


Comprehensive Analysis of CircRNA Expression Profiles in Multiple Tissues of Pigs

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. CircRNA Detection in Multiple Tissues
2.2. CircRNA Clustering Analysis
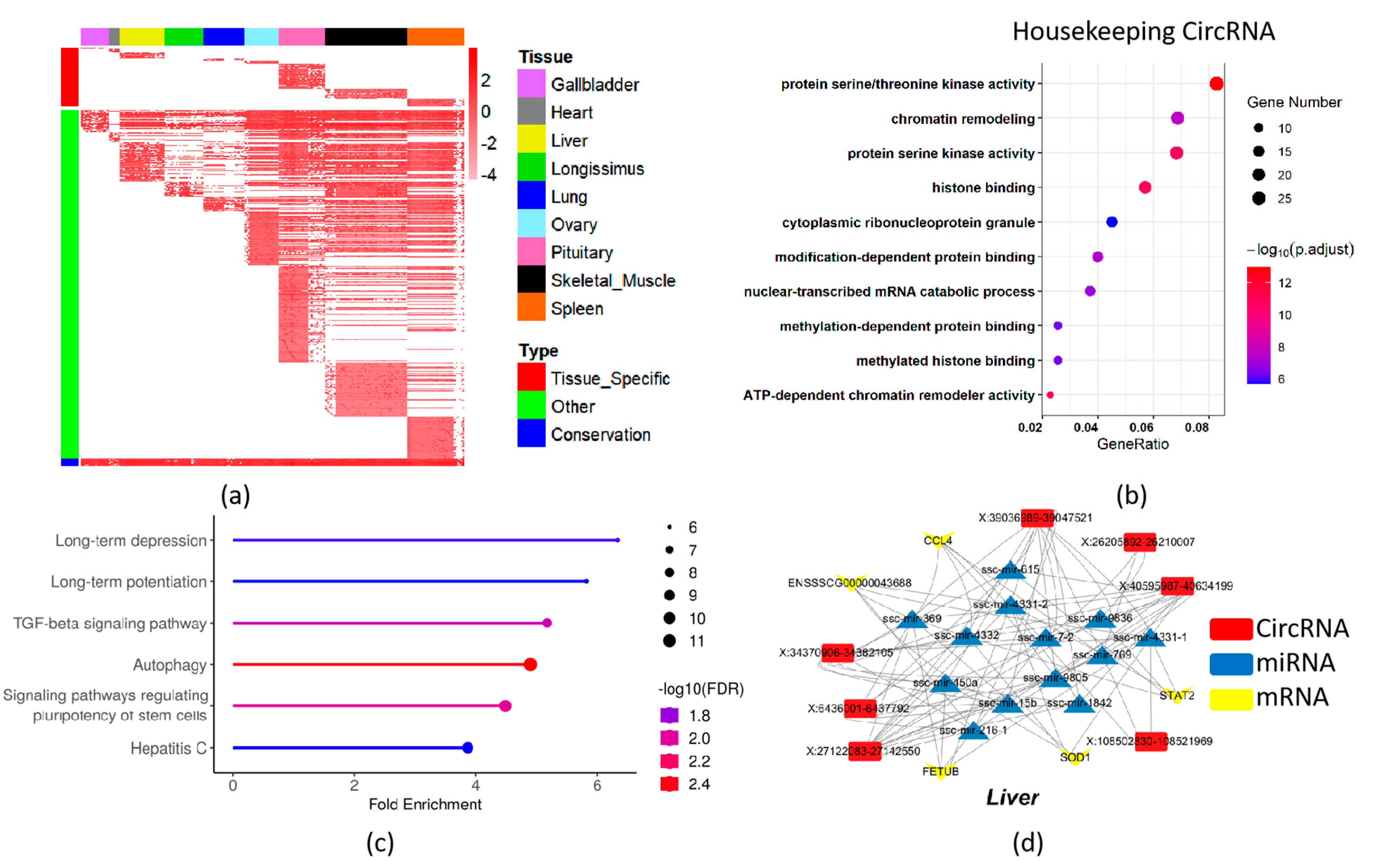
2.3. Tissue-Specific and Housekeeping CircRNA Profile
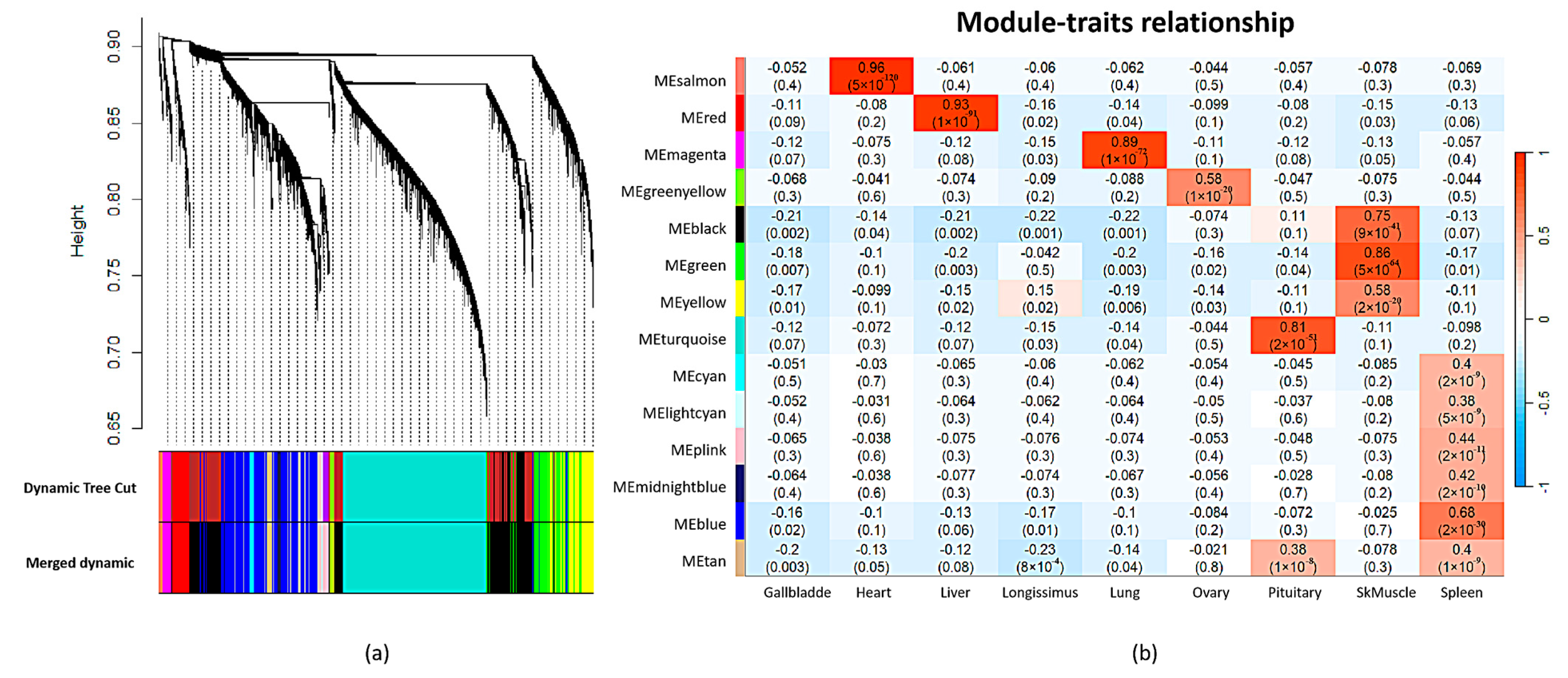
2.4. Co-Expression CircRNA Network across Pig Tissues
2.5. Conservation between Pig and Human
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Quality Control and Alignment
4.3. CircRNA Detection
4.4. CircRNA Profile between Tissues
4.5. Identification of Housekeeping and Tissue-Specific CircRNAs
4.6. GO and KEGG Enrichment
4.7. Competing Endogenous RNA Network Construction
4.8. Co-Expression Network Analysis
4.9. Association Analysis of Traits
4.10. Conservation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanger, H.L.; Klotz, G.; Riesner, D.; Kleinschmidt, G. Viroids are single-strandedcovalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef]
- Lei, M.; Zheng, G.; Ning, Q.; Zheng, J.; Dong, D. Translation and functional roles of circular RNAs in human cancer. Mol. Cancer 2020, 19, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Wang, P.; Fu, X.; Lin, W. Circular RNAs in renal cell carcinoma: Implications for tumorigenesis, diagnosis, and therapy. Mol. Cancer 2020, 19, 149. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Liu, E.; Yang, Z.; Dhaliwal, P.; Yang, B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016, 44, 2846–2858. [Google Scholar] [CrossRef]
- Holdt, L.M.; Kohlmaier, A.; Teupser, D. Molecular roles and function of circular RNAs in eukaryotic cells. Cell. Mol. Life Sci. 2018, 75, 1071–1098. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef]
- Xiao, M.S.; Ai, Y.; Wilusz, J.E. Biogenesis and Functions of Circular RNAs Come into Focus. Trends Cell Biol. 2020, 30, 226–240. [Google Scholar] [CrossRef]
- Liu, L.; Gu, M.; Ma, J.; Wang, Y.; Li, M.; Wang, H.; Yin, X.; Li, X. CircGPR137B/miR-4739/FTO feedback loop suppresses tumorigenesis and metastasis of hepatocellular carcinoma. Mol. Cancer 2022, 21, 149. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, H.Y.; Dai, X.Y.; Zhang, X.; Huang, Y.Z.; Shi, L.; Wei, J.F.; Ding, Q. CircMETTL3, upregulated in a m6A-dependent manner, promotes breast cancer progression. Int. J. Biol. Sci. 2021, 17, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Panda, A.C.; Munk, R.; Grammatikakis, I.; Dudekula, D.B.; De, S.; Kim, J.; Noh, J.H.; Kim, K.M.; Martindale, J.L.; et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017, 14, 361–369. [Google Scholar] [CrossRef]
