
Transcriptome Analysis Reveals the Molecular Mechanism and Responsive Genes of Waterlogging Stress in Actinidia deliciosa Planch Kiwifruit Plants

 ,
,
Abstract
:1. Introduction
2. Results
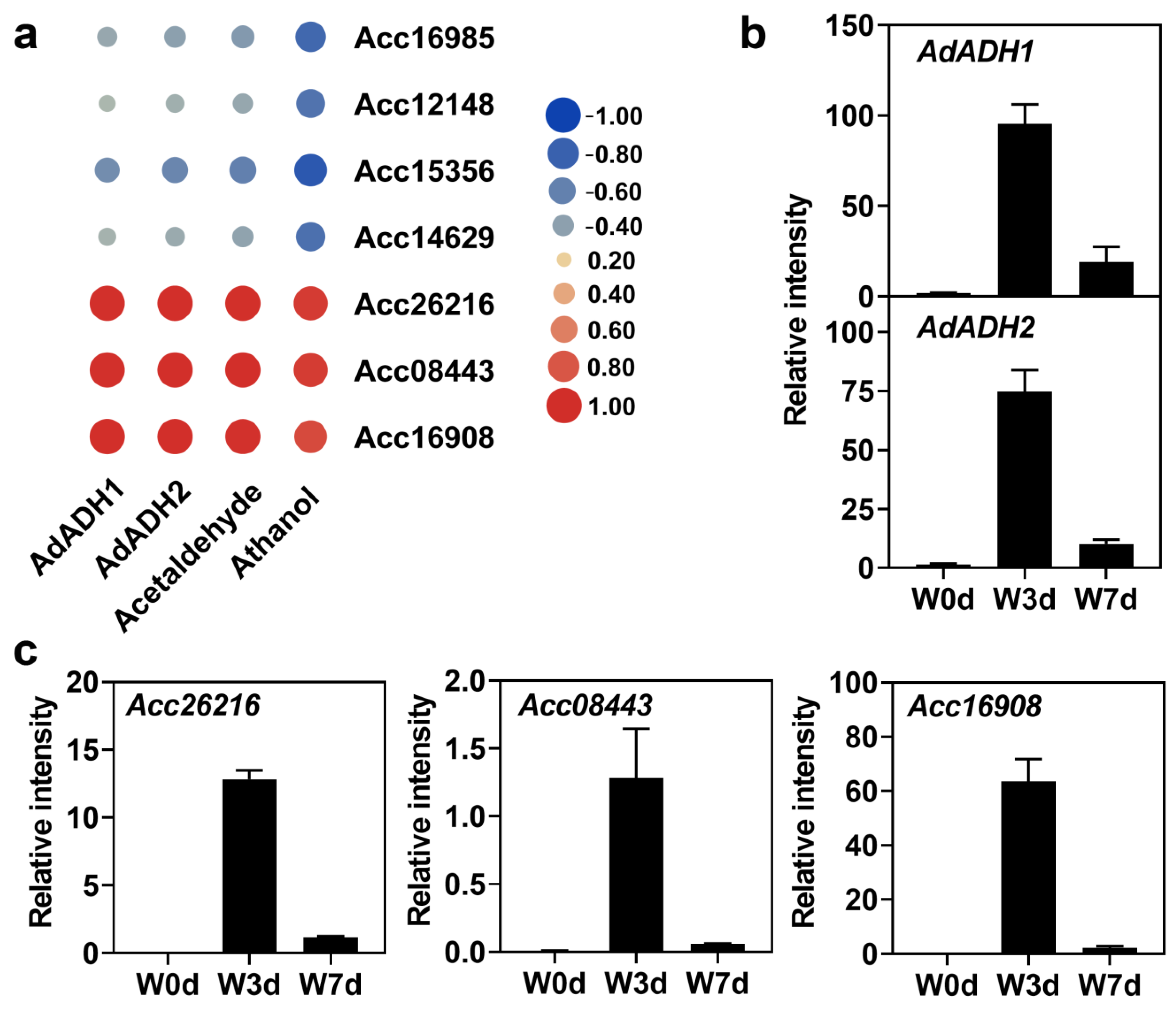
2.1. Accumulation of Acetaldehyde and Ethanol in Kiwifruit Roots
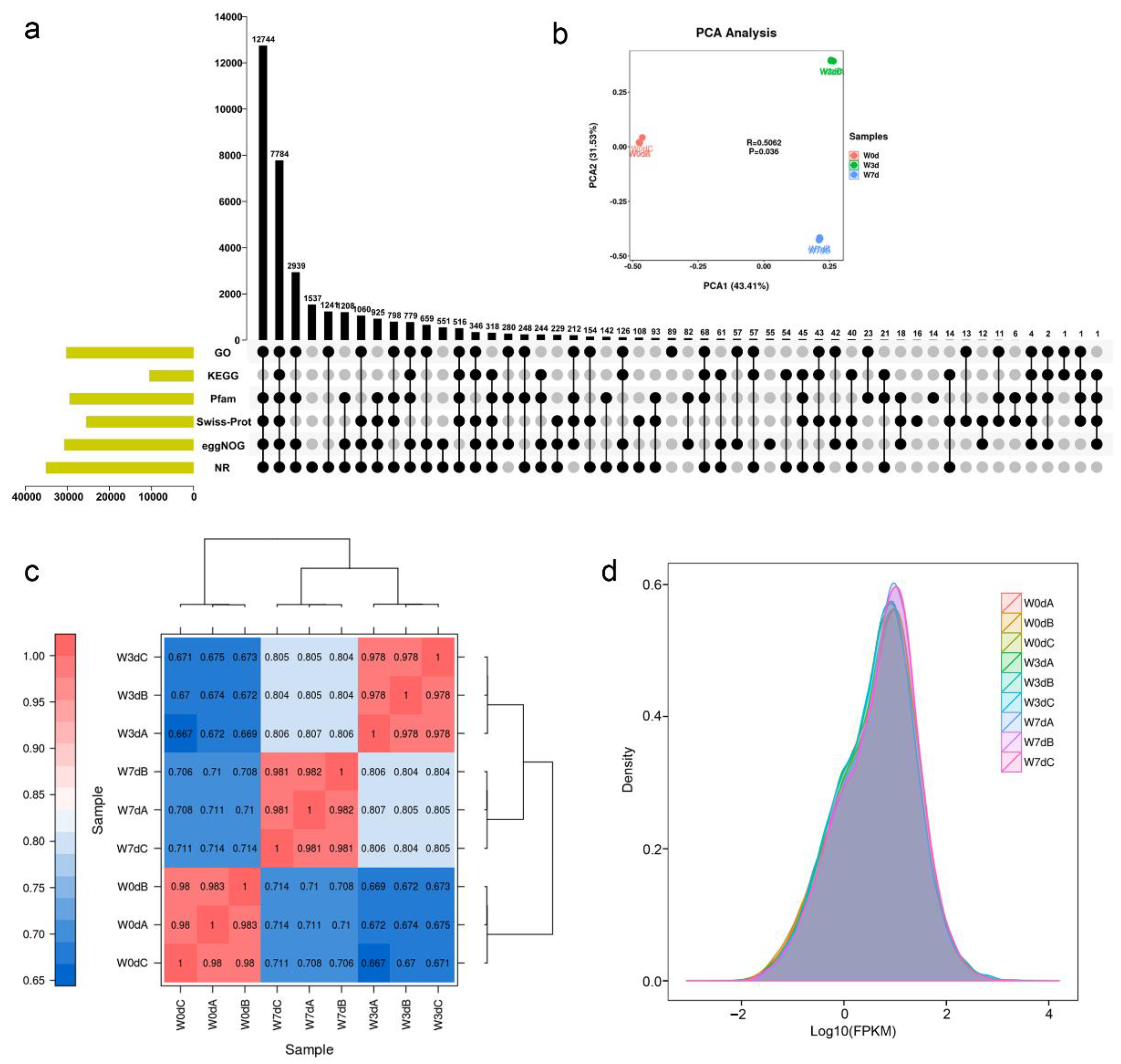
2.2. Analysis of the RNA Sequencing Data
2.3. The Molecular Influence of Waterlogging on Root Tissues
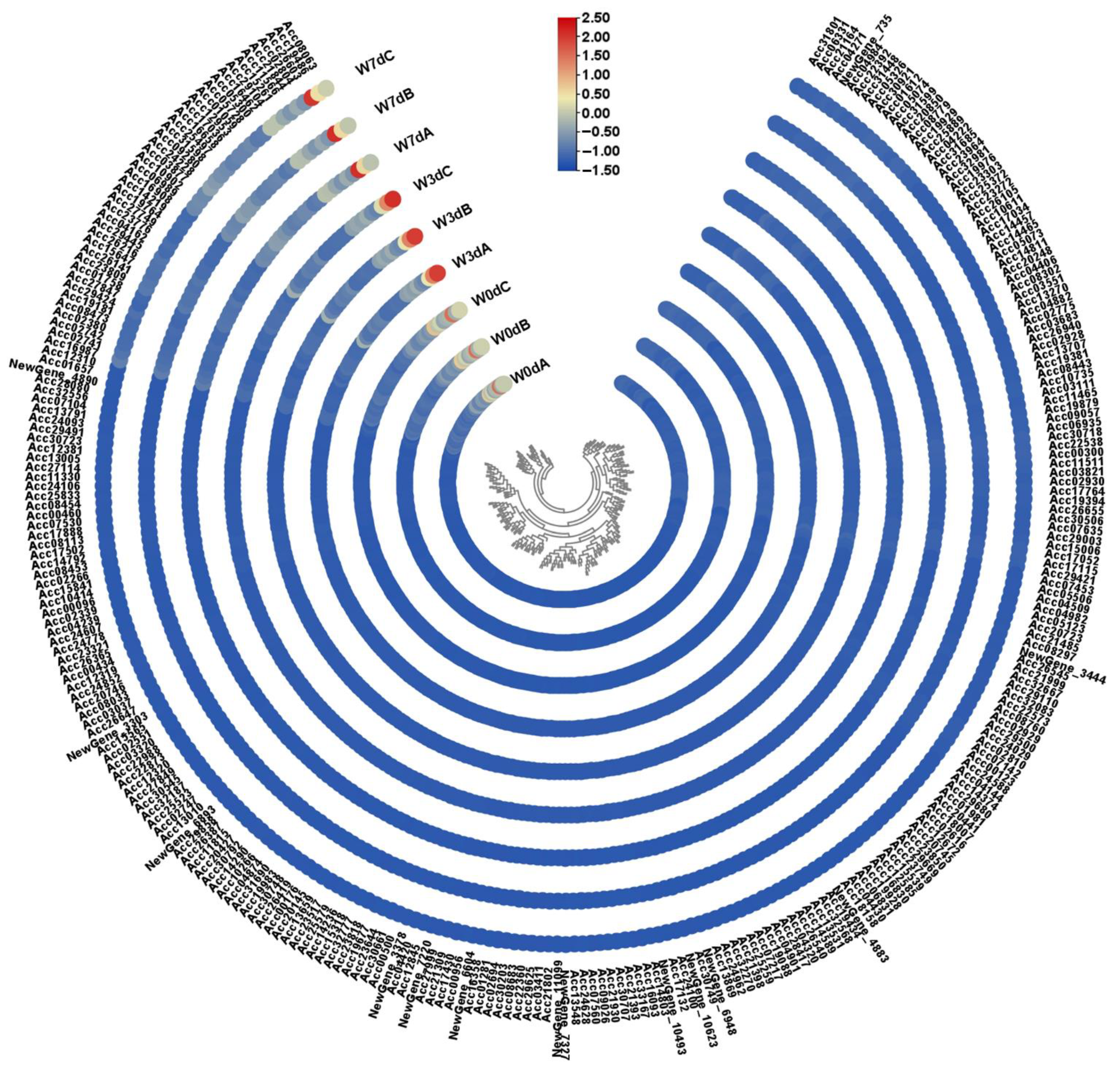
2.4. Differential Gene Expression Analysis
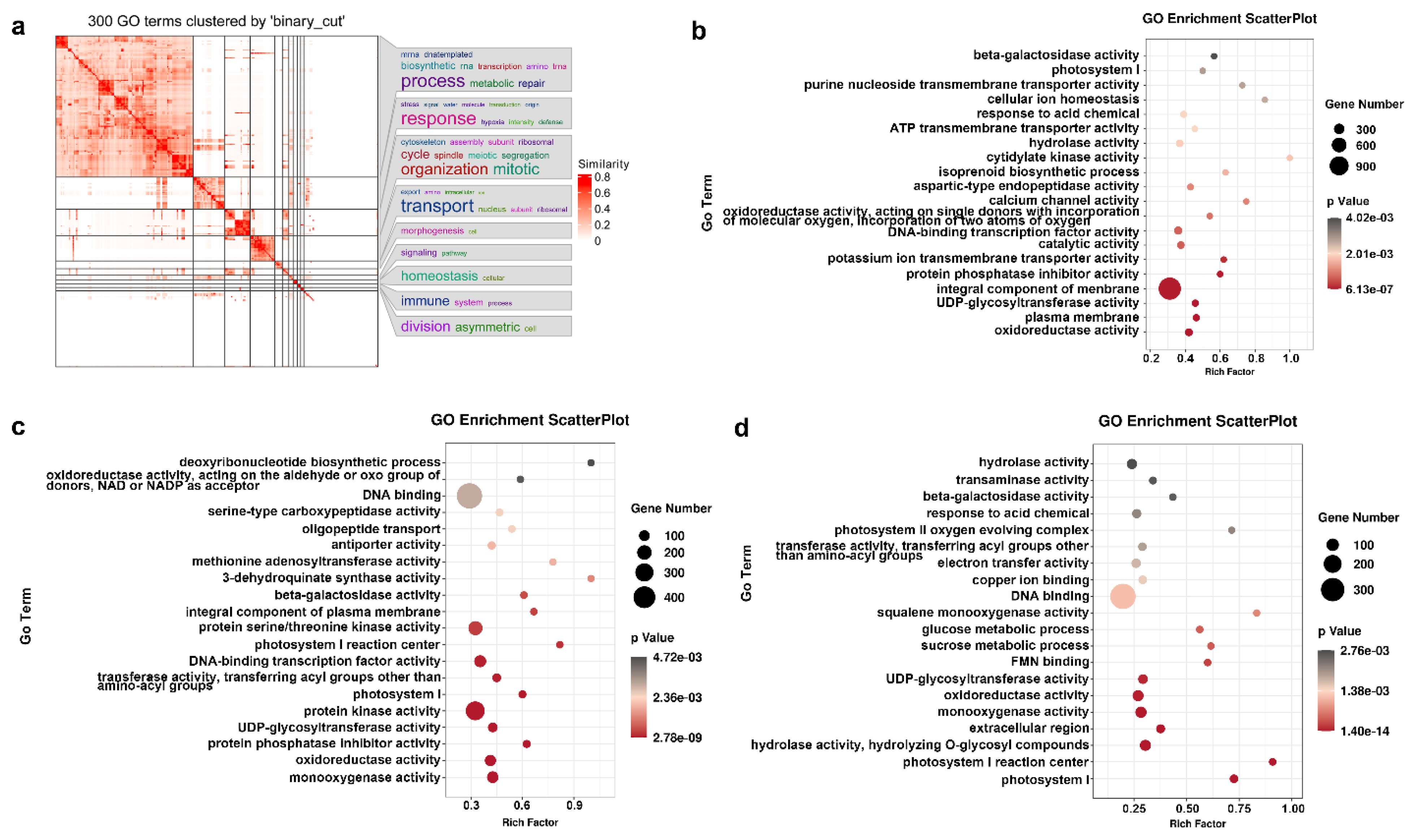
2.5. GO Categories and Enrichment Analysis
2.6. Identification of Candidate Genes under Waterlogging
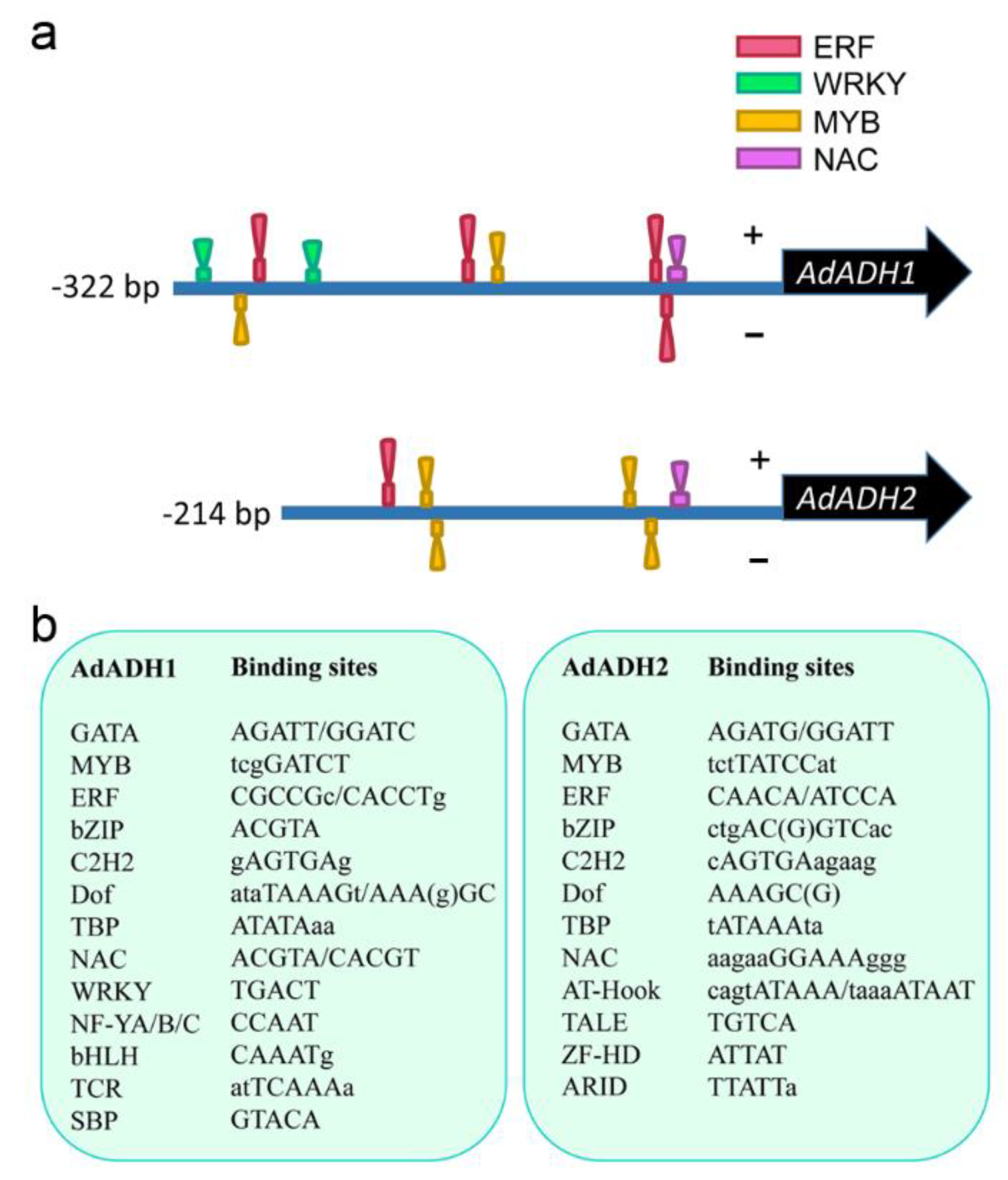
2.7. Analysis of Promoter Sequences of ADH Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Waterlogging Stress Treatment
4.2. Acetaldehyde and Ethanol Contents
4.3. RNA Extraction and Library Preparation
4.4. RNA Sequencing and Assembly
4.5. Differential Gene Expression Analysis
4.6. RT-qPCR Assays
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Setter, T.; Waters, I. Review of prospects for germplasm improvement for waterlogging tolerance in wheat, barley, and oats. Plant Soil 2003, 253, 1–34. [Google Scholar] [CrossRef]
- MacEwan, R.J.; Gardner, W.K.; Ellington, A.; Hopkins, D.G.; Bakker, A.C. Tile andmole drainage for control of waterloggingin duplex soils of southeastern Australia. Aust. J. Exp. Agric. 1992, 32, 865–878. [Google Scholar] [CrossRef]
- Zhou, M.Z. Improvement of plant waterlogging tolerance. In Waterlogging Signalling and Tolerance in Plants; Mancuso, S., Shabala, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 267–285. [Google Scholar]
- Loreti, E.; Valeri, M.C.; Novi, G.; Perata, P. Gene Regulation and Survival under Hypoxia Requires Starch Availability and Metabolism. Plant Physiol. 2017, 176, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Rukh, G.; Ruan, Z.; Xie, X.; Ye, Z.; Liu, D. Effects of hypoxia stress on growth, root respiration, and metabolism of phyllostachys praecox. Life 2022, 12, 808. [Google Scholar] [CrossRef] [PubMed]
