
Tuning the Nucleophilicity and Electrophilicity of Group 10 Elements through Substituent Effects: A DFT Study




Abstract
:1. Introduction
2. Results and Discussion
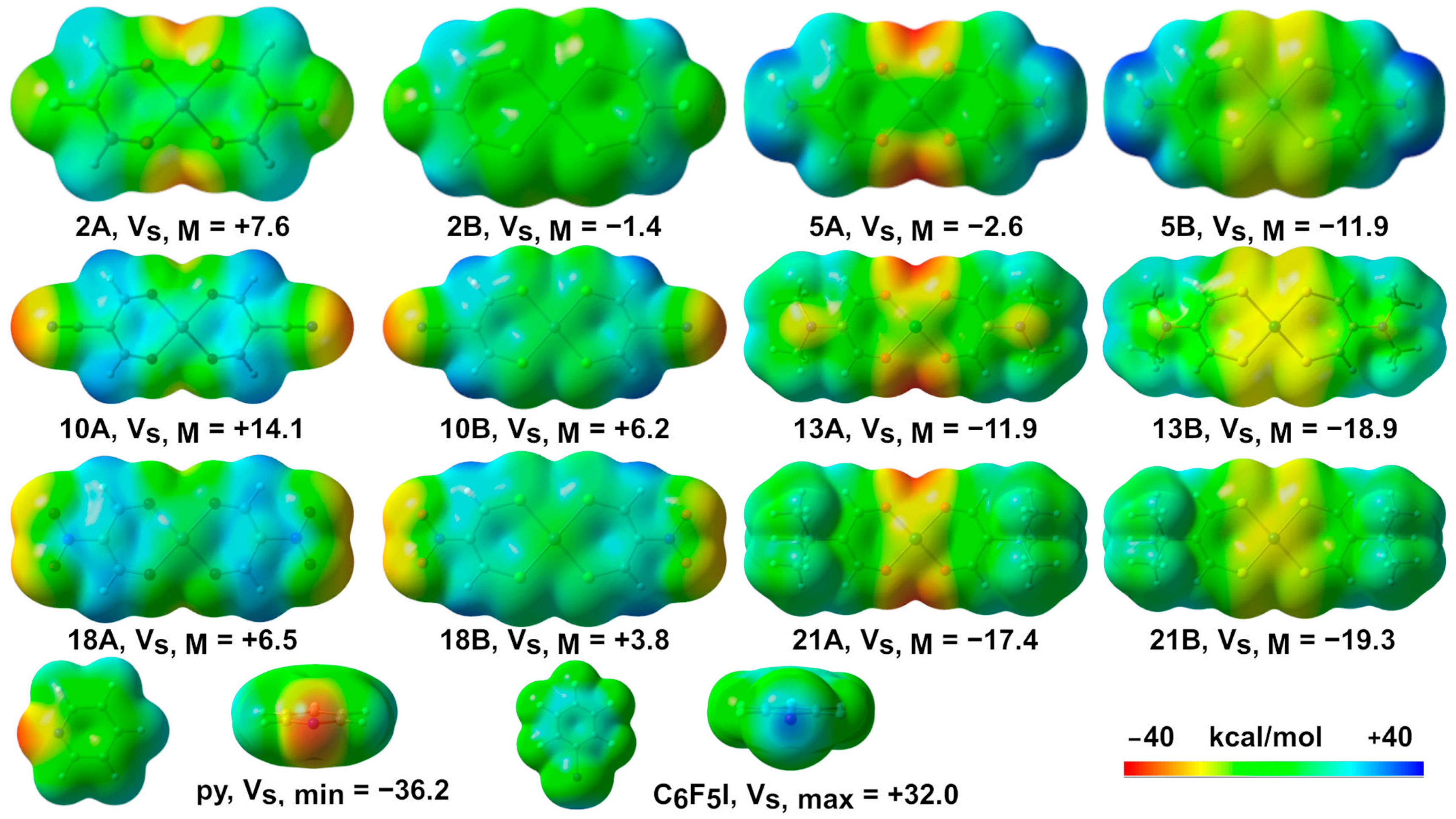
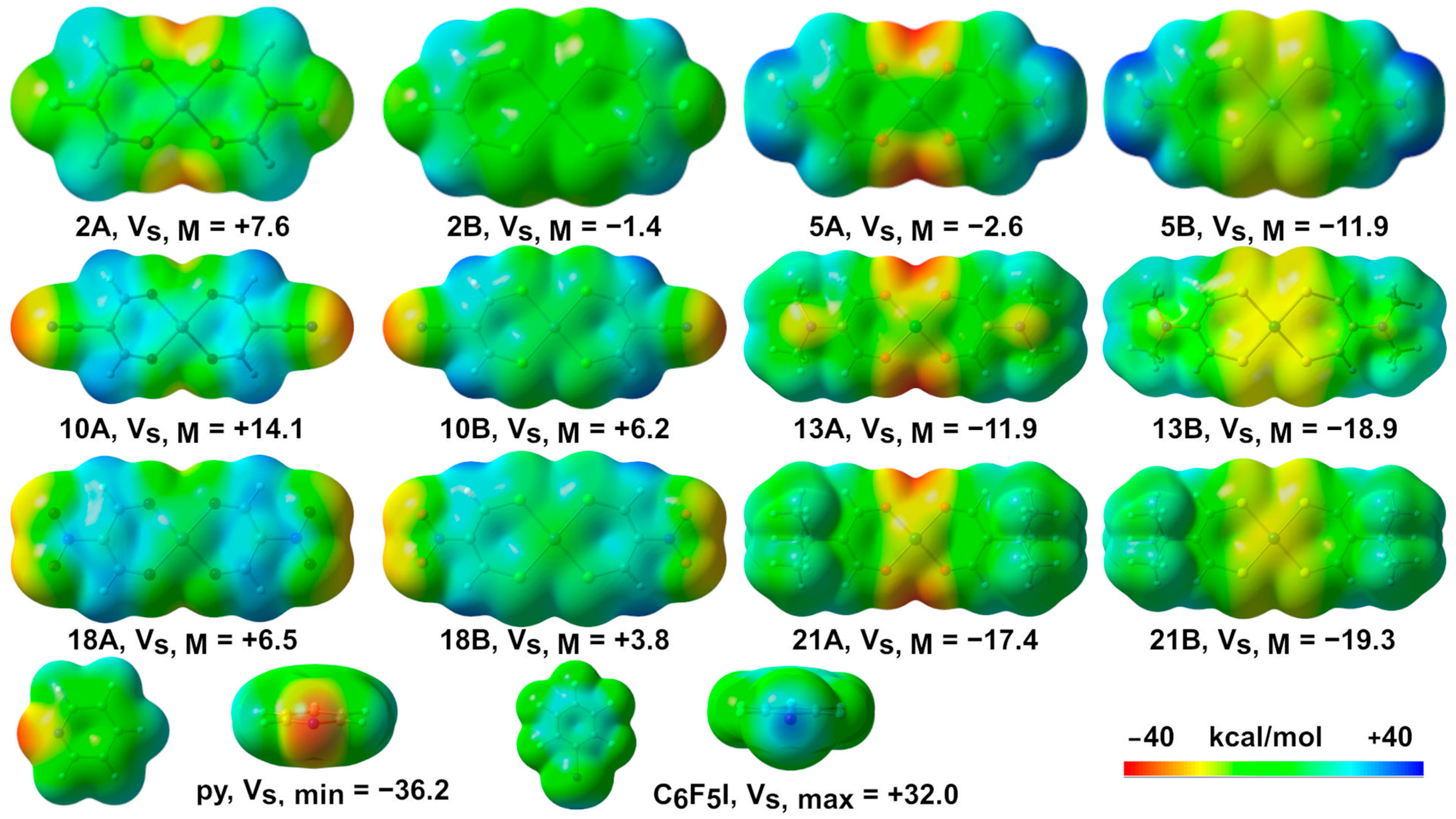
2.1. Molecular Electrostatic Potential Surface Analysis (MEP)
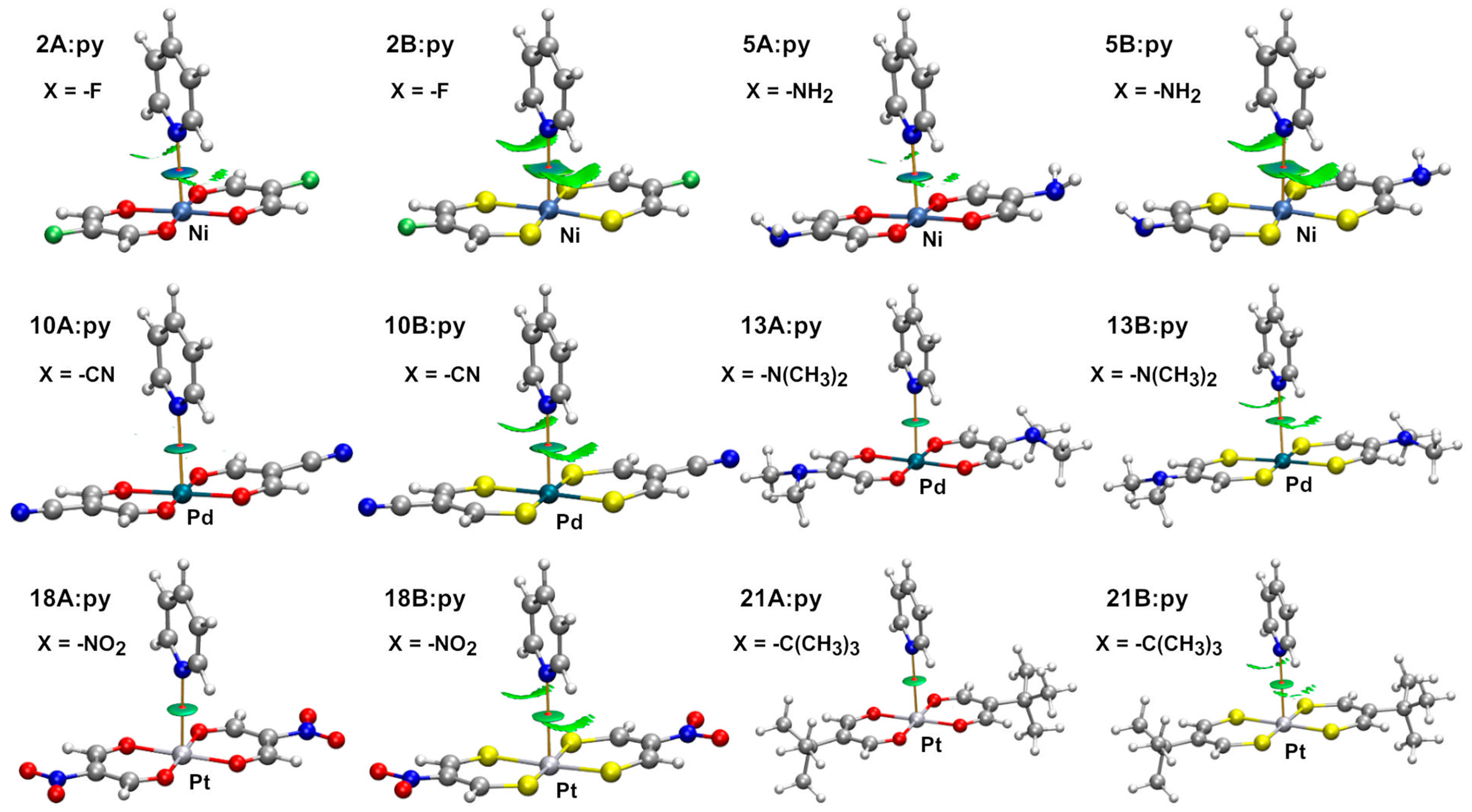
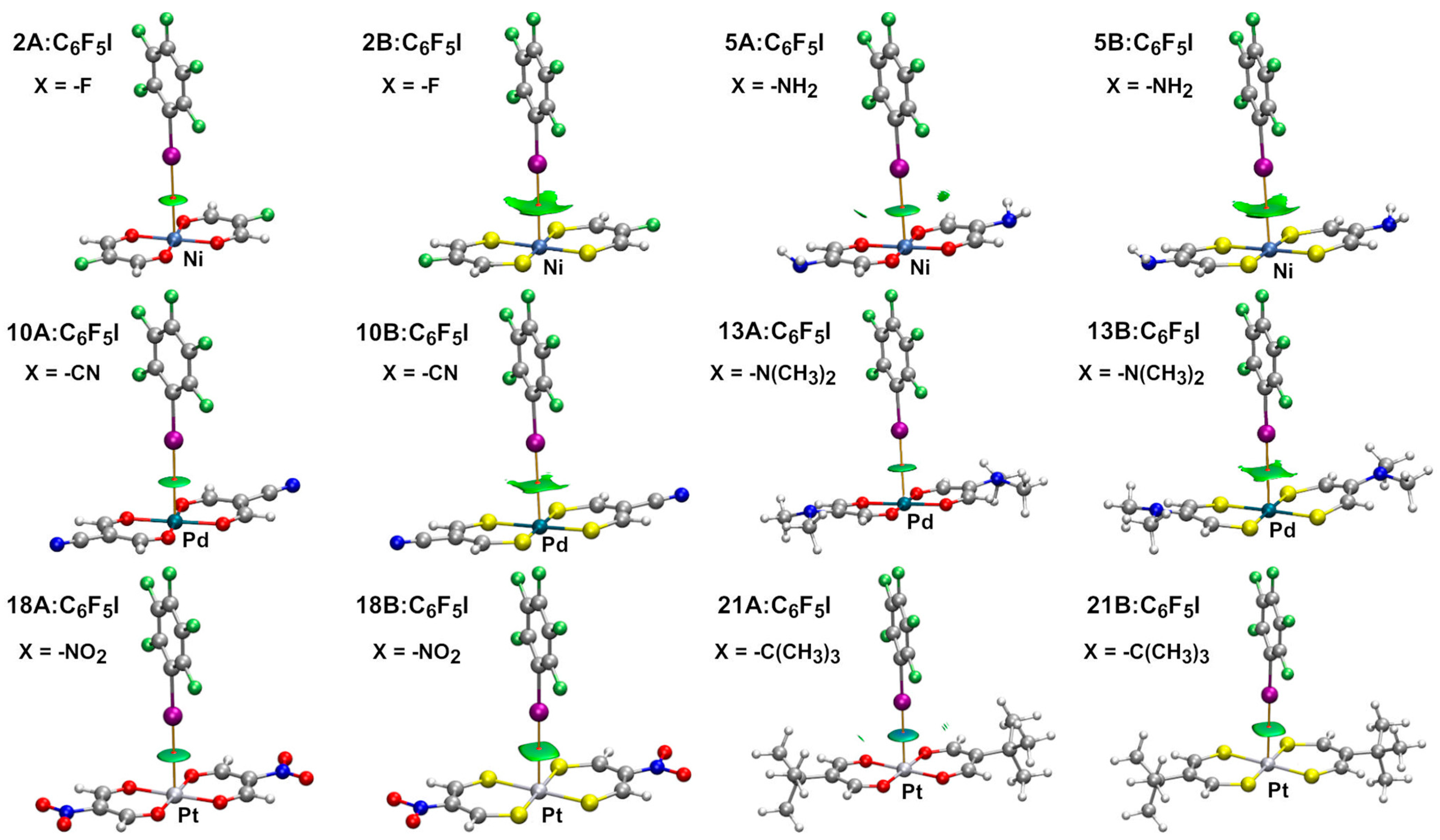
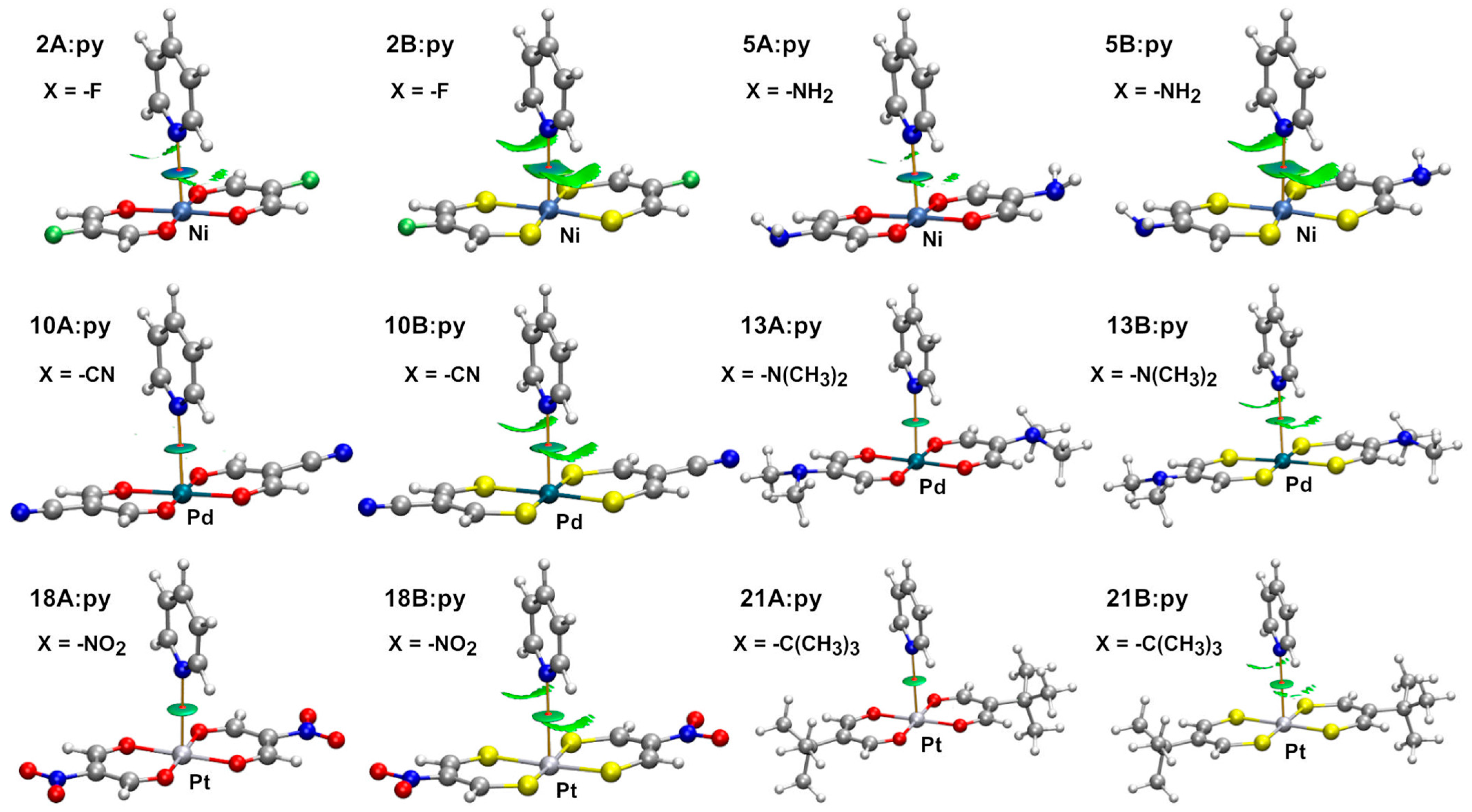
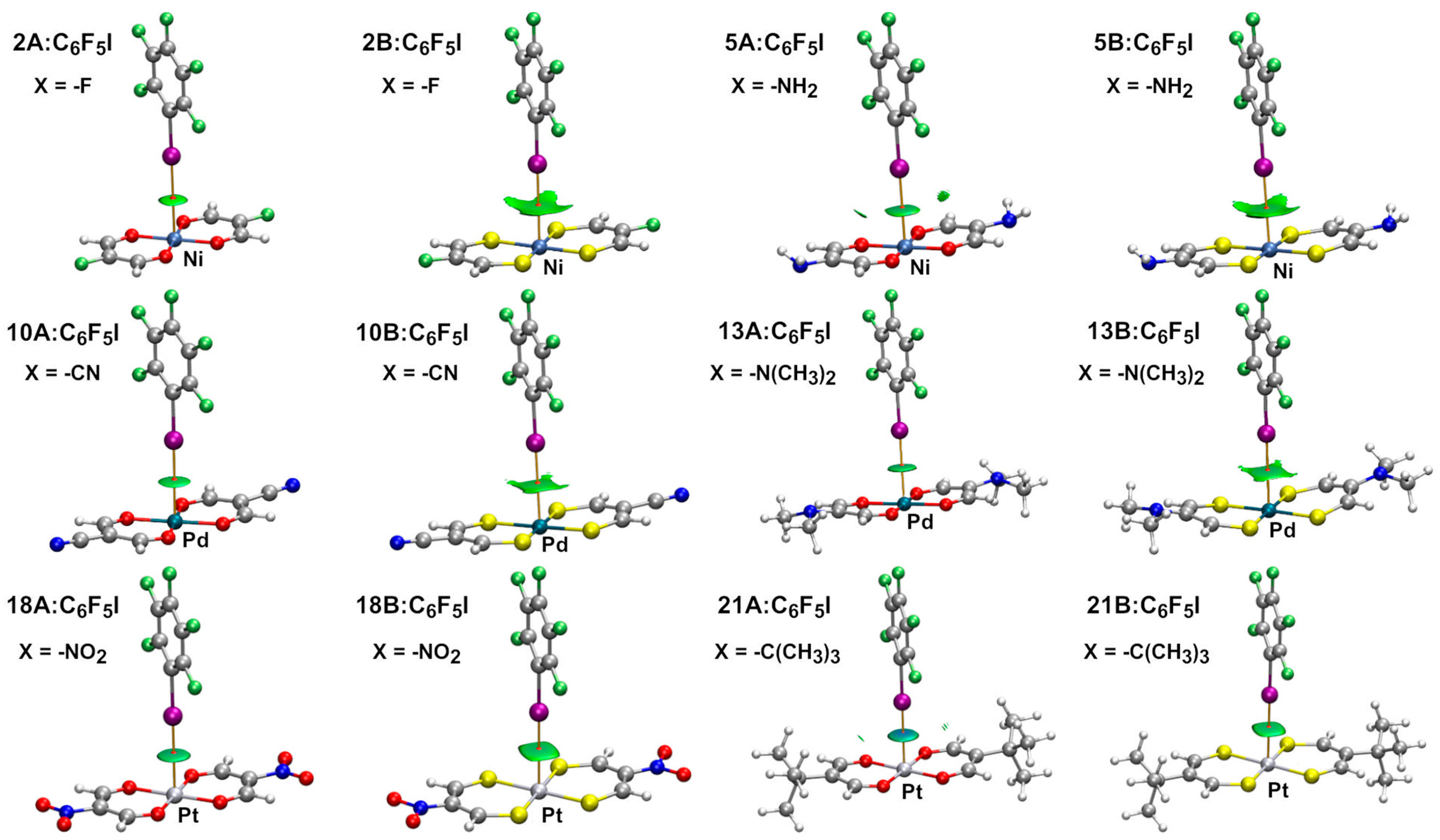
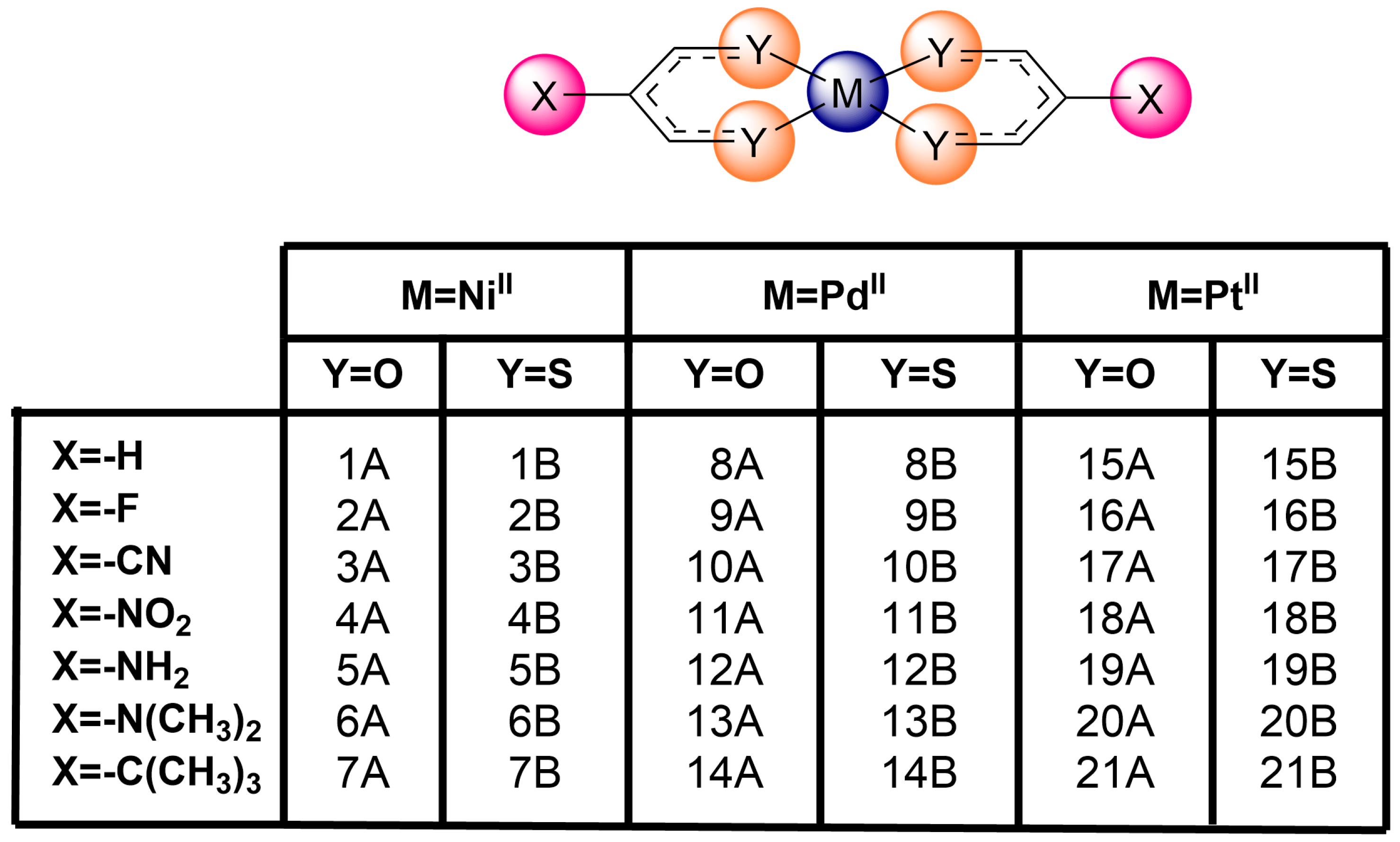
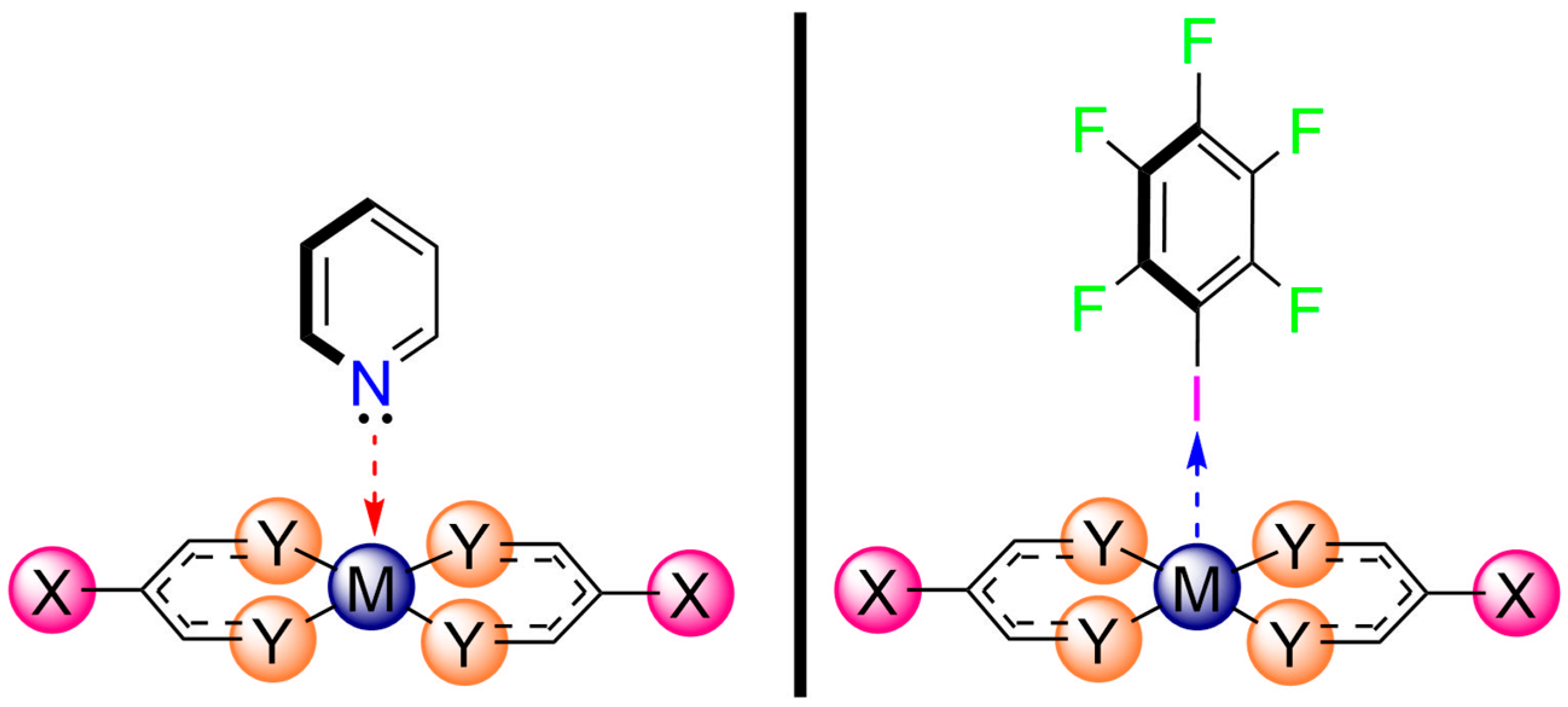
2.2. Dimeric Complexes
2.2.1. Combined Quantum Theory of Atoms in Molecules and Non-Covalent Interaction Plot (AIM/NCIplot) Analyses
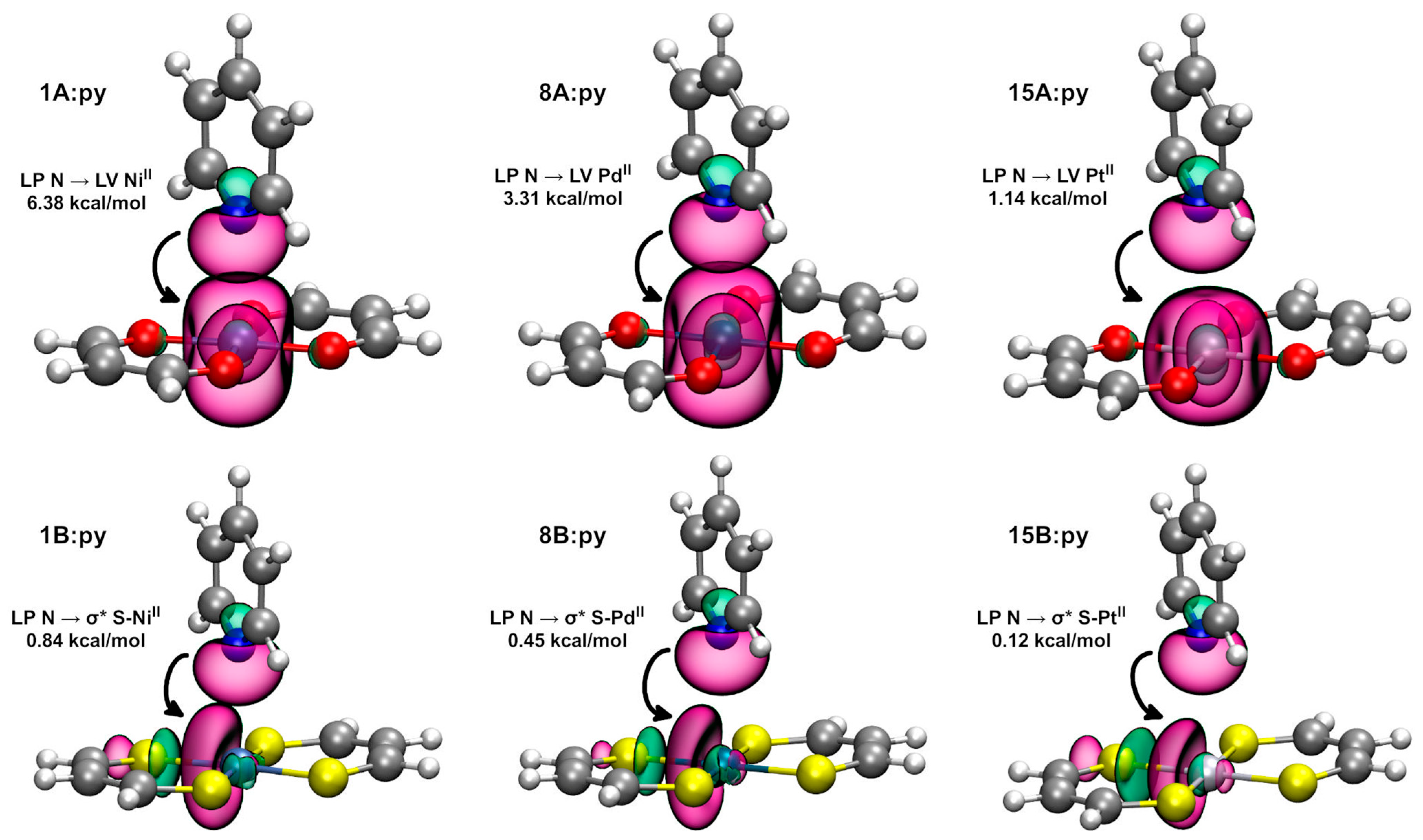
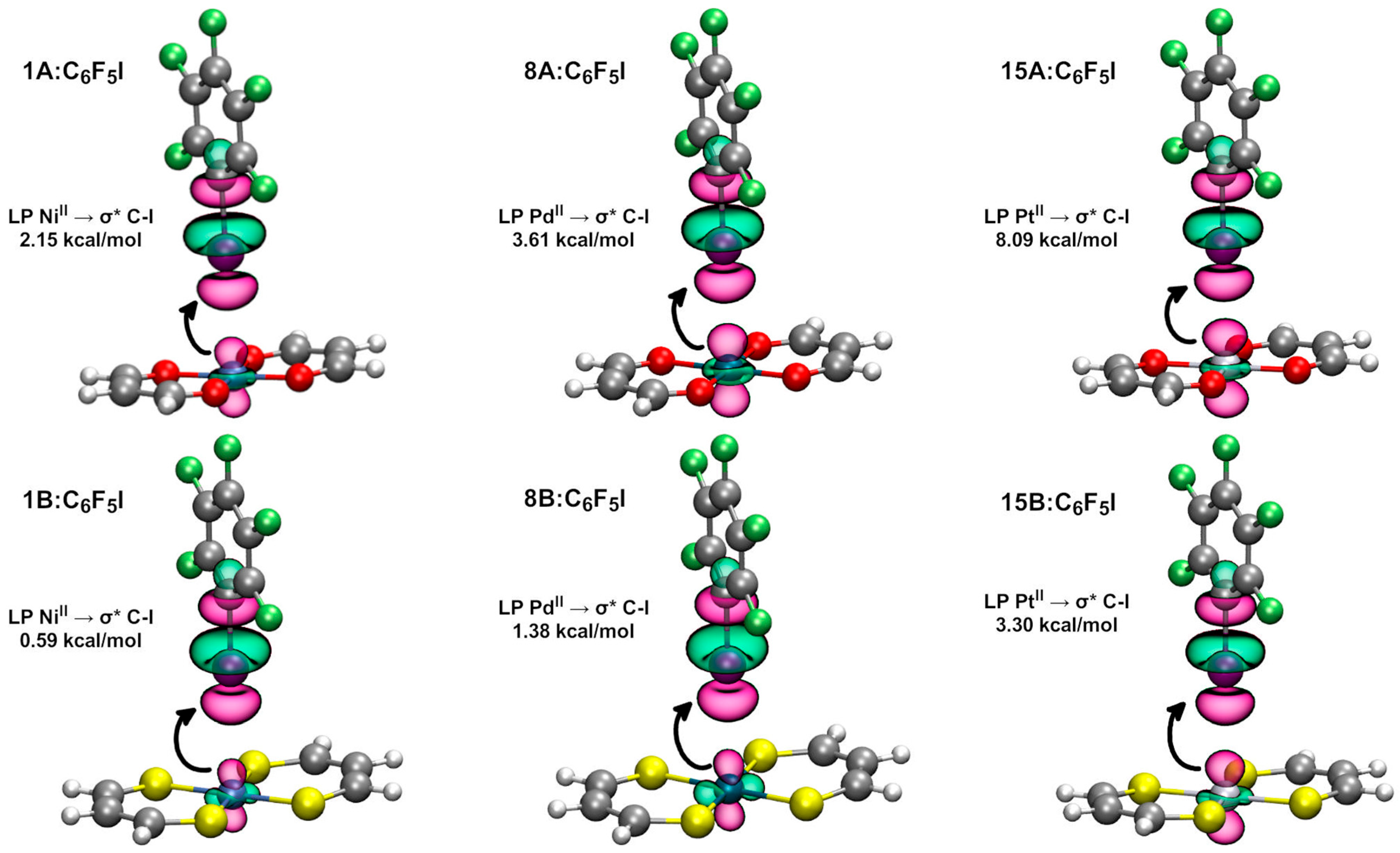
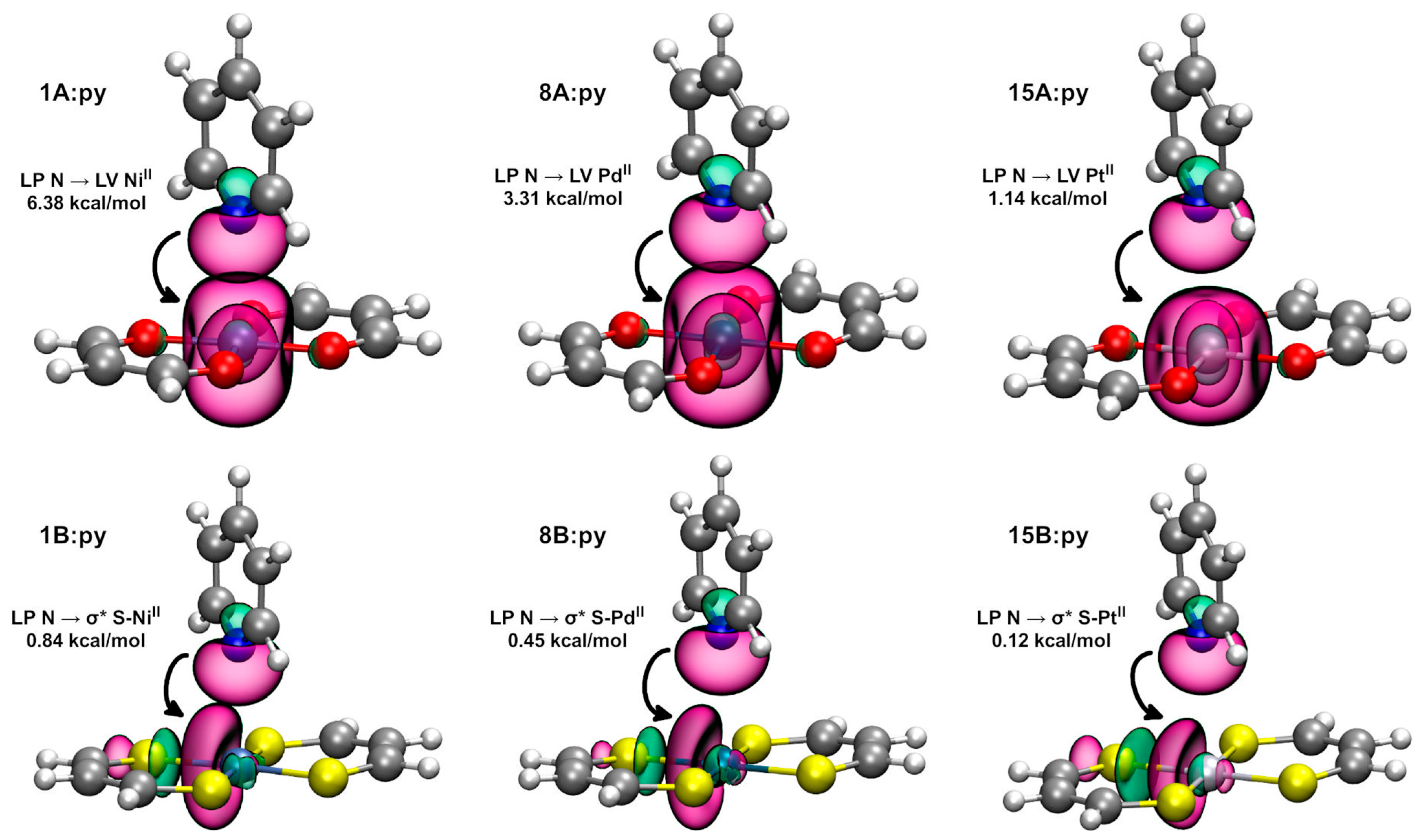
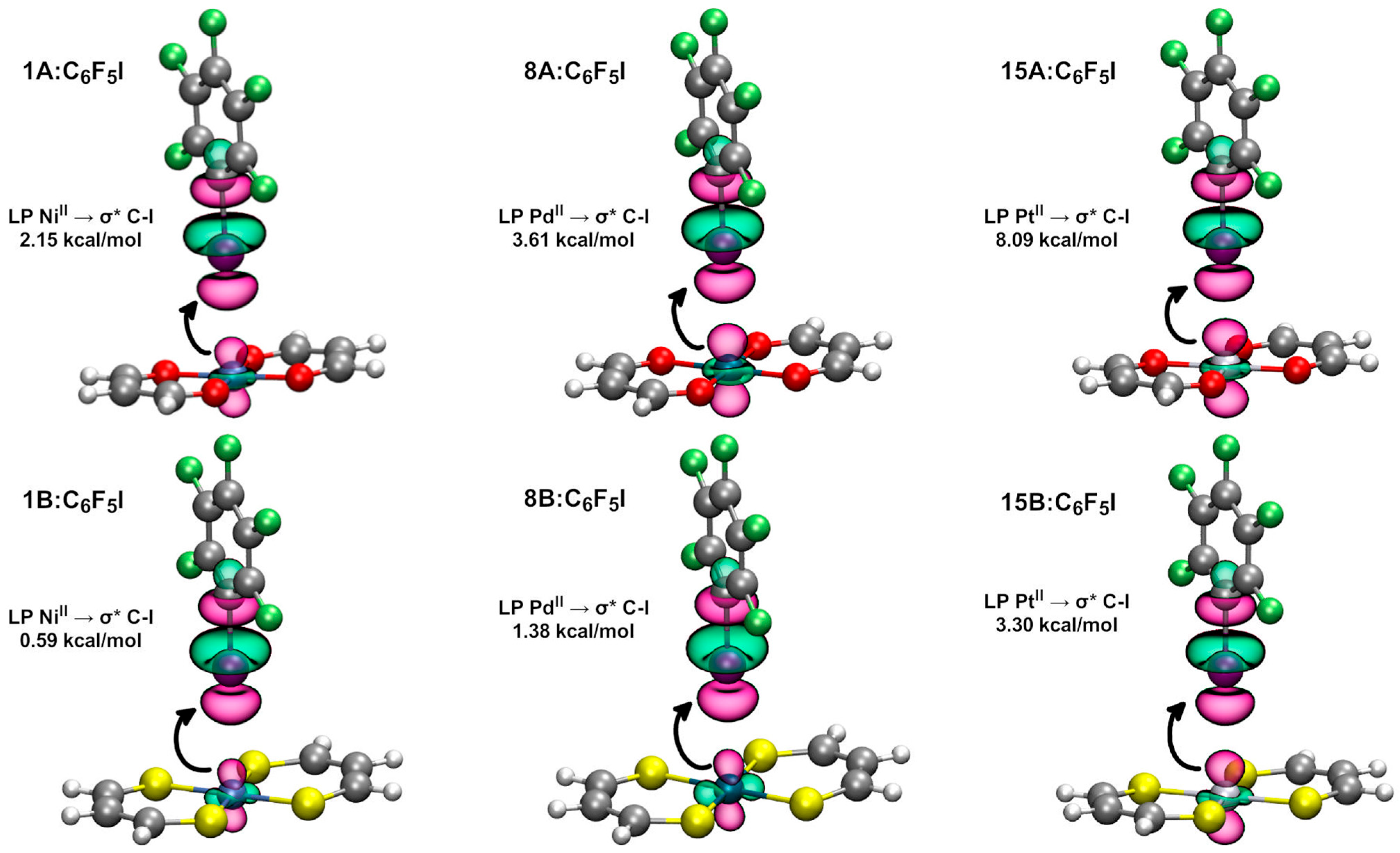
2.2.2. Natural Bond Orbital Analysis
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webber, M.J.; Langer, R. Drug Delivery by Supramolecular Design. Chem. Soc. Rev. 2017, 46, 6600–6620. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen Bonding—A Novel Interaction for Rational Drug Design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Aida, T.; Meijer, E.W.; Stupp, S.I. Functional Supramolecular Polymers. Science 2012, 335, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.J.; Appel, E.A.; Meijer, E.W.; Langer, R. Supramolecular Biomaterials. Nat. Mater. 2015, 15, 13–26. [Google Scholar] [CrossRef]
