
Structural and pKa Estimation of the Amphipathic HR1 in SARS-CoV-2: Insights from Constant pH MD, Linear vs. Nonlinear Normal Mode Analysis


Abstract
1. Introduction


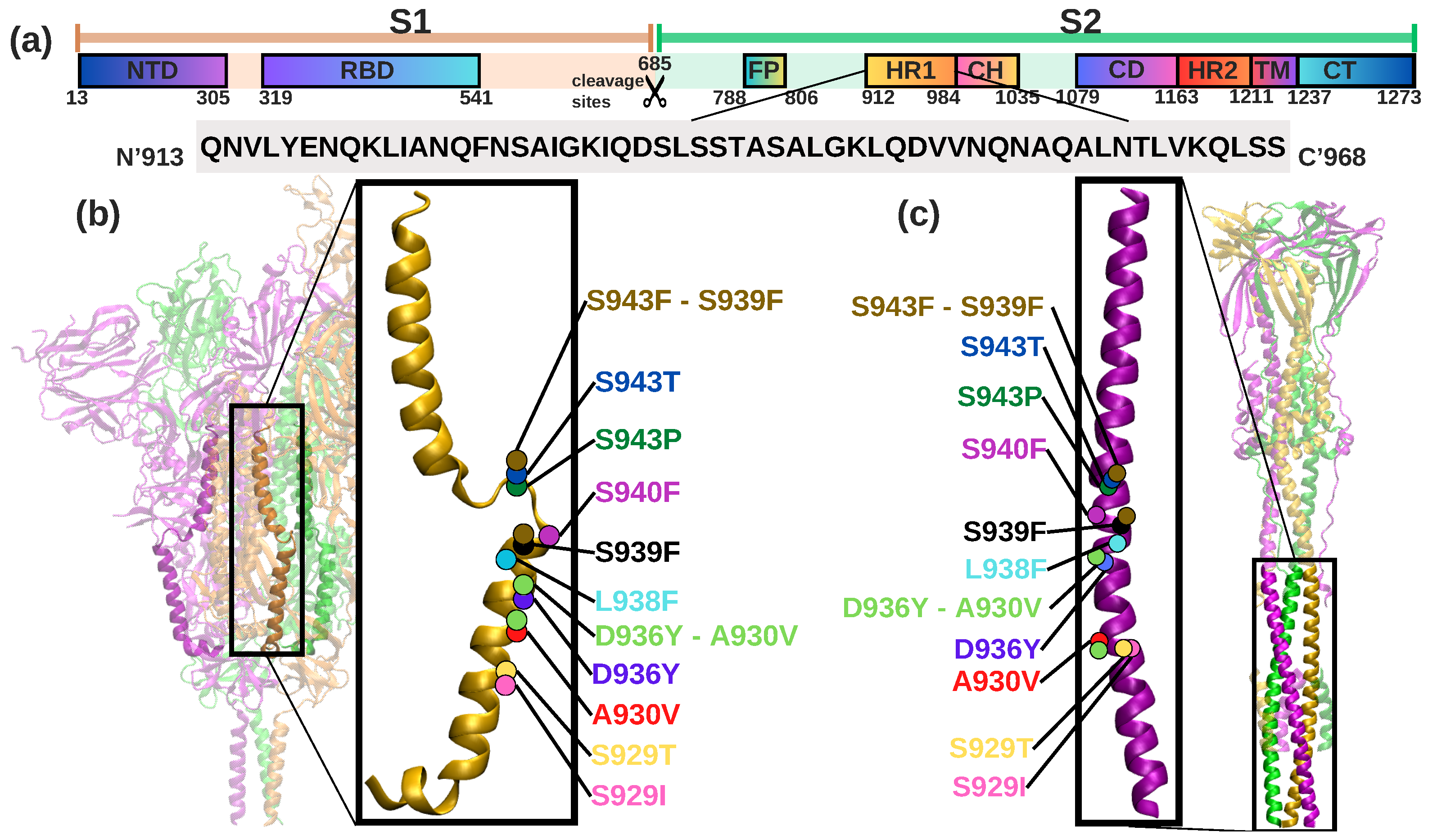{kind=link}
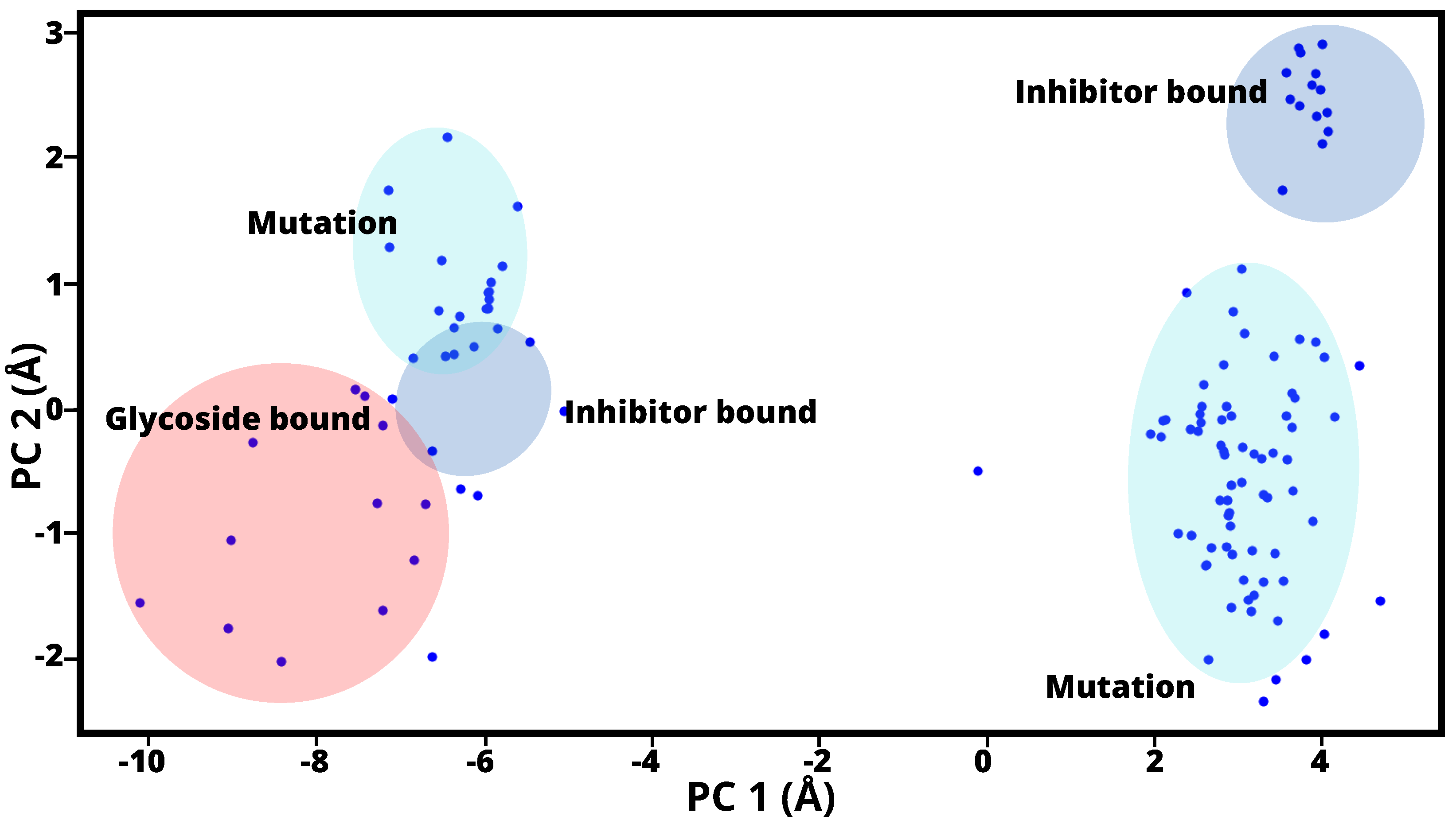{kind=link}
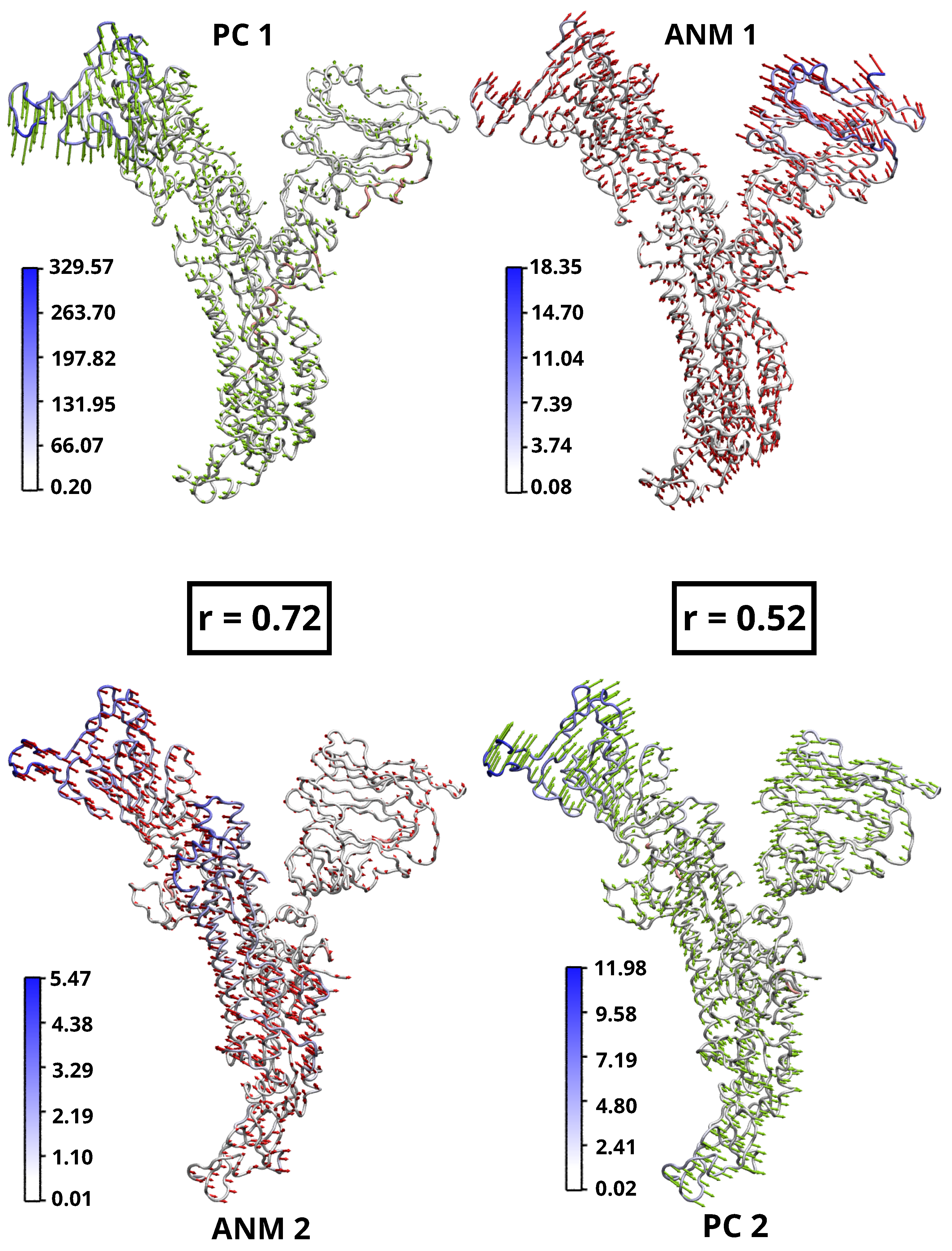{kind=link}
{kind=link}
{kind=link}
{kind=link}
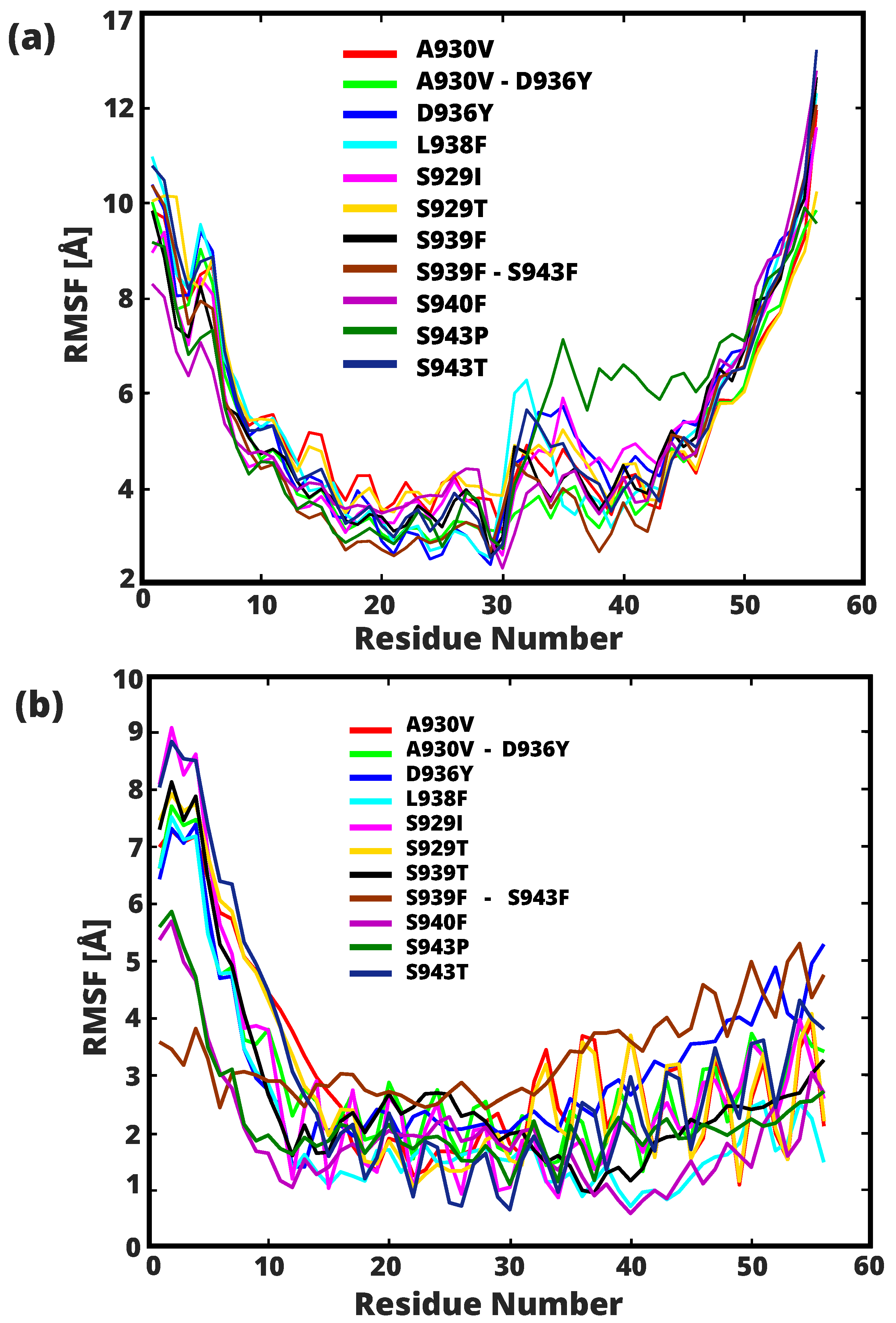{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2. Results and Discussion
2.1. PCA
2.2. Conformational Flexibility and Structural Stability of HR1 Transition
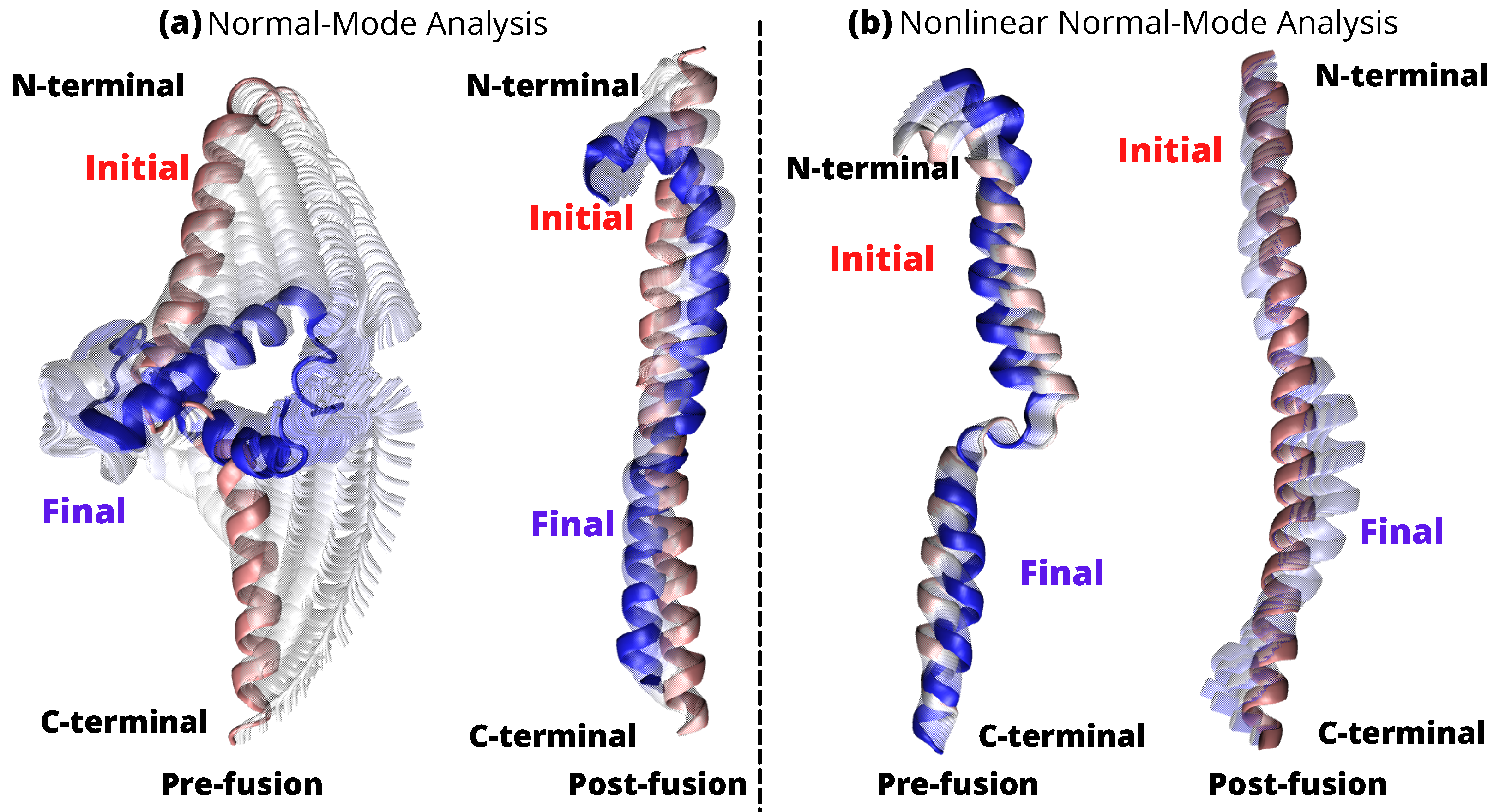
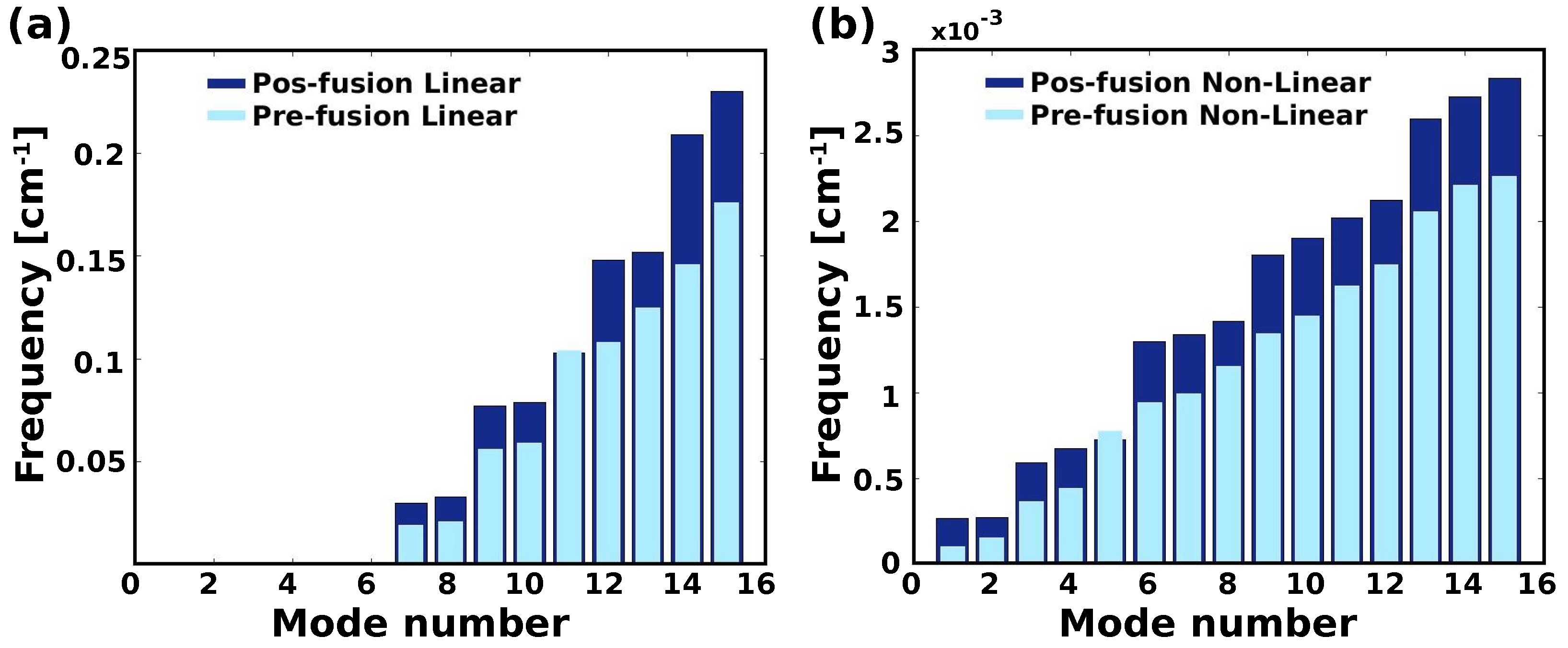
2.3. Linear vs. Nonlinear-NMA of HR1

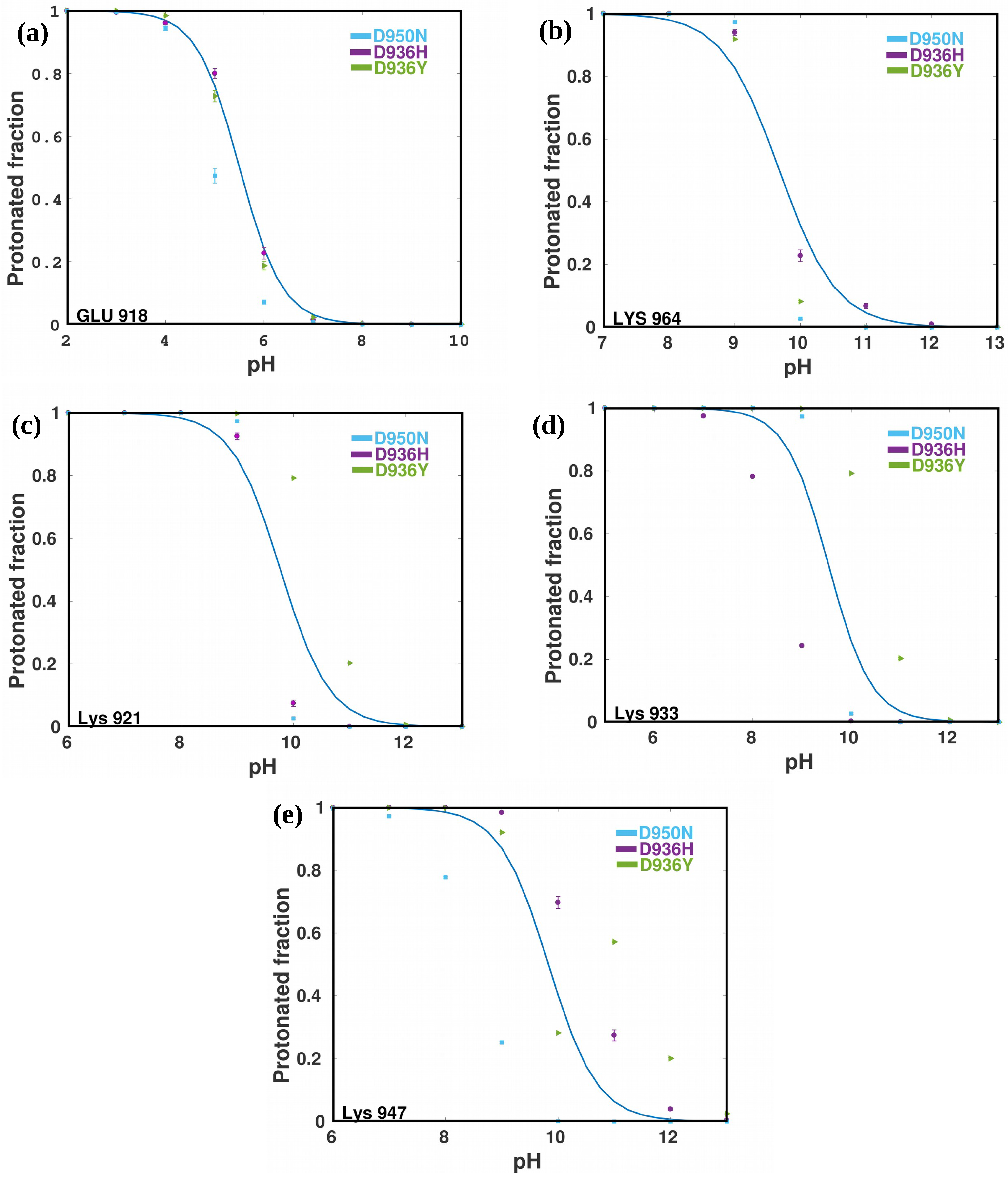
2.4. Prediction of pKa Values For HR1
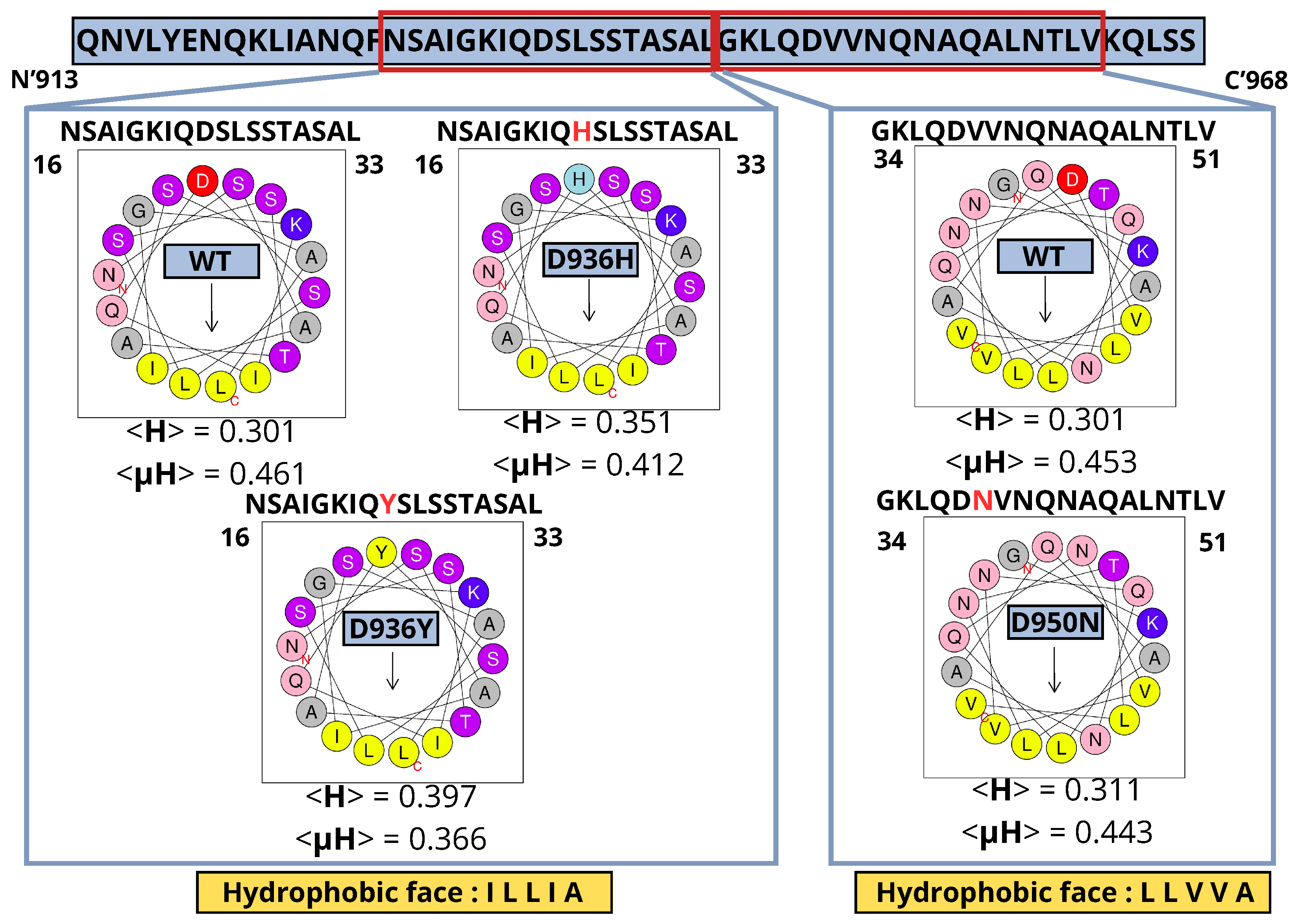
2.5. Presence of an Amphipathic Helix in HR1 Wild Type (WT) and Mutants; D950N, D936Y and D936H
3. Materials and Methods
3.1. Retrieve Dataset
3.2. Preparation of Pre-Fusion State Mutants
3.3. PCA of the Ensemble
3.4. ANM Analysis and Overlap with Modes of PCA
3.5. Linear Normal Mode Analysis (Linear-NMA)
3.6. Nonlinear Normal Mode Analysis (NonLinear NMA)
3.7. cpH-MD Protocol
3.8. Amphipathic Helix Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 Simulations Go Exascale to Predict Dramatic Spike Opening and Cryptic Pockets across the Proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The Endosomal-Lysosomal System: From Acidification and Cargo Sorting to Neurodegeneration. Transl. Neurodegener. 2015, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, Y.; Ruigrok, R.W.H.; Brunner, J. Low-pH Induced Conformational Changes in Viral Fusion Proteins: Implications for the Fusion Mechanism. J. Gen. Virol. 1995, 76, 1541–1556. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Gui, M.; Xiang, Y. Structural Intermediates in the Low pH-Induced Transition of Influenza Hemagglutinin. PLoS Pathog. 2020, 16, e1009062. [Google Scholar] [CrossRef]
- Caffrey, M.; Lavie, A. pH-Dependent Mechanisms of Influenza Infection Mediated by Hemagglutinin. Front. Mol. Biosci. 2021, 8, 777095. [Google Scholar] [CrossRef]
- Gaudin, Y.; Ruigrok, R.W.; Knossow, M.; Flamand, A. Low-pH Conformational Changes of Rabies Virus Glycoprotein and Their Role in Membrane Fusion. J. Virol. 1993, 67, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberger, A.J.B.; Sanyal, A.; Saminathan, A.; Bloyet, L.-M.; Stumpf, S.; Liu, Z.; Ojha, R.; Patjas, M.T.; Geneid, A.; Scanavachi, G.; et al. SARS-CoV-2 Requires Acidic pH to Infect Cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2209514119. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.M.; Dolgova, N.V.; Landreth, S.; Fisette, O.; Pickering, I.J.; George, G.N.; Falzarano, D.; Cygler, M. Disulfide Bonds Play a Critical Role in the Structure and Function of the Receptor-Binding Domain of the SARS-CoV-2 Spike Antigen. J. Mol. Biol. 2022, 434, 167357. [Google Scholar] [CrossRef] [PubMed]
- Birtles, D.; Oh, A.E.; Lee, J. Exploring the pH Dependence of the SARS-CoV-2 Complete Fusion Domain and the Role of Its Unique Structural Features. Protein Sci. 2022, 31, e4390. [Google Scholar] [CrossRef]
- Case, D.A. Normal Mode Analysis of Protein Dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal Mode Analysis of Biomolecular Structures: Functional Mechanisms of Membrane Proteins. Chem. Rev. 2009, 110, 1463–1497. [Google Scholar] [CrossRef]
- Ma, J. Usefulness and Limitations of Normal Mode Analysis in Modeling Dynamics of Biomolecular Complexes. Structure 2005, 13, 373–380. [Google Scholar] [CrossRef]
- Skjaerven, L.; Hollup, S.M.; Reuter, N. Normal Mode Analysis for Proteins. J. Mol. Struct. Theochem 2009, 898, 42–48. [Google Scholar] [CrossRef]
- Mahajan, S.; Sanejouand, Y.-H. On the Relationship between Low-Frequency Normal Modes and the Large-Scale Conformational Changes of Proteins. Arch. Biochem. Biophys. 2015, 567, 59–65. [Google Scholar] [CrossRef]
- Brooks, B.R.; Janezic, D.; Karplus, M. Harmonic Analysis of Large Systems. I. Methodology. J. Comput. Chem. 1995, 16, 1522–1542. [Google Scholar] [CrossRef]
- Mészáros, B.; Park, E.; Malinverni, D.; Sejdiu, B.I.; Immadisetty, K.; Sandhu, M.; Lang, B.; Babu, M.M. Recent Breakthroughs in Computational Structural Biology Harnessing the Power of Sequences and Structures. Curr. Opin. Struct. Biol. 2023, 80, 102608. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Chaudhuri, D.; Datta, J.; Giri, K. Exploring the Intrinsic Dynamics of SARS-CoV-2, SARS-CoV and MERS-CoV Spike Glycoprotein through Normal Mode Analysis Using Anisotropic Network Model. J. Mol. Graph. Model. 2021, 102, 107778. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Grudinin, S. NOLB: Nonlinear Rigid Block Normal-Mode Analysis Method. J. Chem. Theory Comput. 2017, 13, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yin, W.; Jiang, Y.; Xu, H.E. Structure Genomics of SARS-CoV-2 and Its Omicron Variant: Drug Design Templates for COVID-19. Acta Pharmacol. Sin. 2022, 43, 3021–3033. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rao, Z. Structural Biology of SARS-CoV-2 and Implications for Therapeutic Development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Hardenbrook, N.J.; Zhang, P. A Structural View of the SARS-CoV-2 Virus and Its Assembly. Curr. Opin. Virol. 2022, 52, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.; Zhu, G.; Yang, M.; Cao, L.; Xing, X.; Yin, G.; Chan, C.; Qin, C.; Rao, Z.; Wang, X.; et al. Nanometer-Resolution in Situ Structure of the SARS-CoV-2 Postfusion Spike Protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2112703118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, X.; Du, L. Therapeutic Antibodies and Fusion Inhibitors Targeting the Spike Protein of SARS-CoV-2. Expert Opin. Ther. Targets 2020, 25, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Oliva, R.; Shaikh, A.R.; Petta, A.; Vangone, A.; Cavallo, L. D936Y and Other Mutations in the Fusion Core of the SARS-CoV-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect. Molecules 2021, 26, 2622. [Google Scholar] [CrossRef] [PubMed]
- Samal, S.; Khattar, S.K.; Kumar, S.; Collins, P.L.; Samal, S.K. Coordinate Deletion of N-Glycans from the Heptad Repeats of the Fusion F Protein of Newcastle Disease Virus Yields a Hyperfusogenic Virus with Increased Replication, Virulence, and Immunogenicity. J. Virol. 2012, 86, 2501–2511. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Newby, M.L.; Fogarty, C.A.; Allen, J.D.; Butler, J.; Fadda, E.; Crispin, M. Variations within the Glycan Shield of SARS-CoV-2 Impact Viral Spike Dynamics. J. Mol. Biol. 2023, 435, 167928. [Google Scholar] [CrossRef]
- Farhud, D.D.; Mojahed, N. SARS-COV-2 Notable Mutations and Variants: A Review Article. Iran. J. Public Health 2022, 51, 1494. [Google Scholar] [CrossRef]
- Pang, W.; Lu, Y.; Zhao, Y.-B.; Shen, F.; Fan, C.-F.; Wang, Q.; He, W.-Q.; He, X.-Y.; Li, Z.-K.; Chen, T.-T.; et al. A Variant-Proof SARS-CoV-2 Vaccine Targeting HR1 Domain in S2 Subunit of Spike Protein. Cell Res. 2022, 32, 1068–1085. [Google Scholar] [CrossRef]
- Bosch, B.J.; Martina, B.E.E.; van der Zee, R.; Lepault, J.; Haijema, B.J.; Versluis, C.; Heck, A.J.R.; de Groot, R.; Osterhaus, A.D.M.E.; Rottier, P.J.M. Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection Inhibition Using Spike Protein Heptad Repeat-Derived Peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 8455–8460. [Google Scholar] [CrossRef]
