
Transcriptomic Analysis Reveals Functional Interaction of mRNA-lncRNA-miRNA in Trachinotus ovatus Infected by Cryptocaryon irritans

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. Reads Quality Control and Mapping to the Reference Genome
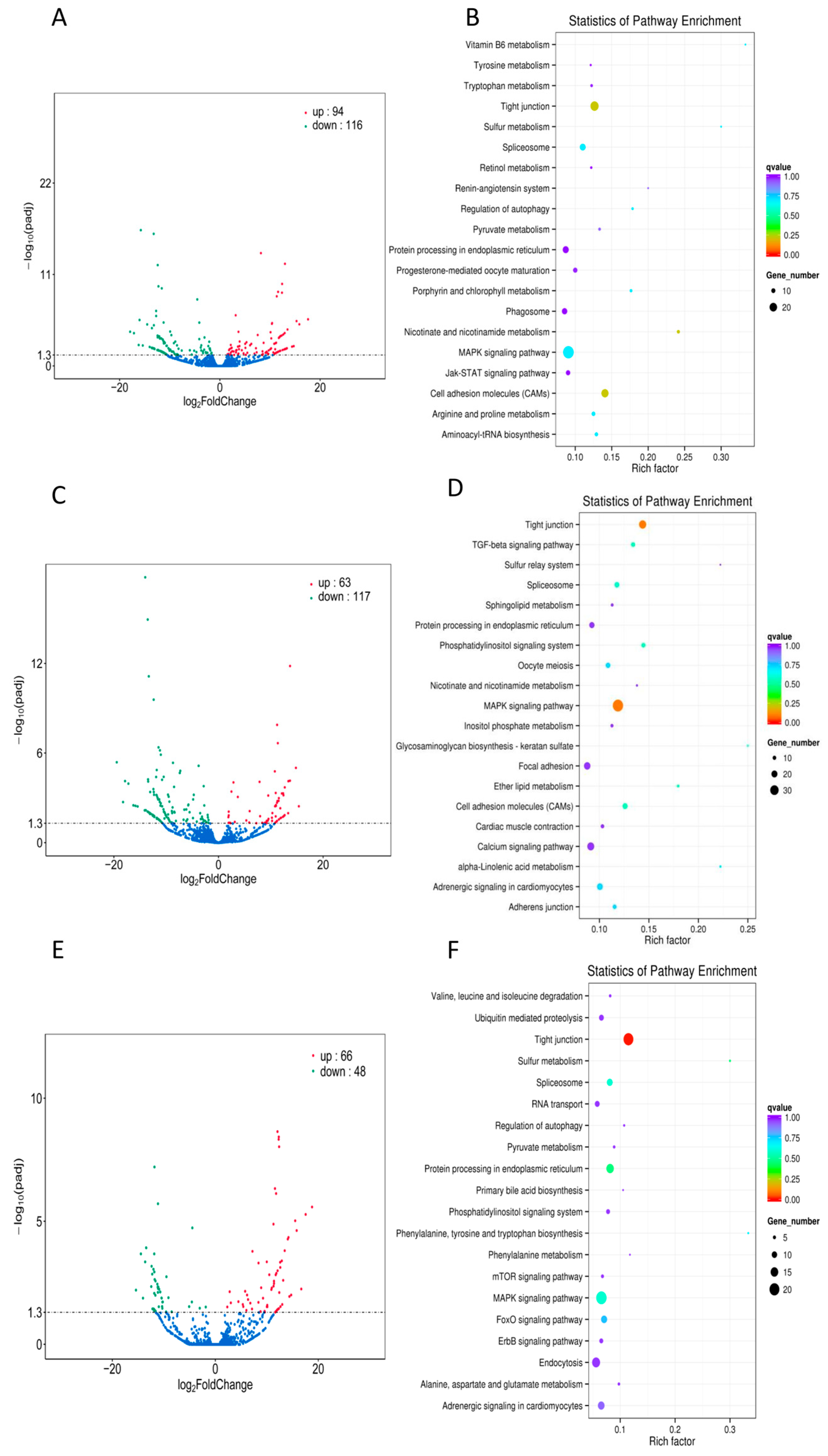
2.2. mRNA Differential Expression Analysis
2.3. Function Enrichment of Differentially Expressed mRNAs
2.4. Target Gene Identification and Functional Analysis of DE lncRNAs
2.5. Target Gene Identification and Functional Analysis of DE miRNAs
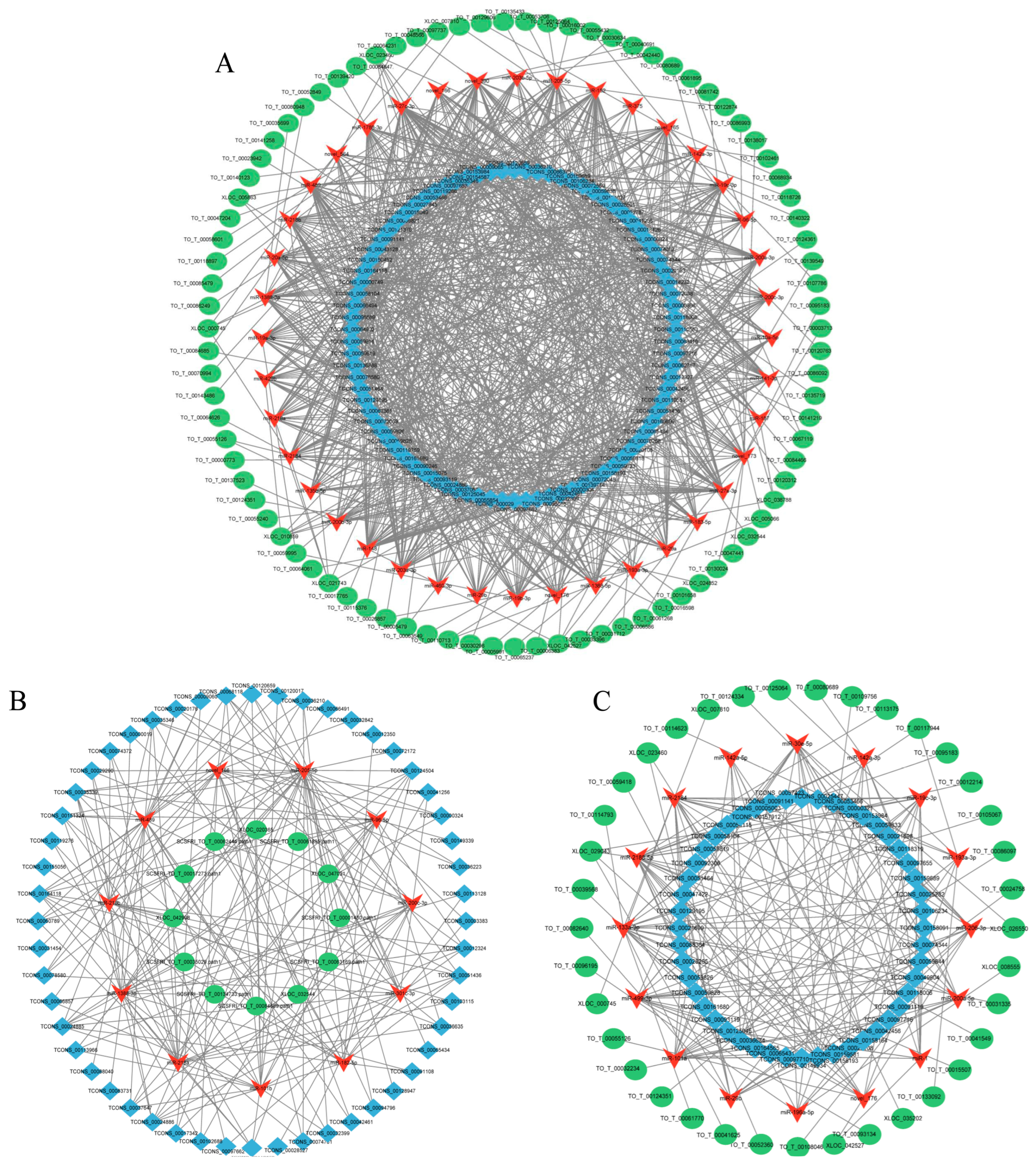
2.6. Construction and Visualization of the lncRNA–miRNA–mRNA Network
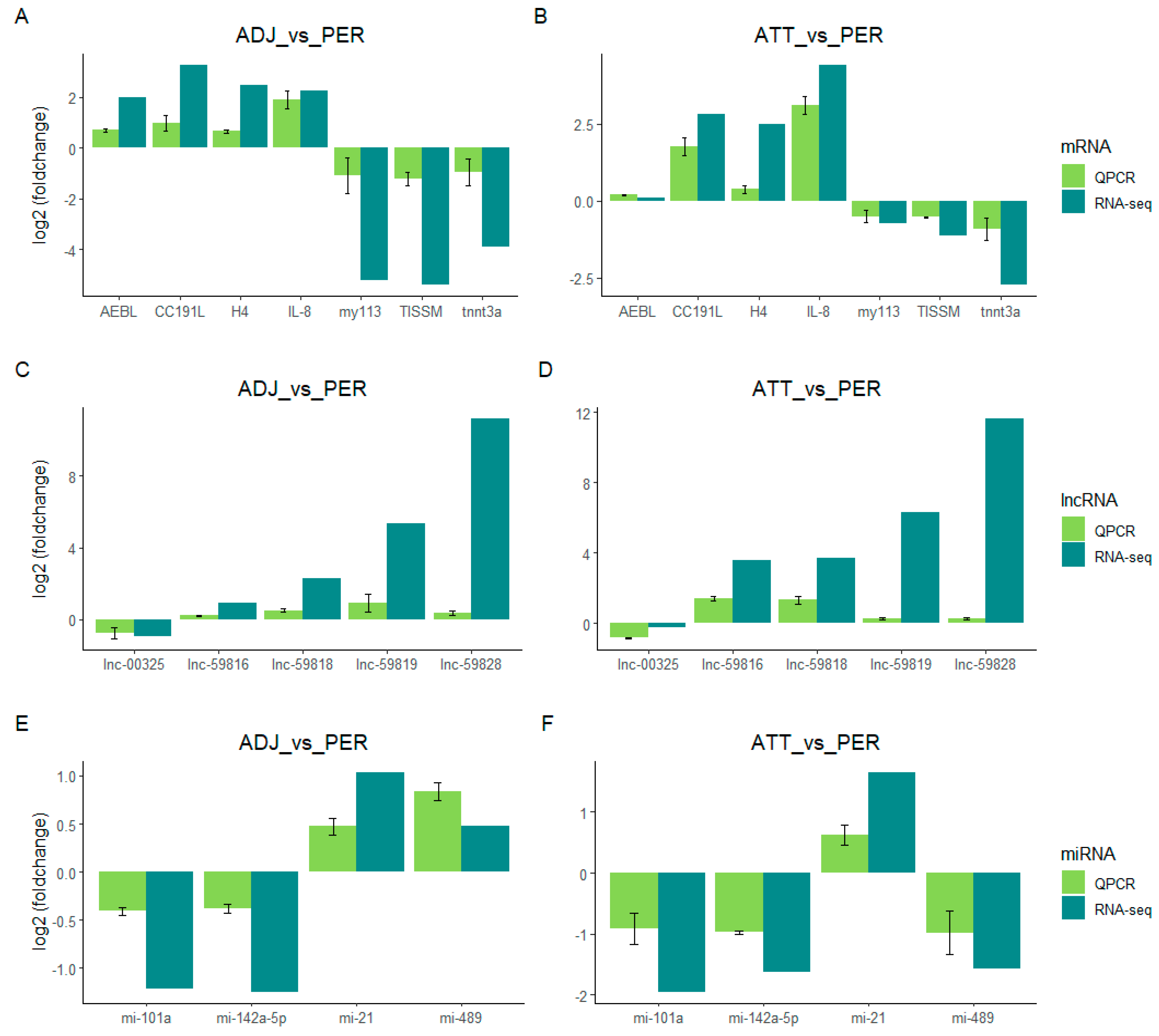
2.7. Validation of RNA-seq by qt-PCR
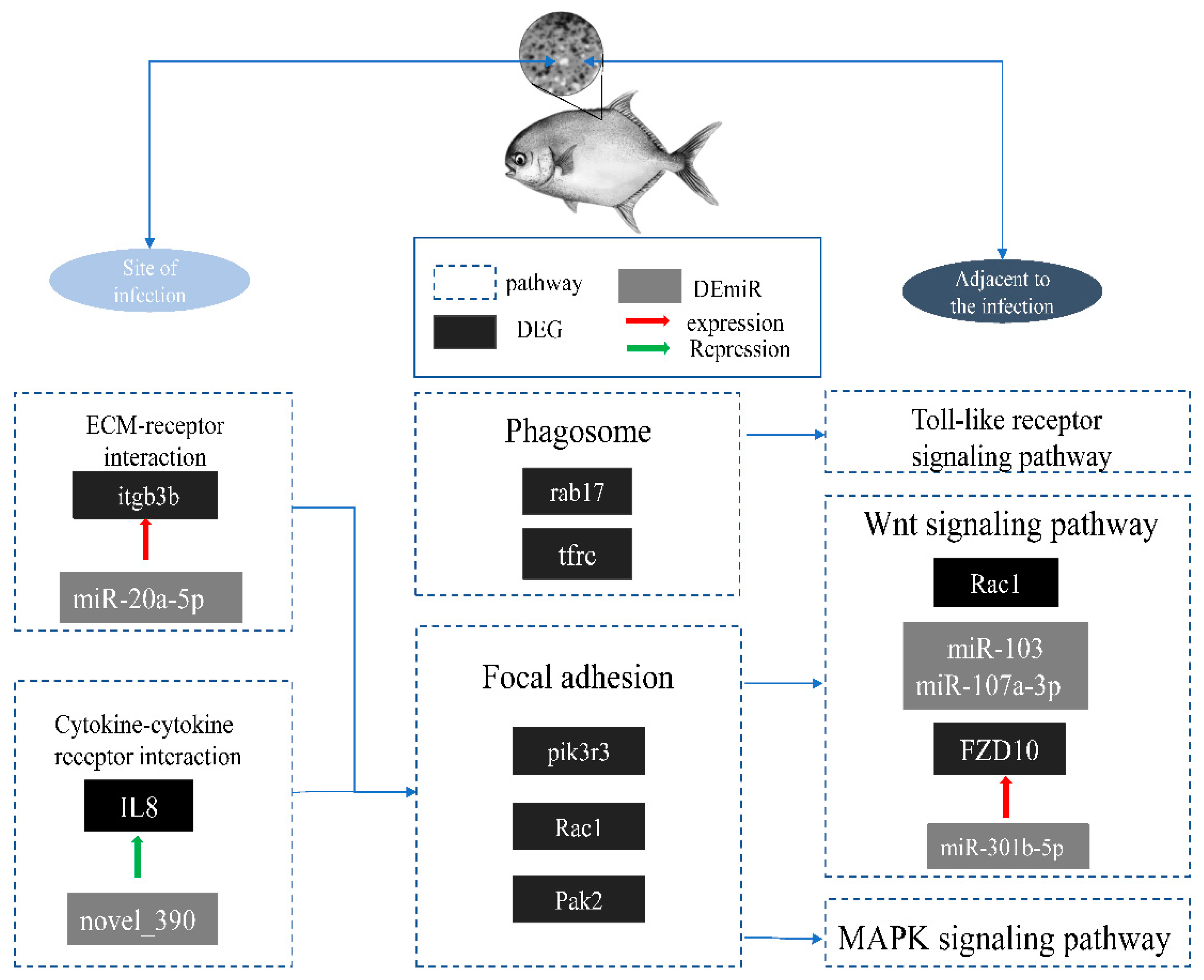
3. Discussion
4. Materials and Methods
4.1. Cryptocaryon irritans and Experimental Fish
4.2. Experimental Methods and Sample Collection
4.3. Total RNA Extraction, Library Construction, and Illumina Sequencing
4.4. Data Analysis and Quality Control
4.5. mRNA Differential Expression and Enrichment Analysis
4.6. LncRNA and miRNA Identification
4.7. Differential Expression and Enrichment Analysis
4.8. Competing Endogenous RNA (ceRNA) Network Construction
4.9. Quantitative Real-Time PCR (qPCR) Verification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.; Jiang, B.; Mo, Z.; Li, A.; Dan, X. Cryptocaryon irritans (Brown, 1951) is a serious threat to aquaculture of marine fish. Rev. Aquac. 2022, 14, 218–236. [Google Scholar] [CrossRef]
- Yin, F.; Liu, W.; Bao, P.; Tang, B. Food intake, survival, and immunity of Nibea albiflora to Cryptocaryon irritans infection. Parasitol. Res. 2018, 117, 2379–2384. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Gong, Q.; Li, Y.; Dan, X.; Sun, P.; Gao, Q.; Shi, Z.; Peng, S.; Li, A. Effects of Cryptocaryon irritans infection on the survival, feeding, respiratory rate and ionic regulation of the marbled rockfish Sebastiscus marmoratus. Parasitology 2014, 141, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, H.; Miao, L.; Fang, W.; Jin, S.; Xie, J.; Zhou, S.; Ma, R.; Li, C. Effect and associated mechanism of copper plates on Cryptocaryon irritans tomonts in large yellow croaker (Larimichthys crocea) farming. Aquaculture 2022, 552, 737981. [Google Scholar] [CrossRef]
- Yin, F.; Liu, W.; Bao, P.; Jin, S.; Qian, D.; Wang, J.; Tang, B. Comparison of the susceptibility and resistance of four marine perciform fishes to Cryptocaryon irritans infection. Fish Shellfish Immunol. 2018, 77, 298–303. [Google Scholar] [CrossRef]
- Maha, I.F.; Xie, X.; Zhou, S.; Yu, Y.; Liu, X.; Zahid, A.; Lei, Y.; Ma, R.; Yin, F.; Qian, D. Skin metabolome reveals immune responses in yellow drum Nibea albiflora to Cryptocaryon irritans infection. Fish Shellfish Immunol. 2019, 94, 661–674. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, X.; Li, R.; Michal, J.J.; Zhang, S.; Dodson, M.V.; Zhang, Z.; Harland, R.M. Whole transcriptome analysis with sequencing: Methods, challenges and potential solutions. Cell. Mol. Life Sci. 2015, 72, 3425–3439. [Google Scholar] [CrossRef]
- Mallory, A.C.; Shkumatava, A. LncRNAs in vertebrates: Advances and challenges. Biochimie 2015, 117, 3–14. [Google Scholar] [CrossRef]
- Obernosterer, G.; Leuschner, P.J.; Alenius, M.; Martinez, J. Post-transcriptional regulation of microRNA expression. RNA 2006, 12, 1161–1167. [Google Scholar] [CrossRef]
- Bizuayehu, T.T.; Babiak, I. MicroRNA in teleost fish. Genome Biol. Evol. 2014, 6, 1911–1937. [Google Scholar] [CrossRef]
- Andreassen, R.; Høyheim, B. miRNAs associated with immune response in teleost fish. Dev. Comp. Immunol. 2017, 75, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-C.; Zhou, Z.-J.; Sun, L. pol-miR-731, a teleost miRNA upregulated by megalocytivirus, negatively regulates virus-induced type I interferon response, apoptosis and cell cycle arrest. Sci. Rep. 2016, 6, 28354. [Google Scholar] [CrossRef] [PubMed]