- Da-Silva, C.J.; Amarante, L.D. Time-course biochemical analyses of soybean plants during waterlogging and reoxygenation. Environ. Exp. Bot. 2020, 180, 104242. [Google Scholar] [CrossRef]
- Yu, F.; Liang, K.; Fang, T.; Zhao, H.; Han, X.; Cai, M.; Qiu, F. A group VII ethylene response factor gene, ZmEREB180, coordinates waterlogging tolerance in maize seedlings. Plant Biotechnol. J. 2019, 17, 2286–2298. [Google Scholar] [CrossRef]
- Men, S.; Chen, H.; Chen, S.; Zheng, S.; Shen, X.; Wang, C.; Yang, Z.; Liu, D. Effects of supplemental nitrogen application on physiological characteristics, dry matter and nitrogen accumulation of winter rapeseed (Brassica napus L.) under waterlogging stress. Sci. Rep. 2020, 10, 10201. [Google Scholar] [CrossRef]
- Yamauchi, T.; Colmer, T.D.; Pedersen, O.; Nakazono, M. Regulation of Root Traits for Internal Aeration and Tolerance to Soil Waterlogging-Flooding Stress. Plant Physiol. 2017, 176, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Milroy, S.P.; Bange, M.P.; Thongbai, P. Cotton leaf nutrient concentrations in response to waterlogging under field conditions. Field Crops Res. 2009, 113, 246–255. [Google Scholar] [CrossRef]
- Shabala, S.; Shabala, L.; Barceló, J.; Poschenrieder, C. Membrane transporters mediating root signalling and adaptive responses to oxygen deprivation and soil flooding. Plant Cell Environ. 2014, 37, 2216–2233. [Google Scholar] [CrossRef]
- Adegoye, G.A.; Olorunwa, O.J.; Alsajri, F.A.; Walne, C.H.; Wijewandana, C.; Kethireddy, S.R.; Reddy, K.N.; Reddy, K.R. Waterlogging Effects on Soybean Physiology and Hyperspectral Reflectance during the Reproductive Stage. Agriculture 2023, 13, 844. [Google Scholar] [CrossRef]
- Singh, B.; Singh, A.; Ram, P.; Singh, P.; Singh, H.; Boamfa, I.; Harren, F.; Santosa, E.; Jackson, M.; Setter, T.; et al. Submergence tolerance in rainfed lowland rice: Physiological basis and prospects for cultivar improvement through marker-aided breeding. Field Crops Res. 2002, 76, 131–152. [Google Scholar] [CrossRef]
