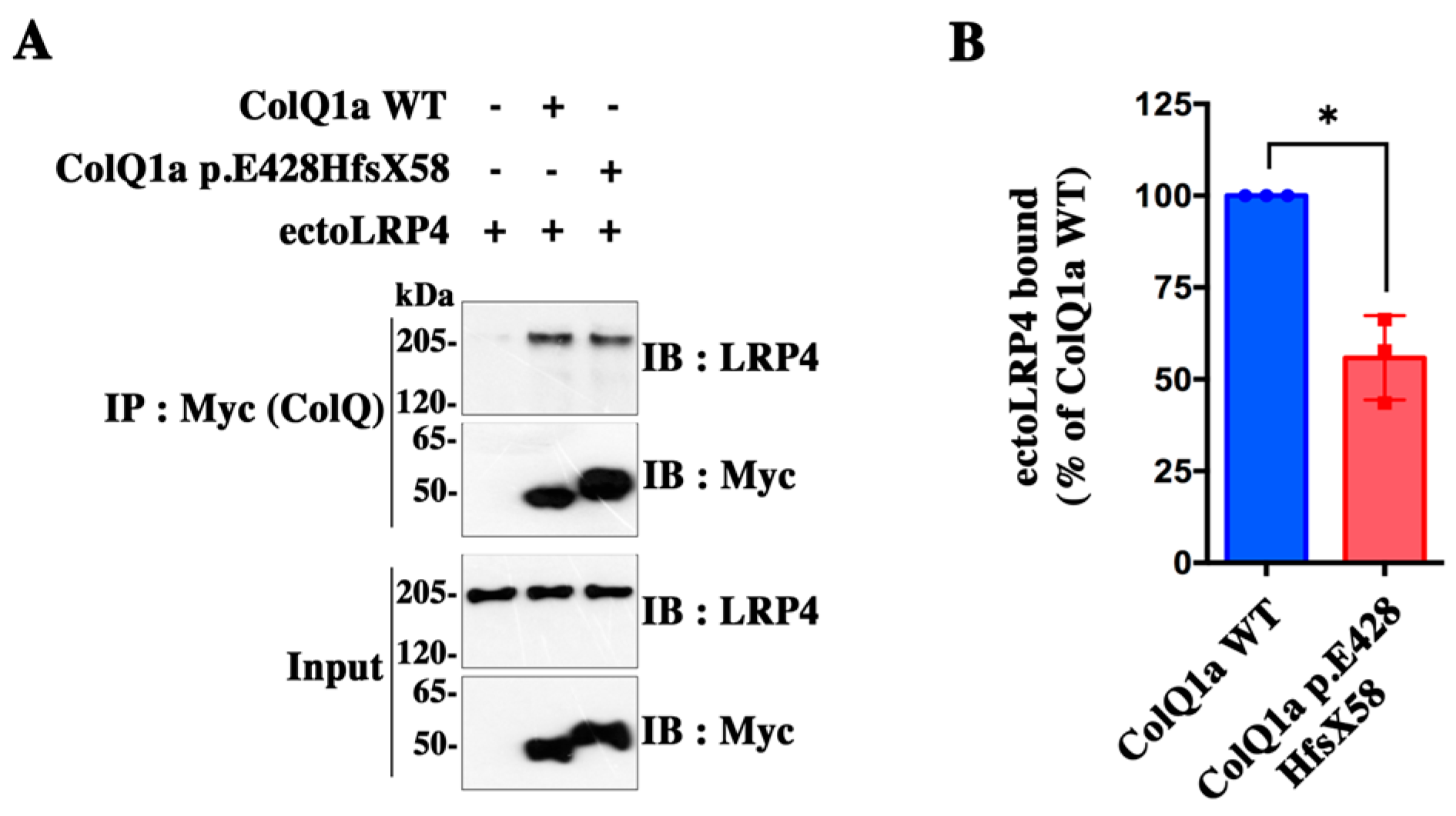
2.4. The Interaction between the ColQ Variant and LRP4 Is Reduced
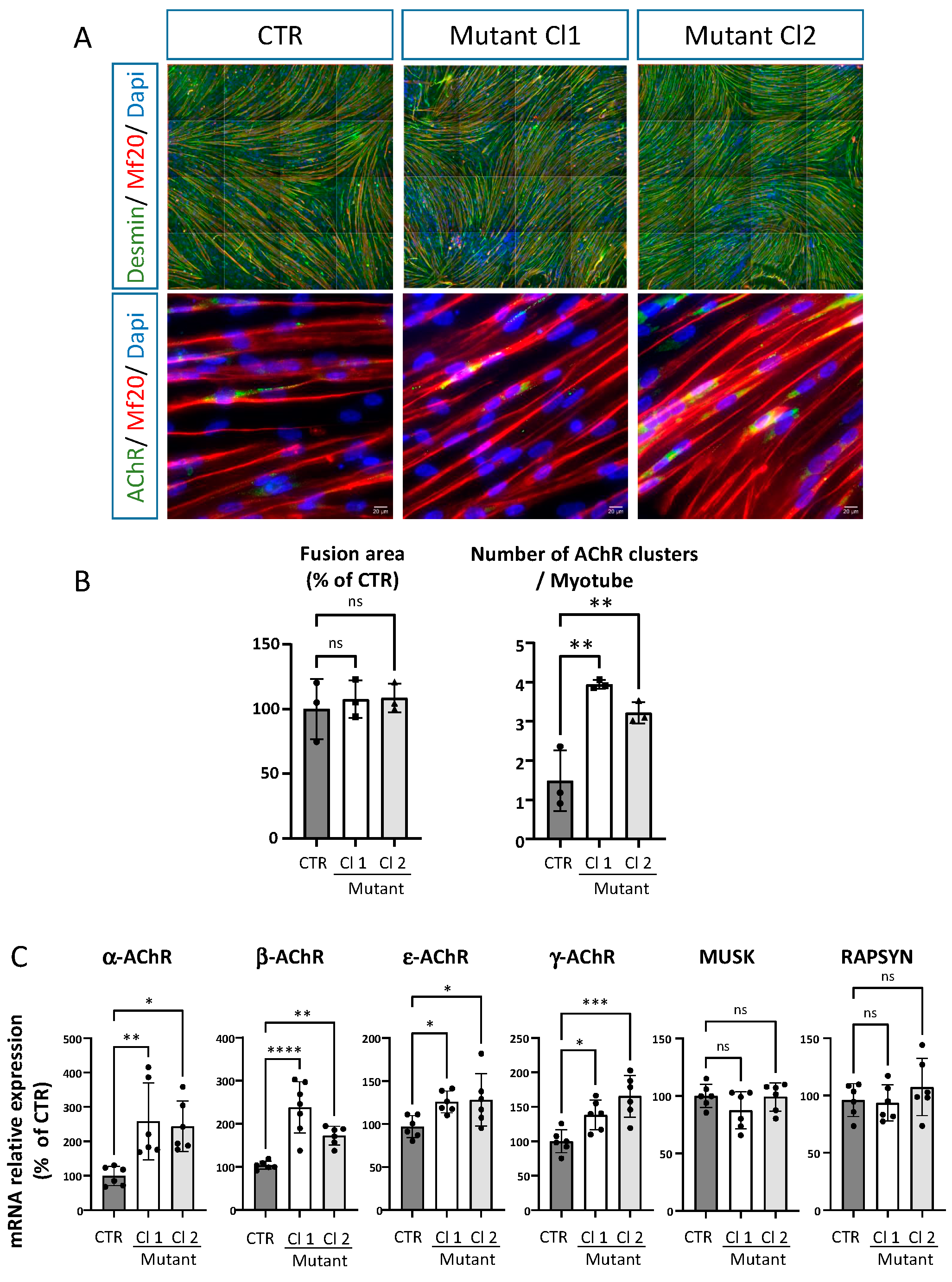
2.5. iPSC Derived Skeletal Muscle Cells from the Patient Reproduce Some of the Phenotypes Observed in Mice ColQ Deficient Muscle Cells
3. Discussion
4. Materials and Methods
4.1. Patient and iPSC
4.2. Constructs
4.3. Cell Culture, Plasmid Transfection and Virus Infection
4.4. RNA Extraction, Real Time RT-qPCR Assay
4.5. AChE Activity and Molecular Form Analysis
4.6. Recombinant Protein Production and Purification
4.7. Western Blot Analysis
4.8. Immunodetection and Microscopy
4.9. Differentiation of iPSC into Myoblasts
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krejci, E.; Thomine, S.; Boschetti, N.; Legay, C.; Sketelj, J.; Massoulié, J. The mammalian gene of acetylcholinesterase-associated collagen. J. Biol. Chem. 1997, 272, 22840–22847. [Google Scholar] [CrossRef] [PubMed]
- Belbeoc’h, S.; Falasca, C.; Leroy, J.; Ayon, A.; Massoulié, J.; Bon, S. Elements of the C-terminal t peptide of acetylcholinesterase that determine amphiphilicity, homomeric and heteromeric associations, secretion and degradation. Eur. J. Biochem. 2004, 271, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Legay, C. Congenital myasthenic syndromes with acetylcholinesterase deficiency, the pathophysiological mechanisms. Ann. N. Y. Acad. Sci. 2018, 1413, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Bon, S.; Ayon, A.; Leroy, J.; Massoulié, J. Trimerization domain of the collagen tail of acetylcholinesterase. Neurochem. Res. 2003, 28, 523–535. [Google Scholar]
- Cartaud, A.; Strochlic, L.; Guerra, M.; Blanchard, B.; Lambergeon, M.; Krejci, E.; Cartaud, J.; Legay, C. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J. Cell Biol. 2004, 165, 505–515. [Google Scholar]