- Najib, A.; Kim, M.S.; Choi, S.H.; Kang, Y.J.; Kim, K.H. Changes in microRNAs expression profile of olive flounder (Paralichthys olivaceus) in response to viral hemorrhagic septicemia virus (VHSV) infection. Fish Shellfish Immunol. 2016, 51, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Manninga, H.; Slanchev, K.; Chien, M.; Russo, J.J.; Ju, J.; Sheridan, R.; John, B.; Marks, D.S.; Gaidatzis, D.; et al. The developmental miRNA profiles of zebrafish as determined by small RNA cloning. J. Bone Jt. Surg. 2005, 19, 1288–1293. [Google Scholar] [CrossRef]
- Zhao, L.; Lu, H.; Meng, Q.; Wang, J.; Wang, W.; Yang, L.; Lin, L. Profilings of MicroRNAs in the liver of common carp (Cyprinus carpio) infected with Flavobacterium columnare. Int. J. Mol. Sci. 2016, 17, 566. [Google Scholar] [CrossRef]
- Gong, G.; Sha, Z.; Chen, S.; Li, C.; Yan, H.; Chen, Y.; Wang, T. Expression profiling analysis of the microRNA response of Cynoglossus semilaevis to Vibrio anguillarum and other stimuli. Mar. Biotechnol. 2015, 17, 338–352. [Google Scholar] [CrossRef]
- Wang, B.; Gan, Z.; Cai, S.; Wang, Z.; Yu, D.; Lin, Z.; Lu, Y.; Wu, Z.; Jian, J. Comprehensive identification and profiling of Nile tilapia (Oreochromis niloticus) microRNAs response to Streptococcus agalactiae infection through high-throughput sequencing. Fish Shellfish Immunol. 2016, 54, 93–106. [Google Scholar] [CrossRef]
- Paneru, B.; Al-Tobasei, R.; Palti, Y.; Wiens, G.D.; Salem, M. Differential expression of long non-coding RNAs in three genetic lines of rainbow trout in response to infection with Flavobacterium psychrophilum. Sci. Rep. 2016, 6, 36032. [Google Scholar] [CrossRef]
- Boltaña, S.; Valenzuela-Miranda, D.; Aguilar, A.; Mackenzie, S.; Gallardo-Escárate, C. Long noncoding RNAs (lncRNAs) dynamics evidence immunomodulation during ISAV-Infected Atlantic salmon (Salmo salar). Sci. Rep. 2016, 6, 22698. [Google Scholar] [CrossRef]