- Stupp, S.I.; Palmer, L.C. Supramolecular Chemistry and Self-Assembly in Organic Materials Design. Chem. Mater. 2014, 26, 507–518. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: A Holistic View. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef]
- Brown, C.J.; Toste, F.D.; Bergman, R.G.; Raymond, K.N. Supramolecular Catalysis in Metal-Ligand Cluster Hosts. Chem. Rev. 2015, 115, 3012–3035. [Google Scholar] [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.Y. Applications of Metal–Organic Frameworks in Heterogeneous Supramolecular Catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [Google Scholar] [CrossRef]
- Sarkar, S.; Peter, S.C. An Overview on Pd-Based Electrocatalysts for the Hydrogen Evolution Reaction. Inorg. Chem. Front. 2018, 5, 2060–2080. [Google Scholar] [CrossRef]
- Ohashi, M.; Saijo, H.; Shibata, M.; Ogoshi, S. Palladium-Catalyzed Base-Free Suzuki–Miyaura Coupling Reactions of Fluorinated Alkenes and Arenes via a Palladium Fluoride Key Intermediate. Eur. J. Org. Chem. 2013, 2013, 443–447. [Google Scholar] [CrossRef]
- Wells, P.B.; Wilkinson, A.G. Platinum Group Metals as Heterogeneous Enantioselective Catalysts. Top. Catal. 1998, 5, 39–50. [Google Scholar] [CrossRef]
- Vij, V.; Sultan, S.; Harzandi, A.M.; Meena, A.; Tiwari, J.N.; Lee, W.G.; Yoon, T.; Kim, K.S. Nickel-Based Electrocatalysts for Energy-Related Applications: Oxygen Reduction, Oxygen Evolution, and Hydrogen Evolution Reactions. ACS Catal. 2017, 7, 7196–7225. [Google Scholar] [CrossRef]
- Abu-Surrah, A.; Kettunen, M. Platinum Group Antitumor Chemistry: Design and Development of New Anticancer Drugs Complementary to Cisplatin. Curr. Med. Chem. 2006, 13, 1337–1357. [Google Scholar] [CrossRef]