- Magneschi, L.; Perata, P. Rice germination and seedling growth in the absence of oxygen. Ann. Bot. 2008, 103, 181–196. [Google Scholar] [CrossRef]
- Zahra, N.; Hafeez, M.B.; Shaukat, K.; Wahid, A.; Hussain, S.; Naseer, R.; Raza, A.; Iqbal, S.; Farooq, M. Hypoxia and Anoxia Stress: Plant responses and tolerance mechanisms. J. Agron. Crop Sci. 2021, 207, 249–284. [Google Scholar] [CrossRef]
- Kürsteiner, O.; Dupuis, I.; Kuhlemeier, C. The Pyruvate decarboxylase1 Gene of Arabidopsis Is Required during Anoxia But Not Other Environmental Stresses. Plant Physiol. 2003, 132, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Y.; Huang, S.-N.; Wang, G.; Xuan, J.-P.; Guo, Z.-R. Overexpression of Actinidia deliciosa pyruvate decarboxylase 1 gene enhances waterlogging stress in transgenic Arabidopsis thaliana. Plant Physiol. Biochem. 2016, 106, 244–252. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Huang, S.-N.; Chen, Y.-H.; Wang, G.; Guo, Z.-R. Identification and characterization of two waterlogging responsive alcohol dehydrogenase genes (AdADH1 and AdADH2) in Actinidia deliciosa. Mol. Breed. 2017, 37, 52. [Google Scholar] [CrossRef]
- Smith, G.S.; Judd, M.J.; Miller, S.A.; Buwalda, J.G. Recovery of kiwifruit vines from transient waterlogging of the root system. New Phytol. 1990, 115, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bai, D.; Zhong, Y.; Abid, M.; Qi, X.; Hu, C.; Fang, J. Physiological Responses of Two Contrasting Kiwifruit (Actinidia spp.) Rootstocks against Waterlogging Stress. Plants 2021, 10, 2586. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Wang, W.Q.; Liu, J.H.; Zhu, C.Q.; Zhong, Y.P.; Zhang, H.Q.; Liu, X.F.; Yin, X.R. Transcription factors AcERF74/75 respond to waterlogging stress and trigger alcoholic fermentation-related genes in kiwifruit. Plant Sci. 2022, 314, 111115. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Gai, C.; Li, X.; Feng, X.; Lai, R.; Song, Y.; Zeng, R.; Chen, D.; Chen, Y. Waterlogging tolerance of Actinidia valvata Dunn is associated with high activities of pyruvate decarboxylase, alcohol dehydrogenase and antioxidant enzymes. Plants 2023, 12, 2872. [Google Scholar] [CrossRef] [PubMed]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Gu, Z.; Hübschmann, D. simplifyEnrichment: A Bioconductor Package for Clustering and Visualizing Functional Enrichment Results. Genom. Proteom. Bioinform. 2023, 21, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.K.; Callis, J.; Jardetzky, O.; Walbot, V.; Freeling, M. Cytoplasmic acidosis as a determinant of flooding intolerance in plants. Proc. Natl. Acad. Sci. USA 1984, 81, 6029–6033. [Google Scholar] [CrossRef] [PubMed]
- Steffens, D.; Hütsch, B.W.; Eschholz, T.; Lošák, T.; Schubert, S. Water logging may inhibit plant growth primarily by nutrient deficiency rather than nutrient toxicity. Plant Soil Environ. 2005, 51, 545–552. [Google Scholar] [CrossRef]
- Xu, X.W.; Wang, H.H.; Qi, X.H.; Xu, Q.; Chen, X.H. Waterlogging-induced increase in fermentation and related gene expression in the root of cucumber (Cucumis sativus L.). Sci. Hortic. 2014, 179, 388–395. [Google Scholar]
- Xu, P.; Liu, Z.; Fan, X.; Gao, J.; Zhang, X.; Zhang, X.; Shen, X. De novo transcriptome sequencing and comparative analysis of differentially expressed genes in Gossypium aridum under salt stress. Gene 2013, 525, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bai, D.; Zhong, Y.; Lin, M.; Sun, L.; Qi, X.; Hu, C.; Fang, J. Full-Length Transcriptome and RNA-Seq Analyses Reveal the Mechanisms Underlying Waterlogging Tolerance in Kiwifruit (Actinidia valvata). Int. J. Mol. Sci. 2022, 23, 3237. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, Z.; Shi, M.; Zhou, Y.; Huo, L.; Li, X.; Xu, K. Comparative transcriptome provides insight into responding mechanism of waterlogging stress in Actinidia valvata Dunn. Gene 2022, 845, 146843. [Google Scholar] [CrossRef]