- Wright, A.-D.G.; Colorni, A. Taxonomic re-assignment of Cryptocaryon irritans, a marine fish parasite. Eur. J. Protistol. 2002, 37, 375–378. [Google Scholar] [CrossRef]
- Jiang, B.; Wang, J.; Luo, H.-L.; Lu, G.-L.; Li, Y.-W.; Li, A.-X. l-amino acid oxidase expression profile and biochemical responses of rabbitfish (Siganus oramin) after exposure to a high dose of Cryptocaryon irritans. Fish Shellfish Immunol. 2017, 69, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Gu, T.; Lu, L.; Cao, Z.; Song, Q.; Wang, Z.; Zhang, Y.; Chang, G.; Xu, Q.; Chen, G. Roles of miRNA-1 and miRNA-133 in the proliferation and differentiation of myoblasts in duck skeletal muscle. J. Cell. Physiol. 2019, 234, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Liu, Y.; Cao, M.; Li, J.; Tian, M.; Zhang, L.; Wang, B.; Liu, X.; Li, C. Transcriptome analysis reveals deep insights into the early immune response of turbot (Scophthalmus maximus) induced by inactivated Aeromonas salmonicida vaccine. Fish Shellfish Immunol. 2021, 119, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, R.; Rangnes, F.; Sivertsen, M.; Chiang, M.; Tran, M.; Worren, M.M. Discovery of miRNAs and their corresponding miRNA genes in atlantic cod (Gadus morhua): Use of stable miRNAs as reference genes reveals subgroups of miRNAs that are highly expressed in particular organs. PLoS ONE 2016, 11, e0153324. [Google Scholar] [CrossRef]
- Liu, S.; Yu, T.; Zhang, Y.; Pan, C.; Cai, L.; Yang, M. Integrated analysis of mRNA and long non-coding RNA expression profiles reveals the potential roles of lncRNA-mRNA network in carp macrophage immune regulation. Vitr. Cell. Dev. Biol.—Anim. 2021, 57, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Chu, Q.; Xu, T. Long noncoding RNA IRL regulates NF-κB-mediated immune responses through suppression of miR-27c-3p-dependent IRAK4 downregulation in teleost fish. J. Biol. Chem. 2021, 296, 100304. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, J.; Wang, G.; Wang, H.; Liu, H. Integrated analysis of lncRNA and mRNA in liver of Megalobrama amblycephala post Aeromonas hydrophila infection. BMC Genom. 2021, 22, 653. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; DePalo, V.A. Anti-inflammatory cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef]
- Steinke, J.; Borish, L. 3. Cytokines and Chemokines. J. Allergy Clin. Immunol. 2006, 117, S441–S445. [Google Scholar] [CrossRef]
- Deng, X.; Tu, Z.; Xiong, M.; Tembo, K.; Zhou, L.; Liu, P.; Pan, S.; Xiong, J.; Yang, X.; Leng, J.; et al. Wnt5a and CCL25 promote adult T-cell acute lymphoblastic leukemia cell migration, invasion and metastasis. Oncotarget 2017, 8, 39033–39047. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, L.; Wang, H.-Q.; Luo, X.-C.; Dan, X.-M.; Li, Y.-W. Molecular cloning and expression analysis of CCL25 and its receptor CCR9s from Epinephelus coioides post Cryptocaryon irritans infection. Fish Shellfish Immunol. 2017, 67, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Song, R.; Wang, X.; Hu, H.; Zhang, Z. Peritoneal bacterial infection repressed the expression of IL17D in Siberia sturgeon a chondrostean fish in the early immune response. Fish Shellfish Immunol. 2017, 64, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Jin, M.; Zhang, Y.; Wei, T.; Bai, Z. Evolution of the IL17 receptor family in chordates: A new subfamily IL17REL. Immunogenetics 2011, 63, 835–845. [Google Scholar] [CrossRef]
- Wang, X.; Li, C.; Thongda, W.; Luo, Y.; Beck, B.; Peatman, E. Characterization and mucosal responses of interleukin 17 family ligand and receptor genes in channel catfish Ictalurus punctatus. Fish Shellfish Immunol. 2014, 38, 47–55. [Google Scholar] [CrossRef]