- Raymond, E.; Chaney, S.G.; Taamma, A.; Cvitkovic, E. Oxaliplatin: A Review of Preclinical and Clinical Studies. Ann. Oncol. 1998, 9, 1053–1071. [Google Scholar] [CrossRef]
- Kaluderovic, N.; Paschke, G.R. Anticancer Metallotherapeutics in Preclinical Development. Curr. Med. Chem. 2011, 18, 4738–4752. [Google Scholar] [CrossRef]
- Kelland, L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Resnati, G.; Frontera, A.; Calabrese, M.; Burguera, S. Group-10 π-Hole⋯dz2[MII] Interactions: A Theoretical Study of Model Systems Inspired by CSD Structures. Dalton Trans. 2023, 52, 5056–5064. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Krykova, M.A.; Ivanov, D.M.; Novikov, A.S.; Sinelshchikova, A.A.; Volostnykh, M.V.; Konovalov, M.A.; Grigoriev, M.S.; Gorbunova, Y.G.; Kukushkin, V.Y. Reverse Arene Sandwich Structures Based upon π-Hole⋅⋅⋅[MII ] (d8 M=Pt, Pd) Interactions, Where Positively Charged Metal Centers Play the Role of a Nucleophile. Angew. Chem. Int. Ed. Engl. 2019, 58, 4164–4168. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Bokach, N.A.; Kukushkin, V.Y.; Frontera, A. Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering. Chem. A Eur. J. 2022, 28, e202103173. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ananyev, I.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. π-Hole··· Dz2[PtII] Interactions with Electron-Deficient Arenes Enhance the Phosphorescence of PtII-Based Luminophores. Inorg. Chem. 2020, 59, 9308–9314. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bikbaeva, Z.M.; Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Bokach, N.A.; Kukushkin, V.Y. Electrophilic-Nucleophilic Dualism of Nickel(II) toward Ni···I Noncovalent Interactions: Semicoordination of Iodine Centers via Electron Belt and Halogen Bonding via σ-Hole. Inorg. Chem. 2017, 56, 13562–13578. [Google Scholar] [CrossRef] [PubMed]
- Yamashina, Y.; Kataoka, Y.; Ura, Y. Inclusion of an Iodine Molecule in a Tiara-Like Octanuclear Palladium Thiolate Complex. Eur. J. Inorg. Chem. 2014, 2014, 4073–4078. [Google Scholar] [CrossRef]
- Baykov, S.V.; Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Boyarskiy, V.P. Pt/PD and I/Br Isostructural Exchange Provides Formation of C−I···PD, C−Br···PT, and C−Br···Pd Metal-Involving Halogen Bonding. Cryst. Growth Des. 2018, 18, 5973–5980. [Google Scholar] [CrossRef]
- Gossage, R.A.; Ryabov, A.D.; Spek, A.L.; Stufkens, D.J.; Van Beek, J.A.M.; Van Eldik, R.; Van Koten, G. Models for the Initial Stages of Oxidative Addition. Synthesis, Characterization, and Mechanistic Investigation of η 1-I 2 Organometallic “pincer” Complexes of Platinum. J. Am. Chem. Soc. 1999, 121, 2488–2497. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Kirina, Y.V.; Kukushkin, V.Y. Halogen Bonding between Metal Centers and Halocarbons. Chem. Commun. 2016, 52, 5565–5568. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Zhang, H.; Wei, F.; Huang, L.; Ke, S.; Fu, J.; Jing, C.; Cheng, J.; Liu, S. Tuning Electron Delocalization of Hydrogen-Bonded Organic Framework Cathode for High-Performance Zinc-Organic Batteries. Nat. Commun. 