- Bianchini, F.; Crivelli, V.; Abernathy, M.E.; Guerra, C.; Palus, M.; Muri, J.; Marcotte, H.; Piralla, A.; Pedotti, M.; De Gasparo, R.; et al. Human Neutralizing Antibodies to Cold Linear Epitopes and Subdomain 1 of the SARS-CoV-2 Spike Glycoprotein. Sci. Immunol. 2023, 8, eade0958. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane Fusion and Immune Evasion by the Spike Protein of SARS-CoV-2 Delta Variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Hart, W.S.; Miller, E.; Andrews, N.J.; Waight, P.; Maini, P.K.; Funk, S.; Thompson, R.N. Generation Time of the Alpha and Delta SARS-CoV-2 Variants: An Epidemiological Analysis. Lancet Infect. Dis. 2022, 22, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- Aljindan, R.Y.; Al-Subaie, A.M.; Al-Ohali, A.I.; Kumar D, T.; Doss C, G.P.; Kamaraj, B. Investigation of Nonsynonymous Mutations in the Spike Protein of SARS-CoV-2 and Its Interaction with the ACE2 Receptor by Molecular Docking and MM/GBSA Approach. Comput. Biol. Med. 2021, 135, 104654. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Kiso, M.; Iida, S.; Uraki, R.; Hirata, Y.; Imai, M.; Suzuki, T.; Yamayoshi, S.; Kawaoka, Y. In SARS-CoV-2 Delta Variants, Spike-P681R and D950N Promote Membrane Fusion, Spike-P681R Enhances Spike Cleavage, but Neither Substitution Affects Pathogenicity in Hamsters. eBioMedicine 2023, 91, 104561. [Google Scholar] [CrossRef]
- Guruprasad, L. Human SARS-CoV-2 Spike Protein Mutations. Proteins 2021, 89, 569–576. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.Org): Delivery of Experimentally-Determined PDB Structures alongside One Million Computed Structure Models of Proteins from Artificial Intelligence/Machine Learning. Nucleic Acids Res. 2022, 51, D488–D508. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Tahir Khan, M.; Saleem, S.; Junaid, M.; Ali, A.; Shujait Ali, S.; Khan, M.; Wei, D.-Q. Structural Insights into the Mechanism of RNA Recognition by the N-Terminal RNA-Binding Domain of the SARS-CoV-2 Nucleocapsid Phosphoprotein. Comput. Struct. Biotechnol. J. 2020, 18, 2174–2184. [Google Scholar] [CrossRef]
- Khan, M.H.R.; Hossain, A. Machine Learning Approaches Reveal That the Number of Tests Do Not Matter to the Prediction of Global Confirmed COVID-19 Cases. Front. Artif. Intell. 2020, 3, 561801. [Google Scholar] [CrossRef]
- Park, S.-J.; Kern, N.; Brown, T.; Lee, J.; Im, W. CHARMM-GUI PDB Manipulator: Various PDB Structural Modifications for Biomolecular Modeling and Simulation. J. Mol. Biol. 2023, 435, 167995. [Google Scholar] [CrossRef]
- Fatoki, T.H.; Ibraheem, O.; Ogunyemi, I.O.; Akinmoladun, A.C.; Ugboko, H.U.; Adeseko, C.J.; Awofisayo, O.A.; Olusegun, S.J.; Enibukun, J.M. Network Analysis, Sequence and Structure Dynamics of Key Proteins of Coronavirus and Human Host, and Molecular Docking of Selected Phytochemicals of Nine Medicinal Plants. J. Biomol. Struct. Dyn. 2020, 39, 6195–6217. [Google Scholar] [CrossRef]