- Sun, J.L.; Jiang, T.; Gu, Y.; Song, F.B.; Wen, X.; Luo, J. Differential immune and metabolic responses underlie differences in the resistance of Siganus oramin and Trachinotus blochii to Cryptocaryon irritans infection. Fish Shellfish Immunol. 2021, 120, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, J.; Su, Y.-Q.; Mao, Y.; Zhang, J.-S.; Wu, C.-W.; Ke, Q.-Z.; Han, K.-H.; Zheng, W.-Q.; Xu, N.-D. Transcriptome analysis of the Larimichthys crocea liver in response to Cryptocaryon irritans. Fish Shellfish Immunol. 2016, 48, 1–11. [Google Scholar] [CrossRef]
- Falasca, M.; Maffucci, T. Role of class II phosphoinositide 3-kinase in cell signalling. Biochem. Soc. Trans. 2007, 35, 211–214. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Fruman, D.A. Pi3ks in lymphocyte signaling and development. Curr. Top. Microbiol. Immunol. 2010, 346, 57–85. [Google Scholar]
- Li, H.X.; Qiang, J.; Song, C.Y.; Xu, P. Acanthopanax senticosus promotes survival of tilapia infected with Streptococcus iniae by regulating the PI3K/AKT and fatty acid metabolism signaling pathway. Front. Physiol. 2021, 12, 699247. [Google Scholar] [CrossRef]
- Huang, X.; Wang, W.; Huang, Y.; Xu, L.; Qin, Q. Involvement of the PI3K and ERK signaling pathways in largemouth bass virus-induced apoptosis and viral replication. Fish Shellfish Immunol. 2014, 41, 371–379. [Google Scholar] [CrossRef]
- Fan, K.; Shen, Y.; Xu, X.; Tao, L.; Bao, T.; Li, J. LncRNA-WAS and lncRNA-C8807 interact with miR-142a-3p to regulate the inflammatory response in grass carp. Fish Shellfish Immunol. 2021, 111, 201–207. [Google Scholar] [CrossRef]
- Dong, W.; Geng, S.; Cui, J.; Gao, W.; Sun, Y.; Xu, T. MicroRNA-103 and microRNA-190 negatively regulate NF-κB-mediated immune responses by targeting IL-1R1 in Miichthys miiuy. Fish Shellfish Immunol. 2022, 123, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zheng, W.; Sun, Y.; Xu, T. microRNA-489 negatively modulates RIG-I signaling pathway via targeting TRAF6 in miiuy croaker after poly(I:C) stimulation. Fish Shellfish Immunol. 2021, 113, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, W.; Zhang, Y.; Cui, Y.; Xu, S.; Li, S. Bisphenol A regulates cytochrome P450 1B1 through miR-27b-3p and induces carp lymphocyte oxidative stress leading to apoptosis. Fish Shellfish Immunol. 