2023, 14, 5235. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Zhang, H.; Wei, F.; Zhang, T.; Wu, Y.; Huang, L.; Fu, J.; Jing, C.; Cheng, J.; et al. Designing Ternary Hydrated Eutectic Electrolyte Capable of Four-Electron Conversion for Advanced Zn–I2 Full Batteries. Energy Environ. Sci. 2023, 16, 4502–4510. [Google Scholar] [CrossRef]
- Xu, H.; Guan, D. Exceptional Anisotropic Noncovalent Interactions in Ultrathin Nanorods: The Terminal σ-Hole. ACS Appl. Mater. Interfaces 2022, 14, 51190–51199. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and Its Applications to Chemical Bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F. Natural Bond Orbital Analysis: A Critical Overview of Relationships to Alternative Bonding Perspectives. J. Comput. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Armbruster, M.K.; Weigend, F.; van Wüllen, C.; Klopper, W. Self-Consistent Treatment of Spin–Orbit Interactions with Efficient Hartree–Fock and Density Functional Methods. Phys. Chem. Chem. Phys. 2008, 10, 1748–1756. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 6.1.1; Semichem Inc.: Shawnee Mission, KS, USA, 2019. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Vs, min | Vs, max | Vs, M | Compound | Vs, min | Vs, max | Vs, M |
|---|---|---|---|---|---|---|---|
| 1A | −37.2 | +19.3 | +0.2 | 1B | −16.7 | +23.2 | −8.0 |
| 2A | −30.9 | +22.7 | +7.6 | 2B | −10.6 | +26.5 | −1.4 |
| 3A | −35.0 | +30.9 | +22.5 | 3B | −33.3 | +32.9 | +11.0 |
| 4A | −27.1 | +27.9 | +24.0 | 4B | −24.9 | +29.1 | +11.9 |