- Nakata, T.; Ito, M.; Azuma, Y.; Otsuka, K.; Noguchi, Y.; Komaki, H.; Okumura, A.; Shiraishi, K.; Masuda, A.; Natsume, J.; et al. Mutations in the C-terminal domain of ColQ in endplate acetylcholinesterase deficiency compromise ColQ-MuSK interaction. Hum. Mutat. 2013, 34, 997–1004. [Google Scholar] [CrossRef]
- Uyen Dao, T.M.; Barbeau, S.; Messéant, J.; Della-Gaspera, B.; Bouceba, T.; Semprez, F.; Legay, C.; Dobbertin, A. The collagen ColQ binds to LRP4 and regulates the activation of the Muscle-Specific Kinase-LRP4 receptor complex by agrin at the neuromuscular junction. J. Biol. Chem. 2023, 299, 104962. [Google Scholar]
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188. [Google Scholar]
- Kimbell, L.M.; Ohno, K.; Engel, A.G. Rotundo RLC-terminal heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. J. Biol. Chem. 2004, 270, 10997–11005. [Google Scholar] [CrossRef]
- Ohno, K.; Ohkawara, B.; Shen, X.M.; Selcen, D.; Engel, A.G. Clinical and Pathological features of Congenital Myasthenic Syndromes caused by 35 genes—A comprehensive review. Int. J. Mol. Sci. 2023, 24, 3730. [Google Scholar]
- Donger, C.; Krejci, E.; Serradell, A.P.; Eymard, B.; Bon, S.; Nicole, S.; Chateau, D.; Gary, F.; Fardeau, M.; Massoulié, J.; et al. Mutation in the human acetylcholinesterase-associated collagen gene, COLQ, is responsible for congenital myasthenic syndrome with end-plate acetylcholinesterase deficiency (Type Ic). Am. J. Hum. Genet. 1998, 63, 967–975. [Google Scholar] [CrossRef]
- Ohno, K.; Brengman, J.; Tsujino, A.; Engel, A.G. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 9654–9659. [Google Scholar] [CrossRef]
- Sigoillot, S.M.; Bourgeois, F.; Lambergeon, M.; Strochlic, L.; Legay, C. ColQ controls postsynaptic differentiation at the neuromuscular junction. J. Neurosci. 2010, 30, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Sigoillot, S.M.; Bourgeois, F.; Karmouch, J.; Molgó, J.; Dobbertin, A.; Chevalier, C.; Houlgatte, R.; Léger, J.; Legay, C. Neuromuscular junction immaturity and muscle atrophy are hallmarks of the ColQ-deficient mouse, a model of congenital myasthenic syndrome with acetylcholinesterase deficiency. FASEB J. 2016, 30, 2382–2399. [Google Scholar] [CrossRef] [PubMed]