- Liu, F.; VanToai, T.; Moy, L.P.; Bock, G.; Linford, L.D.; Quackenbush, J. Global Transcription Profiling Reveals Comprehensive Insights into Hypoxic Response in Arabidopsis. Plant Physiol. 2005, 137, 1115–1129. [Google Scholar] [CrossRef]
- Zhang, P.; Lyu, D.; Jia, L.; He, J.; Qin, S. Physiological and de novo transcriptome analysis of the fermentation mechanism of Cerasus sachalinensis roots in response to short-term waterlogging. BMC Genom. 2017, 18, 649. [Google Scholar] [CrossRef]
- Qiao, D.; Zhang, Y.; Xiong, X.; Li, M.; Cai, K.; Luo, H.; Zeng, B. Transcriptome analysis on responses of orchardgrass (Dactylis glomerata L.) leaves to a short term flooding. Hereditas 2020, 157, 20. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Vankova, R.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.-S.P. Cytokinins: Metabolism and function in plant adaptation to environmental stresses. Trends Plant Sci. 2012, 17, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef]
- Ding, Y.; Shi, Y.; Yang, S. Advances and challenges in uncovering cold tolerance regulatory mechanisms in plants. New Phytol. 2019, 222, 1690–1704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gao, C.; Xu, L.; Niu, H.; Liu, Q.; Huang, Y.; Lv, G.; Yang, H.; Li, M. Melatonin and Indole-3-Acetic Acid Synergistically Regulate Plant Growth and Stress Resistance. Cells 2022, 11, 3250. [Google Scholar] [CrossRef]
- Cheng, X.-X.; Yu, M.; Zhang, N.; Zhou, Z.-Q.; Xu, Q.-T.; Mei, F.-Z.; Qu, L.-H. Reactive oxygen species regulate programmed cell death progress of endosperm in winter wheat (Triticum aestivum L.) under waterlogging. Protoplasma 2015, 253, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Xu, Y.; Li, H.; Chen, X.; Song, Z. Antioxidant activation, cell wall reinforcement, and reactive oxygen species regulation promote resistance to waterlogging stress in hot pepper (Capsicum annuum L.). BMC Plant Biol. 2022, 22, 425. [Google Scholar] [CrossRef]