| 5A | −39.7 | +34.5 | −2.6 | 5B | −24.5 | +38.9 | −11.9 |
| 6A | −40.8 | +18.3 | −5.6 | 6B | −21.9 | +22.6 | −16.0 |
| 7A | −40.0 | +17.2 | −3.2 | 7B | −19.8 | +20.9 | −12.4 |
| 8A | −35.9 | +20.0 | −6.4 | 8B | −11.4 | +13.8 | −11.1 |
| 9A | −29.1 | +23.5 | +0.6 | 9B | −9.7 | +26.8 | −5.6 |
| 10A | −34.7 | +31.3 | +14.1 | 10B | −33.5 | +33.0 | +6.2 |
| 11A | −26.5 | +28.0 | +15.2 | 11B | −28.6 | +32.8 | +9.4 |
| 12A | −37.7 | +35.3 | −8.0 | 12B | −24.7 | +39.0 | −15.0 |
| 13A | −39.2 | +19.0 | −11.9 | 13B | −19.4 | +22.9 | −18.9 |
| 14A | −38.4 | +18.2 | −9.7 | 14B | −17.9 | +30.6 | −15.7 |
| 15A | −34.0 | +20.6 | −14.5 | 15B | −18.0 | +23.7 | −18.0 |
| 16A | −27.2 | +24.2 | −7.5 | 16B | −11.6 | +28.0 | −11.6 |
| 17A | −34.3 | +31.9 | +5.3 | 17B | −33.3 | +32.7 | +1.9 |
| 18A | −26.1 | +28.6 | +6.5 | 18B | −25.3 | +32.0 | +3.8 |
| 19A | −35.9 | +35.8 | −16.0 | 19B | −24.5 | +39.1 | −18.8 |
| 20A | −37.4 | +19.4 | −19.4 | 20B | −22.7 | +23.0 | −22.7 |
| 21A | −36.4 | +18.6 | −17.4 | 21B | −19.3 | +21.2 | −19.3 |
| py (C5H5N) | −36.2 | +21.6 | / | C6F5I | −7.0 | +32.0 | / |
| Dimer | dN: ⋯ M (Å) | Interaction Energy (kcal/mol) | Dimer | dN ⋯ M (Å) | Interaction Energy (kcal/mol) |
|---|---|---|---|---|---|
| 1A:py | 2.616 | −6.6 | 1B:py | 2.752 | −5.5 |
| 2A:py | 2.592 | −7.7 | 2B:py | 2.737 | −6.2 |
| 3A:py | 2.542 | −9.9 | 3B:py | 2.689 | −7.9 |
| 4A:py | 2.533 | −10.3 | 4B:py | 2.676 | −8.2 |
| 5A:py | 2.645 | −6.2 | 5B:py | 2.789 | −5.0 |
| 6A:py | 2.665 | −6.1 | 6B:py | 2.846 | −4.7 |
| 7A:py | 2.645 | −6.2 | 7B:py | 2.749 | −5.5 |