- Krejci, E.; Legay, C.; Thomine, S.; Sketelj, J.; Massoulié, J. Differences in expression of acetylcholinesterase and collagen Q control the distribution and oligomerization of the collagen-tailed forms in fast and slow muscles. J. Neurosci. 1999, 19, 10672–10679. [Google Scholar] [CrossRef]
- Feng, G.; Krejci, E.; Molgo, J.; Cunningham, J.M.; Massoulié, J.; Sanes, J.R. Genetic analysis of collagen Q: Roles in acetylcholinesterase and butyrylcholinesterase assembly and in synaptic structure and function. J. Cell Biol. 1999, 144, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, S.; Desprat, R.; Eymard, B.; Martinat, C.; Lemaitre, J.M.; Legay, C. Generation of a human induced pluripotent stem cell line (iPSC) from peripheral blood mononuclear cells of a patient with a myasthenic syndrome due to mutation in COLQ. Stem Cell Res. 2020, 49, 102106. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, J.; Lara, M.; Ng, F.; Gochez, D.A.; Lee, D.C.; Logia, S.P.; Nguyen, J.; Maselli, R.A. COOH-terminal collagen Q (COLQ) mutants causing human deficiency of endplate acetylcholinesterase impair the interaction of ColQ with proteins of the basal lamina. Hum. Genet. 2014, 133, 599–616. [Google Scholar] [CrossRef]
- Simon, S.; Krejci, E.; Massoulié, J. A four-to-one association between peptide motifs: Four C-terminal domains from cholinesterase assemble with one proline-rich attachment domain (PRAD) in the secretory pathway. EMBO J. 1998, 17, 6178–6187. [Google Scholar] [CrossRef]
- Bon, S.; Coussen, F.; Massoulié, J. Quaternary associations of acetylcholinesterase. II. The polyproline attachment domain of the collagen tail. J. Biol. Chem. 1997, 272, 3016–3021. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, H.; Carvalho, S.; Schmitt, C.; Massoulié, J.; Bon, S. Acetylcholinesterases associates differently with its anchoring proteins ColQ and PRiMA. J. Biol. Chem. 2008, 283, 20722–20732. [Google Scholar] [CrossRef] [PubMed]