- Alberio, R. Regulation of Cell Fate Decisions in Early Mammalian Embryos. Annu. Rev. Anim. Biosci. 2020, 8, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Baxter, T.; Muir, W.M.; Groenen, M.A.; Schook, L.B. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int. J. Biol. Sci. 2007, 3, 153–165. [Google Scholar] [CrossRef]
- Li, M.; Tian, S.; Jin, L.; Zhou, G.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.; Chen, L.; Ma, J.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013, 45, 1431–1438. [Google Scholar] [CrossRef]
- Li, M.; Zhang, N.; Li, J.; Ji, M.; Zhao, T.; An, J.; Cai, C.; Yang, Y.; Gao, P.; Cao, G.; et al. CircRNA Profiling of Skeletal Muscle in Two Pig Breeds Reveals CircIGF1R Regulates Myoblast Differentiation via miR-16. Int. J. Mol. Sci. 2023, 24, 3779. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.F.; Ma, Q.; Mo, D.L.; Sun, J.J.; Ren, Q.L.; Zhang, J.Q.; Lu, Q.X.; Xing, B.S. Identification and characterization of circRNAs related to meat quality during embryonic development of the longissimus dorsi muscle in two pig breeds. Front. Genet. 2022, 13, 1019687. [Google Scholar] [CrossRef]
- Zhuang, X.; Lin, Z.; Xie, F.; Luo, J.; Chen, T.; Xi, Q.; Zhang, Y.; Sun, J. Identification of circRNA-associated ceRNA networks using longissimus thoracis of pigs of different breeds and growth stages. BMC Genom. 2022, 23, 294. [Google Scholar] [CrossRef]
- Niu, X.; Huang, Y.; Lu, H.; Li, S.; Huang, S.; Ran, X.; Wang, J. CircRNAs in Xiang pig ovaries among diestrus and estrus stages. Porc. Health Manag. 2022, 8, 29. [Google Scholar] [CrossRef]
- Mester-Tonczar, J.; Winkler, J.; Einzinger, P.; Hasimbegovic, E.; Kastner, N.; Lukovic, D.; Zlabinger, K.; Spannbauer, A.; Traxler, D.; Batkai, S.; et al. Association between Circular RNA CDR1as and Post-Infarction Cardiac Function in Pig Ischemic Heart Failure: Influence of the Anti-Fibrotic Natural Compounds Bufalin and Lycorine. Biomolecules 2020, 10, 1180. [Google Scholar] [CrossRef]