- Jiang, L.; Yang, H. Prometryne-induced oxidative stress and impact on antioxidant enzymes in wheat. Ecotoxicol. Environ. Saf. 2009, 72, 1687–1693. [Google Scholar] [CrossRef]
- Kreuzwieser, J.; Rennenberg, H. Molecular and physiological responses of trees to waterlogging stress. Plant Cell Environ. 2014, 37, 2245–2259. [Google Scholar] [CrossRef]
- Cho, H.; Loreti, E.; Shih, M.; Perata, P. Energy and sugar signaling during hypoxia. New Phytol. 2019, 229, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Chen, T.; Wang, X.; Cao, J.; Li, X.; Xu, X.; Chen, L.; Xia, Q.; Dong, Y.; Huang, L.; et al. Physiological and Expressional Regulation on Photosynthesis, Starch and Sucrose Metabolism Response to Waterlogging Stress in Peanut. Front. Plant Sci. 2021, 12, 601771. [Google Scholar]
- Voesenek, L.A.C.J.; Bailey-Serres, J. Flood adaptive traits and processes: An overview. New Phytol. 2015, 206, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, R.; Mustroph, A.; Boonman, A.; Akman, M.; Ammerlaan, A.M.; Breit, T.; Schranz, M.E.; Voesenek, L.A.; van Tienderen, P.H. Root Transcript Profiling of Two Rorippa Species Reveals Gene Clusters Associated with Extreme Submergence Tolerance. Plant Physiol. 2013, 163, 1277–1292. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Nagai, K.; Furukawa, S.; Song, X.-J.; Kawano, R.; Sakakibara, H.; Wu, J.; Matsumoto, T.; Yoshimura, A.; Kitano, H.; et al. The ethylene response factors SNORKEL1 and SNORKEL2 allow rice to adapt to deep water. Nature 2009, 460, 1026–1030. [Google Scholar] [CrossRef]
- Licausi, F.; Kosmacz, M.; Weits, D.A.; Giuntoli, B.; Giorgi, F.M.; Voesenek, L.A.C.J.; Perata, P.; van Dongen, J.T. Oxygen sensing in plants is mediated by an N-end rule pathway for protein destabilization. Nature 2011, 479, 419–422. [Google Scholar] [CrossRef]
- Hsu, F.-C.; Chou, M.-Y.; Chou, S.-J.; Li, Y.-R.; Peng, H.-P.; Shih, M.-C. Submergence Confers Immunity Mediated by the WRKY22 Transcription Factor in Arabidopsis. Plant Cell 2013, 25, 2699–2713. [Google Scholar] [CrossRef]