- Wang, B. Adjusting Extracellular pH to Prevent Entry of SARS-CoV-2 into Human Cells. Genome 2021, 64, 595–598. [Google Scholar]
- Kumawat, N.; Tucs, A.; Bera, S.; Chuev, G.N.; Fedotova, M.V.; Tsuda, K.; Kruchinin, S.E.; Sljoka, A.; Hakraborty, A. Prefusion Conformation of SARS-CoV-2 Receptor-Binding Domain Favours Interactions with Human Receptor ACE2; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2021. [Google Scholar]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Xie, Y.; Guo, W.; Lopez-Hernadez, A.; Teng, S.; Li, L. The pH Effects on SARS-CoV and SARS-CoV-2 Spike Proteins in the Process of Binding to hACE2. Pathogens 2022, 11, 238. [Google Scholar] [CrossRef]
- Abdella, S.; Abid, F.; Youssef, S.H.; Kim, S.; Afinjuomo, F.; Malinga, C.; Song, Y.; Garg, S. pH and Its Applications in Targeted Drug Delivery. Drug Discov. Today 2023, 28, 103414. [Google Scholar] [CrossRef] [PubMed]
- Isom, D.G.; Castañeda, C.A.; Cannon, B.R.; García-Moreno E., B. Large Shifts in pK a Values of Lysine Residues Buried inside a Protein. Proc. Natl. Acad. Sci. USA 2011, 108, 5260–5265. [Google Scholar] [CrossRef]
- Al Adem, K.; Ferreira, J.C.; Fadl, S.; Rabeh, W.M. pH Profiles of 3-Chymotrypsin-like Protease (3CLpro) from SARS-CoV-2 Elucidate Its Catalytic Mechanism and a Histidine Residue Critical for Activity. J. Biol. Chem. 2023, 299, 102790. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef] [PubMed]
- Heximer, S.P.; Lim, H.; Bernard, J.L.; Blumer, K.J. Mechanisms Governing Subcellular Localization and Function of Human RGS2. J. Biol. Chem. 2001, 276, 14195–14203. [Google Scholar] [CrossRef]
- Su, Y.; Li, S.; Hong, M. Cationic Membrane Peptides: Atomic-Level Insight of Structure–Activity Relationships from Solid-State NMR. Amino Acids 2012, 44, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.M.; Aiken, E.J.; Beres, K.A.; Hahn, A.R.; Kamin, S.J.; Hagness, S.C.; Booske, J.H.; Murphy, W.L. Cationic Peptide Exposure Enhances Pulsed-Electric-Field-Mediated Membrane Disruption. PLoS ONE 2014, 9, e92528. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Calavia, P.; González-Acosta, S.; Otazo-Pérez, A.; López, M.R.; Morales-delaNuez, A.; Pérez de la Lastra, J.M. Teleost Piscidins—In Silico Perspective of Natural Peptide Antibiotics from Marine Sources. Antibiotics 2023, 12, 855. [Google Scholar] [CrossRef]