- Groenen, M.A. A decade of pig genome sequencing: A window on pig domestication and evolution. Genet. Sel. Evol. 2016, 48, 23. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Jin, J.; Xu, Z.; Guo, Y.; Wang, X.; Wang, S.; Zhang, J.; Zuo, H.; Bai, W.; Peng, Y.; et al. lncMGPF is a novel positive regulator of muscle growth and regeneration. J. Cachexia Sarcopenia Muscle 2020, 11, 1723–1746. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Liu, M.; Liu, Q.; Li, H.; Qu, Y.; Gao, X.; Huang, C.; Luo, G.; Cao, G.; Xu, D. miRNA let-7 family regulated by NEAT1 and ARID3A/NF-κB inhibits PRRSV-2 replication in vitro and in vivo. PLoS Pathog. 2022, 18, e1010820. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A database for functional circular RNAs. Database 2019, 2019, baz003. [Google Scholar] [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Jin, L.; Tang, Q.; Hu, S.; Chen, Z.; Zhou, X.; Zeng, B.; Wang, Y.; He, M.; Li, Y.; Gui, L.; et al. A pig BodyMap transcriptome reveals diverse tissue physiologies and evolutionary dynamics of transcription. Nat. Commun. 2021, 12, 3715. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Hansen, T.B.; Venø, M.T.; Damgaard, C.K.; Kjems, J. Comparison of circular RNA prediction tools. Nucleic Acids Res. 2016, 44, e58. [Google Scholar] [CrossRef]
- Pan, X.; Gong, W.; He, Y.; Li, N.; Zhang, H.; Zhang, Z.; Li, J.; Yuan, X. Ovary-derived circular RNAs profile analysis during the onset of puberty in gilts. BMC Genomics 2021, 22, 445. [Google Scholar] [CrossRef]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef]
- Tian, J.; Fu, Y.; Li, Q.; Xu, Y.; Xi, X.; Zheng, Y.; Yu, L.; Wang, Z.; Yu, B.; Tian, J. Differential Expression and Bioinformatics Analysis of CircRNA in PDGF-BB-Induced Vascular Smooth Muscle Cells. Front. Genet. 2020, 11, 530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, X.; Pei, J.; Chu, M.; Ding, X.; Wu, X.; Liang, C.; Yan, P. CircRNA Expression Profile during Yak Adipocyte Differentiation and Screen Potential circRNAs for Adipocyte Differentiation. Genes 2020, 11, 414. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Xiang, Y.; Ko, J.; Li, S.; Jing, Y.; Zhu, X.; Ye, Y.; Zhang, Z.; Mills, T.; Feng, J.; et al. Comprehensive characterization of circular RNAs in ~ 1000 human cancer cell lines. Genome Med. 2019, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, M.; van Opbergen, C.J.M.; Bagwan, N.; Vissing, C.R.; Marrón-Liñares, G.M.; Zhang, M.; Torres Vega, E.; Sorrentino, A.; Drici, L.; Sulek, K.; et al. Loss of Nuclear Envelope Integrity and Increased Oxidant Production Cause DNA Damage in Adult Hearts Deficient in PKP2: A Molecular Substrate of ARVC. Circulation 2022, 146, 851–867. [Google Scholar] [CrossRef]