- Rauf, M.; Arif, M.; Fisahn, J.; Xue, G.-P.; Balazadeh, S.; Mueller-Roeber, B. NAC Transcription Factor SPEEDY HYPONASTIC GROWTH Regulates Flooding-Induced Leaf Movement in Arabidopsis. Plant Cell 2013, 25, 4941–4955. [Google Scholar] [CrossRef] [PubMed]
- Reynoso, M.A.; Kajala, K.; Bajic, M.; West, D.A.; Pauluzzi, G.; Yao, A.I.; Hatch, K.; Zumstein, K.; Woodhouse, M.; Rodriguez-Medina, J.; et al. Evolutionary flexibility in flooding response circuitry in angiosperms. Science 2019, 365, 1291–1295. [Google Scholar] [CrossRef]
- Tang, H.; Bi, H.; Liu, B.; Lou, S.; Song, Y.; Tong, S.; Chen, N.; Jiang, Y.; Liu, J.; Liu, H. WRKY33 interacts with WRKY12 protein to up-regulate RAP2.2 during submergence induced hypoxia response in Arabidopsis thaliana. New Phytol. 2021, 229, 106–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Wang, J.; Wu, Y.Y.; Li, D.W.; Allan, A.C.; Yin, X.R. Genome-wide analysis of coding and non-coding RNA reveals a conserved miR164-NAC regulatory pathway for fruit ripening. New Phytol. 2020, 225, 1618–1634. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Moss, S.M.A.; Zeng, L.; Espley, R.V.; Wang, T.; Lin-Wang, K.; Fu, B.; Schwinn, K.E.; Allan, A.C.; Yin, X. The red flesh of kiwifruit is differentially controlled by specific activation–repression systems. New Phytol. 2022, 235, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Pilkington, S.M.; Crowhurst, R.; Hilario, E.; Nardozza, S.; Fraser, L.; Peng, Y.; Gunaseelan, K.; Simpson, R.; Tahir, J.; Deroles, S.C.; et al. A manually annotated Actinidia chinensis var. chinensis (kiwifruit) genome highlights the challenges associated with draft genomes and gene prediction in plants. BMC Genom. 2018, 19, 257. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene. | Combinations | Reference Genome | Predicted Length (bp) | Obtained Length (bp) |
|---|---|---|---|---|
| AdADH1 | FP1 + RP1 (FP1 + RP2) | ‘Hayward’ | 2000 (1970) | No |
| FP2 + RP1 (FP1 + RP2) | ‘Red5’ | 2000 (1970) | No | |
| FP3 + RP1 (FP3 + RP2) | ‘Hayward’ | 1342 (1312) | No | |