- Guo, X.; Steinkühler, J.; Marin, M.; Li, X.; Lu, W.; Dimova, R.; Melikyan, G.B. Interferon-Induced Transmembrane Protein 3 Blocks Fusion of Diverse Enveloped Viruses by Altering Mechanical Properties of Cell Membranes. ACS Nano 2021, 15, 8155–8170. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Bahar, I. The Intrinsic Dynamics of Enzymes Plays a Dominant Role in Determining the Structural Changes Induced upon Inhibitor Binding. Proc. Natl. Acad. Sci. USA 2009, 106, 14349–14354. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Islam, S.M.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K.; et al. CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Nonstandard Residues. Adv. Protein Chem. Struct. Biol. 2014, 96, 235–265. [Google Scholar]
- Stoddard, S.V.; Wallace, F.E.; Stoddard, S.D.; Cheng, Q.; Acosta, D.; Barzani, S.; Bobay, M.; Briant, J.; Cisneros, C.; Feinstein, S.; et al. In Silico Design of Peptide-Based SARS-CoV-2 Fusion Inhibitors That Target WT and Mutant Versions of SARS-CoV-2 HR1 Domains. Biophysica 2021, 1, 311–327. [Google Scholar] [CrossRef]
- Gopalan, V.; Chandran, A.; Arumugam, K.; Sundaram, M.; Velladurai, S.; Govindan, K.; Azhagesan, N.; Jeyavel, P.; Dhandapani, P.; Sivasubramanian, S.; et al. Distribution and Functional Analyses of Mutations in Spike Protein and Phylogenic Diversity of SARS-CoV-2 Variants Emerged during the Year 2021 in India. J. Glob. Infect. Dis. 2023, 15, 43–51. [Google Scholar] [CrossRef]
- Yang, K.; Wang, C.; White, K.I.; Pfuetzner, R.A.; Esquivies, L.; Brunger, A.T. Structural Conservation among Variants of the SARS-CoV-2 Spike Postfusion Bundle. Proc. Natl. Acad. Sci. USA 2022, 119, e2119467119. [Google Scholar] [CrossRef]
- Cosar, B.; Karagulleoglu, Z.Y.; Unal, S.; Ince, A.T.; Uncuoglu, D.B.; Tuncer, G.; Kilinc, B.R.; Ozkan, Y.E.; Ozkoc, H.C.; Demir, I.N.; et al. SARS-CoV-2 Mutations and Their Viral Variants. Cytokine Growth Factor Rev. 2022, 63, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhuang, H. Profiling and Characterization of SARS-CoV-2 Mutants’ Infectivity and Antigenicity. Sig. Transduct. Target. Ther. 2020, 5, 185. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Ribeiro, J.V.; Bernardi, R.C.; Rudack, T.; Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Schulten, K. QwikMD—Integrative Molecular Dynamics Toolkit for Novices and Experts. Sci. Rep. 2016, 6, 26536. [Google Scholar] [CrossRef]