- Dobbertin, A.; Hrabovska, A.; Dembele, K.; Camp, S.; Taylor, P.; Krejci, E.; Bernard, V. Targeting of acetylcholinesterase in neurons in vivo: A dual processing function for the proline-rich membrane anchor subunit and the attachment domain on the catalytic subunit. J. Neurosci. 2009, 29, 4519–4530. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, V.; Müller, J.S.; Vilchez, J.J.; Salih, M.A.; Kabiraj, M.M.; D’Amico, A.; Bertini, E.; Wölfle, J.; Schreiner, F.; Kurlemann, G.; et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain 2008, 131, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Cetin, H.; Beeson, D.; Vincent, A.; Webster, R. The Structure, Function, and Physiology of the Fetal and Adult Acetylcholine Receptor in muscle. Front. Mol. Neurosci. 2020, 13, 581097. [Google Scholar] [CrossRef]
- Ohno, K.; Engel, A.G.; Brengman, J.M.; Shen, X.M.; Heidenreich, F.; Vincent, A.; Milone, M.; Tan, E.; Demirci, M.; Walsh, P.; et al. The spectrum of mutations causing end-plate acetylchlolinesterase deficiency. Ann. Neurol. 2000, 47, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Wargon, I.; Richard, P.; Kuntzer, T.; Sternberg, D.; Nafissi, S.; Gaudon, K.; Lebail, A.; Bauche, S.; Hantaï, D.; Fournier, E.; et al. Long-term follow-up of patients with congenital myasthenic syndrome caused by COLQ mutations. Neuromuscul. Disord. 2012, 22, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Legay, C.; Bon, S.; Massoulié, J. Expression of a cDNA encoding the glycolipid-anchored form of rat acetylcholinesterase. FEBS Lett. 1993, 315, 163–166. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Mérien, A.; Tahraoui-Bories, J.; Cailleret, M.; Dupont, J.B.; Leteur, C.; Polentes, J.; Carteron, A.; Polvèche, H.; Concordet, J.P.; Pinset, C.; et al. CRISPR gene editing in pluripotent stem cells reveals the function of MBNL proteins during human in vitro myogenesis. Hum. Mol. Genet. 2021, 31, 41–56. [Google Scholar] [CrossRef]