- Cherian, A.V.; Fukuda, R.; Augustine, S.M.; Maischein, H.M.; Stainier, D.Y. N-cadherin relocalization during cardiac trabeculation. Proc. Natl. Acad. Sci. USA 2016, 113, 7569–7574. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, H.; Zhang, X.; Jiang, X.; Liu, Y.; Zhang, H.; Peng, Y.; Li, D.; Yu, Y.; Zhang, J.; et al. N6-methyladenosine modification governs liver glycogenesis by stabilizing the glycogen synthase 2 mRNA. Nat. Commun. 2022, 13, 7038. [Google Scholar] [CrossRef]
- Schartner, V.; Romero, N.B.; Donkervoort, S.; Treves, S.; Munot, P.; Pierson, T.M.; Dabaj, I.; Malfatti, E.; Zaharieva, I.T.; Zorzato, F.; et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 2017, 133, 517–533. [Google Scholar] [CrossRef]
- McKelvey, M.C.; Abladey, A.A.; Small, D.M.; Doherty, D.F.; Williams, R.; Scott, A.; Spek, C.A.; Borensztajn, K.S.; Holsinger, L.; Booth, R.; et al. Cathepsin S Contributes to Lung Inflammation in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2022, 205, 769–782. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, B.; Xu, X.; Zhang, J.; Tong, X.; Wang, Y.; Dong, Z.; Zhang, X.; Shen, N.; Zhai, Y.; et al. PBX1 expression in uterine natural killer cells drives fetal growth. Sci. Transl. Med. 2020, 12, eaax1798. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Sorrentino, S.; Medalia, O.; Korkhov, V.M. The structure of a membrane adenylyl cyclase bound to an activated stimulatory G protein. Science 2019, 364, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; MacArthur, D.G.; Gulbin, J.P.; Hahn, A.G.; Beggs, A.H.; Easteal, S.; North, K. ACTN3 genotype is associated with human elite athletic performance. Am. J. Hum. Genet. 2003, 73, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Glawe, J.D.; Patrick, D.R.; Huang, M.; Sharp, C.D.; Barlow, S.C.; Kevil, C.G. Genetic deficiency of Itgb2 or ItgaL prevents autoimmune diabetes through distinctly different mechanisms in NOD/LtJ mice. Diabetes 2009, 58, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.R.; Holmes, H.; Rakshit, K.; Javeed, N.; Her, T.K.; Stiller, A.A.; Sen, S.; Shull, G.E.; Prakash, Y.S.; Romero, M.F.; et al. Electrogenic sodium bicarbonate cotransporter NBCe1 regulates pancreatic β cell function in type 2 diabetes. J. Clin. Investig. 2021, 131, e142365. [Google Scholar] [CrossRef]