- Meireles, L.; Gur, M.; Bakan, A.; Bahar, I. Pre-Existing Soft Modes of Motion Uniquely Defined by Native Contact Topology Facilitate Ligand Binding to Proteins. Protein Sci. 2011, 20, 1645–1658. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Kim, M.K.; Jernigan, R.L.; Chirikjian, G.S. Efficient Generation of Feasible Pathways for Protein Conformational Transitions. Biophys. J. 2002, 83, 1620–1630. [Google Scholar] [CrossRef]
- Lindahl, E.; Azuara, C.; Koehl, P.; Delarue, M. NOMAD-Ref: Visualization, Deformation and Refinement of Macromolecular Structures Based on All-Atom Normal Mode Analysis. Nucleic Acids Res. 2006, 34, W52–W56. [Google Scholar] [CrossRef]
- Radak, B.K.; Chipot, C.; Suh, D.; Jo, S.; Jiang, W.; Phillips, J.C.; Schulten, K.; Roux, B. Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput. 2017, 13, 5933–5944. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A Web Server to Screen Sequences with Specific α-Helical Properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]










| ANM1 | ANM2 | ANM3 | ANM4 | ANM5 | ANM6 | ANM7 | ANM8 | |
|---|---|---|---|---|---|---|---|---|
| () | () | (2.56 ) | (3.15 ) | (6.56 ) | (1.27 ) | (2.06 ) | (2.83 ) | |
| PC1 (85.11) | 0.42 | 0.72 | 0.03 | 0.31 | 0.07 | 0.06 | 0.07 | 0.01 |
| PC2 (8.17) | 0.52 | 0.25 | 0.30 | 0.22 | 0.09 | 0.48 | 0.25 | 0.29 |
| PC3 (2.11) | 0.05 | 0.28 | 0.42 | 0.03 | 0.17 | 0.07 | 0.02 | 0.02 |
| PC4 (1.68) | 0.27 | 0.35 | 0.28 | 0.02 | 0.15 | 0.41 | 0.26 | 0.06 |
| PC5 (0.54) | 0.21 | 0.04 | 0.37 | 0.23 | 0.47 | 0.20 | 0.03 | 0.30 |
| PC6 (0.52) | 0.19 | 0.11 | 0.14 | 0.16 | 0.19 | 0.17 | 0.53 | 0.48 |
| PC7 (0.34) | 0.01 | 0.27 | 0.09 | 0.39 | 0.19 | 0.02 | 0.19 | 0.18 |
| PC8 (0.23) | 0.04 | 0.09 | 0.09 | 0.15 | 0.19 | 0.12 | 0.13 | 0.17 |
| Mutation | Group | pka | pka | pka |
|---|---|---|---|---|
| D950N | ASP 936 | 5.26 | 3.8604 ± 0.0511 | 4.4001 ± 0.0523 |
| GLU 918 | 5.90 | 4.8754 ± 0.0565 | 4.9693 ± 0.0430 | |
| LYS 921 | 9.77 | 11.1691 ± 0.0752 | 9.5000 ± 0.0553 | |
| LYS 933 | 9.54 | 12.1578 ± 0.1182 | 9.5000 ± 0.0553 | |
| LYS 947 | 9.83 | 10.3714 ± 0.0079 | 8.5296 ± 0.0237 | |
| LYS 964 | 9.68 | 10.4851 ± 0.0529 | 9.5000 ± 0.0553 | |
| D936H | ASP 950 | 5.50 | 3.2198 ± 0.0511 | 4.5541 ± 0.0380 |
| HIS 936 | 6.702 | 5.1570 ± 0.0016 | 5.8596 ± 0.0629 | |
| GLU 918 | 5.90 | 3.8604 ± 0.0298 | 5.4325 ± 0.0746 | |
| LYS 921 | 9.77 | 10.3372 ± 0.0170 | 9.5179 ± 0.0213 | |
| LYS 933 | 9.53 | 9.9543 ± 0.0023 | 8.5244 ± 0.0228 | |
| LYS 947 | 9.82 | 11.4573 ± 0.0209 | 10.5839 ± 0.0767 | |
| LYS 964 | 9.68 | 10.4612 ± 0.0755 | 9.7427 ± 0.1314 | |
| D936Y | ASP 950 | 5.51 | 3.6691 ± 0.0866 | 4.0672 ± 0.0809 |