- Tahraoui-Bories, J.; Mérien, A.; González-Barriga, A.; Lainé, J.; Leteur, C.; Polvèche, H.; Carteron, A.; De Lamotte, J.D.; Nicoleau, C.; Polentes, J.; et al. MBNL-dependent impaired development within the neuromuscular system in myotonic dystrophy type 1. Neuropathol. Appl. Neurobiol. 2023, 49, e12876. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbeau, S.; Semprez, F.; Dobbertin, A.; Merriadec, L.; Roussange, F.; Eymard, B.; Sternberg, D.; Fournier, E.; Karasoy, H.; Martinat, C.; et al. Molecular Analysis of a Congenital Myasthenic Syndrome Due to a Pathogenic Variant Affecting the C-Terminus of ColQ. Int. J. Mol. Sci. 2023, 24, 16217. https://doi.org/10.3390/ijms242216217
Barbeau S, Semprez F, Dobbertin A, Merriadec L, Roussange F, Eymard B, Sternberg D, Fournier E, Karasoy H, Martinat C, et al. Molecular Analysis of a Congenital Myasthenic Syndrome Due to a Pathogenic Variant Affecting the C-Terminus of ColQ. International Journal of Molecular Sciences. 2023; 24(22):16217. https://doi.org/10.3390/ijms242216217
Chicago/Turabian StyleBarbeau, Susie, Fannie Semprez, Alexandre Dobbertin, Laurine Merriadec, Florine Roussange, Bruno Eymard, Damien Sternberg, Emmanuel Fournier, Hanice Karasoy, Cécile Martinat, and et al. 2023. "Molecular Analysis of a Congenital Myasthenic Syndrome Due to a Pathogenic Variant Affecting the C-Terminus of ColQ" International Journal of Molecular Sciences 24, no. 22: 16217. https://doi.org/10.3390/ijms242216217
APA StyleBarbeau, S., Semprez, F., Dobbertin, A., Merriadec, L., Roussange, F., Eymard, B., Sternberg, D., Fournier, E., Karasoy, H., Martinat, C., & Legay, C. (2023). Molecular Analysis of a Congenital Myasthenic Syndrome Due to a Pathogenic Variant Affecting the C-Terminus of ColQ. International Journal of Molecular Sciences, 24(22), 16217. https://doi.org/10.3390/ijms242216217