2020, 102, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.K.; Han, D.S. Toll-like receptors signaling and regulation of immune response. Prog. Biochem. Biophys. 2006, 33, 820–827. [Google Scholar]
- Cen, X.; Liu, S.; Cheng, K. The role of toll-like receptor in inflammation and tumor immunity. Front. Pharmacol. 2018, 9, 878. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Pasare, C.; Medzhitov, R. Toll-like receptors: Linking innate and adaptive immunity. Microbes Infect. 2004, 6, 1382–1387. [Google Scholar] [CrossRef]
- Chang, Z.L. Important aspects of Toll-like receptors, ligands and their signaling pathways. Inflamm. Res. 2010, 59, 791–808. [Google Scholar] [CrossRef]
- Li, Y.W.; Dan, X.M.; Zhang, T.W.; Luo, X.C.; Li, A.X. Immune-related genes expression profile in orange-spotted grouper during exposure to Cryptocaryon irritans. Parasite Immunol. 2011, 33, 679–987. [Google Scholar] [CrossRef]
- Dan, X.; Li, A.; Lin, X.; Teng, N.; Zhu, X. A standardized method to propagate Cryptocaryon irritans on a susceptible host pompano Trachinotus ovatus. Aquaculture 2006, 258, 127–133. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.-C.; Konaté, M.M.; Chen, L.; Das, B.; Karlovich, C.; Williams, P.M.; Evrard, Y.A.; Doroshow, J.H.; McShane, L.M. TPM, FPKM, or Normalized Counts? A comparative study of quantification measures for the analysis of RNA-seq data from the NCI patient-derived models repository. J. Transl. Med. 2021, 19, 269. [Google Scholar] [CrossRef] [PubMed]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.-Y.; Dillies, M.-A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample_Name | Raw_Reads | Clean_Reads | Error Rate (%) | Q20 (%) | Q30 (%) | GC_Content (%) | Total Mapped (%) |
|---|---|---|---|---|---|---|---|
| ADJ_1 | 82,430,898 | 81,556,240 | 0.03 | 97.41 | 93.01 | 47.74 | 86.81 |