- Andersson, E.R.; Chivukula, I.V.; Hankeova, S.; Sjöqvist, M.; Tsoi, Y.L.; Ramsköld, D.; Masek, J.; Elmansuri, A.; Hoogendoorn, A.; Vazquez, E.; et al. Mouse Model of Alagille Syndrome and Mechanisms of Jagged1 Missense Mutations. Gastroenterology 2018, 154, 1080–1095. [Google Scholar] [CrossRef]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Devis-Jauregui, L.; Eritja, N.; Davis, M.L.; Matias-Guiu, X.; Llobet-Navàs, D. Autophagy in the physiological endometrium and cancer. Autophagy 2021, 17, 1077–1095. [Google Scholar] [CrossRef]
- Huang, Y.H.; Cai, K.; Xu, P.P.; Wang, L.; Huang, C.X.; Fang, Y.; Cheng, S.; Sun, X.J.; Liu, F.; Huang, J.Y.; et al. CREBBP/EP300 mutations promoted tumor progression in diffuse large B-cell lymphoma through altering tumor-associated macrophage polarization via FBXW7-NOTCH-CCL2/CSF1 axis. Signal Transduct. Target. Ther. 2021, 6, 10. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Zhang, J. AdipoR1-mediated miR-3908 inhibits glioblastoma tumorigenicity through downregulation of STAT2 associated with the AMPK/SIRT1 pathway. Oncol. Rep. 2017, 37, 3387–3396. [Google Scholar] [CrossRef]
- Gong, L.P.; Chen, J.N.; Dong, M.; Xiao, Z.D.; Feng, Z.Y.; Pan, Y.H.; Zhang, Y.; Du, Y.; Zhang, J.Y.; Bi, Y.H.; et al. Epstein-Barr virus-derived circular RNA LMP2A induces stemness in EBV-associated gastric cancer. EMBO Rep. 2020, 21, e49689. [Google Scholar] [CrossRef]
- Guo, J.U.; Agarwal, V.; Guo, H.; Bartel, D.P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; Gao, Y.; Yin, H.; Bai, Z.; Liu, S.; Zeng, H.; Bai, L.; Cai, Z.; Zhao, B.; Li, X.; et al. A compendium of genetic regulatory effects across pig tissues. bioRxiv 2022. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chen, C.Y.; Mai, T.L.; Chuang, C.F.; Chen, Y.C.; Gupta, S.K.; Yen, L.; Wang, Y.D.; Chuang, T.J. Genome-wide, integrative analysis of circular RNA dysregulation and the corresponding circular RNA-microRNA-mRNA regulatory axes in autism. Genome Res. 2020, 30, 375–391. [Google Scholar] [CrossRef]
- Kobak, D.; Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 2019, 10, 5416. [Google Scholar] [CrossRef]