| 8A:py | 2.956 | −4.3 | 8B:py | 3.052 | −3.9 |
| 9A:py | 2.925 | −5.2 | 9B:py | 3.029 | −4.5 |
| 10A:py | 2.884 | −7.0 | 10B:py | 2.967 | −5.9 |
| 11A:py | 2.878 | −7.3 | 11B:py | 3.019 | −5.9 |
| 12A:py | 2.981 | −4.0 | 12B:py | 3.088 | −3.5 |
| 13A:py | 2.994 | −3.9 | 13B:py | 3.151 | −3.3 |
| 14A:py | 2.980 | −4.2 | 14B:py | 3.115 | −3.4 |
| 15A:py | 3.223 | −2.6 | 15B:py | 3.276 | −3.0 |
| 16A:py | 3.186 | −3.4 | 16B:py | 3.251 | −3.6 |
| 17A:py | 3.128 | −4.9 | 17B:py | 3.193 | −4.8 |
| 18A:py | 3.118 | −5.1 | 18B:py | 3.271 | −4.9 |
| 19A:py | 3.251 | −2.4 | 19B:py | 3.308 | −2.6 |
| 20A:py | 3.261 | −2.2 | 20B:py | 3.357 | −2.2 |
| 21A:py | 3.287 | −2.4 | 21B:py | 3.328 | −2.7 |
| Dimer | dI ⋯ M (Å) | Interaction Energy (kcal/mol) | Dimer | dI ⋯ M (Å) | Interaction Energy (kcal/mol) |
|---|---|---|---|---|---|
| 1A:C6F5I | 3.313 | −3.7 | 1B:C6F5I | 3.521 | −5.1 |
| 2A:C6F5I | 3.751 | −3.0 | 2B:C6F5I | 3.543 | −4.8 |
| 3A:C6F5I | 3.754 | −2.5 | 3B:C6F5I | 3.567 | −4.3 |
| 4A:C6F5I | 3.768 | −2.4 | 4B:C6F5I | 3.583 | −4.1 |
| 5A:C6F5I | 3.262 | −4.4 | 5B:C6F5I | 3.438 | −5.8 |
| 6A:C6F5I | 3.286 | −4.7 | 6B:C6F5I | 3.502 | −5.9 |
| 7A:C6F5I | 3.296 | −4.5 | 7B:C6F5I | 3.420 | −5.9 |
| 8A:C6F5I | 3.395 | −4.8 | 8B:C6F5I | 3.503 | −6.0 |
| 9A:C6F5I | 3.443 | −4.4 | 9B:C6F5I | 3.524 | −5.7 |
| 10A:C6F5I | 3.498 | −3.8 | 10B:C6F5I | 3.541 | −5.2 |
| 11A:C6F5I | 3.493 | −3.8 | 11B:C6F5I | 3.473 | −5.7 |
| 12A:C6F5I | 3.361 | −5.4 | 12B:C6F5I | 3.480 | −6.6 |
| 13A:C6F5I | 3.376 | −5.7 | 13B:C6F5I | 3.457 | −7.2 |