| FP4 + RP1 (FP4 + RP2) | ‘Hayward’/‘Red5’ | 422/392 | No | |
| FP5 + RP1 (FP5 + RP2) | ‘Hayward’/‘Red5’ | 346 (316)/351 (321) | 322 (No) | |
| AdADH2 | FP1 + RP1 (FP1 + RP2) | ‘Hayward’ | 2000 (1976) | No |
| FP2 + RP1 (FP1 + RP2) | ‘Red5’ | 2000 (1976) | No | |
| FP3 + RP1 (FP3 + RP2) | ‘Hayward’/‘Red5’ | 1556 (1532)/1295 (1271) | No | |
| FP4 + RP1 (FP4 + RP2) | ‘Hayward’/‘Red5’ | 1140 (1116)/862 (838) | No | |
| FP5 + RP1 (FP5 + RP2) | ‘Hayward’/‘Red5’ | 864 (840)/586 (562) | No | |
| FP6 + RP1 (FP6 + RP2) | ‘Hayward’/‘Red5’ | 645 (621)/379 (355) | No | |
| FP7 + RP1 (FP7 + RP2) | ‘Hayward’/‘Red5’ | 235 (211)/230 (206) | 214 (No) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, M.; Huang, K.; Zhang, C.; Xi, D.; Luo, H.; Pei, J.; Ruan, R.; Liu, H. Transcriptome Analysis Reveals the Molecular Mechanism and Responsive Genes of Waterlogging Stress in Actinidia deliciosa Planch Kiwifruit Plants. Int. J. Mol. Sci. 2023, 24, 15887. https://doi.org/10.3390/ijms242115887
Xing M, Huang K, Zhang C, Xi D, Luo H, Pei J, Ruan R, Liu H. Transcriptome Analysis Reveals the Molecular Mechanism and Responsive Genes of Waterlogging Stress in Actinidia deliciosa Planch Kiwifruit Plants. International Journal of Molecular Sciences. 2023; 24(21):15887. https://doi.org/10.3390/ijms242115887
Chicago/Turabian StyleXing, Mengyun, Kangkang Huang, Chen Zhang, Dujun Xi, Huifeng Luo, Jiabo Pei, Ruoxin Ruan, and Hui Liu. 2023. "Transcriptome Analysis Reveals the Molecular Mechanism and Responsive Genes of Waterlogging Stress in Actinidia deliciosa Planch Kiwifruit Plants" International Journal of Molecular Sciences 24, no. 21: 15887. https://doi.org/10.3390/ijms242115887
APA StyleXing, M., Huang, K., Zhang, C., Xi, D., Luo, H., Pei, J., Ruan, R., & Liu, H. (2023). Transcriptome Analysis Reveals the Molecular Mechanism and Responsive Genes of Waterlogging Stress in Actinidia deliciosa Planch Kiwifruit Plants. International Journal of Molecular Sciences, 24(21), 15887. https://doi.org/10.3390/ijms242115887