| ADJ_2 | 83,301,486 | 82,361,856 | 0.03 | 97.53 | 93.3 | 50.48 | 84.03 |
| ADJ_3 | 82,206,822 | 81,388,816 | 0.03 | 97.25 | 92.65 | 46.61 | 89.64 |
| ATT_1 | 81,229,824 | 80,361,428 | 0.03 | 97.2 | 92.58 | 46.69 | 85.75 |
| ATT_2 | 83,299,108 | 82,251,852 | 0.03 | 96.93 | 92.12 | 46.41 | 86.45 |
| ATT_3 | 80,793,446 | 79,959,968 | 0.03 | 97.3 | 92.8 | 46.67 | 88.20 |
| PRE_1 | 81,092,584 | 80,206,522 | 0.03 | 97.1 | 92.41 | 48.72 | 85.43 |
| PRE_2 | 85,308,302 | 84,316,544 | 0.03 | 97.32 | 92.93 | 47.53 | 85.69 |
| PRE_3 | 93,181,024 | 91,990,894 | 0.03 | 97.05 | 92.45 | 46.79 | 84.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; San, L.; Guo, H.; Zhu, K.; Zhang, N.; Yang, J.; Liu, B.; Hou, J.; Zhang, D. Transcriptomic Analysis Reveals Functional Interaction of mRNA-lncRNA-miRNA in Trachinotus ovatus Infected by Cryptocaryon irritans. Int. J. Mol. Sci. 2023, 24, 15886. https://doi.org/10.3390/ijms242115886
Liu B, San L, Guo H, Zhu K, Zhang N, Yang J, Liu B, Hou J, Zhang D. Transcriptomic Analysis Reveals Functional Interaction of mRNA-lncRNA-miRNA in Trachinotus ovatus Infected by Cryptocaryon irritans. International Journal of Molecular Sciences. 2023; 24(21):15886. https://doi.org/10.3390/ijms242115886
Chicago/Turabian StyleLiu, Baosuo, Lize San, Huayang Guo, Kecheng Zhu, Nan Zhang, Jingwen Yang, Bo Liu, Jilun Hou, and Dianchang Zhang. 2023. "Transcriptomic Analysis Reveals Functional Interaction of mRNA-lncRNA-miRNA in Trachinotus ovatus Infected by Cryptocaryon irritans" International Journal of Molecular Sciences 24, no. 21: 15886. https://doi.org/10.3390/ijms242115886
APA StyleLiu, B., San, L., Guo, H., Zhu, K., Zhang, N., Yang, J., Liu, B., Hou, J., & Zhang, D. (2023). Transcriptomic Analysis Reveals Functional Interaction of mRNA-lncRNA-miRNA in Trachinotus ovatus Infected by Cryptocaryon irritans. International Journal of Molecular Sciences, 24(21), 15886. https://doi.org/10.3390/ijms242115886