| GLU 918 | 5.90 | 5.7565 ± 0.0755 | 5.6504 ± 0.1109 | |
| LYS 921 | 9.77 | 10.8356 ± 0.0671 | 10.4949 ± 0.0192 | |
| LYS 933 | 9.53 | 10.1694 ± 0.0919 | 10.4949 ± 0.0192 | |
| LYS 947 | 9.82 | 10.9075 ± 0.0345 | 10.4167 ± 0.5285 | |
| LYS 964 | 9.68 | 11.1651 ± 0.0880 | 9.5009 ± 0.0007 |
| Inhibitor bound | 6XCM | 7CWS | 7DZX | 7L02 | 7ONA | 7JWB | 7BYR | 7EJ4 |
| 7DCC | 7A25 | 6Z43 | 7AKD | 7K85 | 7CWL | 7C2L | 7DL1 | |
| 7N9T | 7CAI | 7KMK | 7E8C | 7LRT | 7MKL | 7CHH | 7KL9 | |
| 7L3N | 7R8M | 7SC1 | 7CAC | 6NB6 | 7OAN | 7FAE | 7NS6 | |
| 7LD1 | 7P40 | 7K8S | 7E3K | 7E5R | 7VNC | |||
| Glucoside bound | 6VYB | 6VXX | 6X79 | 7CN9 | 6XLU | 6X6P | 7BNN | 6XF5 |
| 7KDG | 6VSD | 6ZB4 | 7KDJ | 6ZOW | 7JJI | 7E7B | 6XR8 | |
| 7E7D | 6ZP0 | 6ZP1 | 7KDK | 7MTE | 7KD1 | 7A4N | 6ZWV | |
| 7DX1 | 7KRQ | 6ZOY | 6ZOX | 6XS6 | 7LWW | 6XKL | 7MJ9 | |
| 7LWI | 7TLC | 7N1U | 7CAB | 7LYK | 7M8K | 7N1Q | 7EDF | |
| 7K9H | 7CN4 | 7CN8 | 6ZGE | 5X58 | 6CRW | 7SOB | 6X2A | |
| 7TLA | 7KJ2 | 7LAA | 7LQV | 6ACC | 7BBH | 6ZGF | 7SO9 | |
| 7SBP | ||||||||
| Mutation | 7V8C | 7SBK | 7TOU | 7SBS | 7VX1 | 7Q6E | 7V76 | 7LWS |
| 7V7N | 7OD3 | 7SXW | 7SXV | 7V78 | 7FEM | 7SXU | 7V7D | |
| 7N8H | 7SXS | 7SXT | 7W92 | 7MJG | 7SXR | 7VX9 | 7KDI | |
| 7FCD | 7EAZ | 7BNM | 6ZP2 | |||||
| 7DZW | 6ZGG | 6X29 | 7T9J | 7QO7 | 7WK2 | 7TB4 | 7WK4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yánez Arcos, D.L.; Thirumuruganandham, S.P. Structural and pKa Estimation of the Amphipathic HR1 in SARS-CoV-2: Insights from Constant pH MD, Linear vs. Nonlinear Normal Mode Analysis. Int. J. Mol. Sci. 2023, 24, 16190. https://doi.org/10.3390/ijms242216190
Yánez Arcos DL, Thirumuruganandham SP. Structural and pKa Estimation of the Amphipathic HR1 in SARS-CoV-2: Insights from Constant pH MD, Linear vs. Nonlinear Normal Mode Analysis. International Journal of Molecular Sciences. 2023; 24(22):16190. https://doi.org/10.3390/ijms242216190
Chicago/Turabian StyleYánez Arcos, Dayanara Lissette, and Saravana Prakash Thirumuruganandham. 2023. "Structural and pKa Estimation of the Amphipathic HR1 in SARS-CoV-2: Insights from Constant pH MD, Linear vs. Nonlinear Normal Mode Analysis" International Journal of Molecular Sciences 24, no. 22: 16190. https://doi.org/10.3390/ijms242216190
APA StyleYánez Arcos, D. L., & Thirumuruganandham, S. P. (2023). Structural and pKa Estimation of the Amphipathic HR1 in SARS-CoV-2: Insights from Constant pH MD, Linear vs. Nonlinear Normal Mode Analysis. International Journal of Molecular Sciences, 24(22), 16190. https://doi.org/10.3390/ijms242216190