- Spearman, C. The proof and measurement of association between two things. Int. J. Epidemiol. 2010, 39, 1137–1150. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, T.; Niu, Q.; Xu, L.; Chen, Y.; Gao, X.; Gao, H.; Zhang, L.; Liu, G.E.; Li, J.; et al. Transcriptional atlas analysis from multiple tissues reveals the expression specificity patterns in beef cattle. BMC Biol. 2022, 20, 79. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Pan, X.; Cai, J.; Wang, Y.; Xu, D.; Jiang, Y.; Gong, W.; Tian, Y.; Shen, Q.; Zhang, Z.; Yuan, X.; et al. Expression Profile of Housekeeping Genes and Tissue-Specific Genes in Multiple Tissues of Pigs. Animals 2022, 12, 3539. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinformatics 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Liang, W.; Sun, F.; Zhao, Y.; Shan, L.; Lou, H. Identification of Susceptibility Modules and Genes for Cardiovascular Disease in Diabetic Patients Using WGCNA Analysis. J. Diabetes Res. 2020, 2020, 4178639. [Google Scholar] [CrossRef]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Frequency | |
|---|---|---|
| Detected CircRNAs | 24,745 | / |
| Aligned CircRNAs | 19,078 | 77.10% |
| Conserved CircRNAs | 4774 | 19.29% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Q.; Gong, W.; Pan, X.; Cai, J.; Jiang, Y.; He, M.; Zhao, S.; Li, Y.; Yuan, X.; Li, J. Comprehensive Analysis of CircRNA Expression Profiles in Multiple Tissues of Pigs. Int. J. Mol. Sci. 2023, 24, 16205. https://doi.org/10.3390/ijms242216205
Shen Q, Gong W, Pan X, Cai J, Jiang Y, He M, Zhao S, Li Y, Yuan X, Li J. Comprehensive Analysis of CircRNA Expression Profiles in Multiple Tissues of Pigs. International Journal of Molecular Sciences. 2023; 24(22):16205. https://doi.org/10.3390/ijms242216205
Chicago/Turabian StyleShen, Qingpeng, Wentao Gong, Xiangchun Pan, Jiali Cai, Yao Jiang, Mingran He, Shanghui Zhao, Yipeng Li, Xiaolong Yuan, and Jiaqi Li. 2023. "Comprehensive Analysis of CircRNA Expression Profiles in Multiple Tissues of Pigs" International Journal of Molecular Sciences 24, no. 22: 16205. https://doi.org/10.3390/ijms242216205
APA StyleShen, Q., Gong, W., Pan, X., Cai, J., Jiang, Y., He, M., Zhao, S., Li, Y., Yuan, X., & Li, J. (2023). Comprehensive Analysis of CircRNA Expression Profiles in Multiple Tissues of Pigs. International Journal of Molecular Sciences, 24(22), 16205. https://doi.org/10.3390/ijms242216205