| 14A:C6F5I | 3.324 | −5.8 | 14B:C6F5I | 3.461 | −6.8 |
| 15A:C6F5I | 3.318 | −6.2 | 15B:C6F5I | 3.523 | −6.6 |
| 16A:C6F5I | 3.366 | −5.6 | 16B:C6F5I | 3.545 | −6.2 |
| 17A:C6F5I | 3.437 | −4.8 | 17B:C6F5I | 3.571 | −5.6 |
| 18A:C6F5I | 3.443 | −4.7 | 18B:C6F5I | 3.533 | −6.1 |
| 19A:C6F5I | 3.274 | −6.8 | 19B:C6F5I | 3.502 | −7.2 |
| 20A:C6F5I | 3.295 | −7.0 | 20B:C6F5I | 3.520 | −7.4 |
| 21A:C6F5I | 3.261 | −7.2 | 21B:C6F5I | 3.552 | −7.2 |
| Dimer | E (2) LP N → LV (MII) (kcal/mol) | Dimer | E (2) LP N → σ* S-(MII) (kcal/mol) * |
|---|---|---|---|
| 1A:py | 6.38 | 1B:py | 0.84 |
| 2A:py | 6.99 | 2B:py | 0.90 |
| 3A:py | 8.12 | 3B:py | 1.02 |
| 4A:py | 8.37 | 4B:py | 1.05 |
| 5A:py | 5.82 | 5B:py | 0.79 |
| 6A:py | 5.55 | 6B:py | 0.70 |
| 7A:py | 5.78 | 7B:py | 0.88 |
| 8A:py | 3.31 | 8B:py | 0.45 |
| 9A:py | 3.77 | 9B:py | 0.50 |
| 10A:py | 4.37 | 10B:py | 0.64 |
| 11A:py | 4.47 | 11B:py | 0.56 |
| 12A:py | 2.96 | 12B:py | 0.39 |
| 13A:py | 2.79 | 13B:py | 0.32 |
| 14A:py | 2.87 | 14B:py | 0.36 |
| 15A:py | 1.14 | 15B:py | 0.12 |
| 16A:py | 1.34 | 16B:py | 0.13 |
| 17A:py | 1.77 | 17B:py | 0.17 |
| 18A:py | 1.81 | 18B:py | 0.08 |
| 19A:py | 1.00 | 19B:py | 0.11 |
| 20A:py | 0.99 | 20B:py | 0.10 |
| 21A:py | 0.89 | 21B:py | 0.11 |
| Dimer | E (2) LP (MII) → σ* C-I (kcal/mol) | Dimer | E (2) LP (MII) → σ* C-I (kcal/mol) * |
|---|---|---|---|
| 1A:C6F5I | 2.15 | 1B:C6F5I | 0.59 (0.15) |
| 2A:C6F5I | 0.78 | 2B:C6F5I | 0.54 (0.13) |
| 3A:C6F5I | 0.72 | 3B:C6F5I | 0.48 (0.11) |
| 4A:C6F5I | 0.69 | 4B:C6F5I | 0.47 (0.11) |
| 5A:C6F5I | 2.47 | 5B:C6F5I | 0.73 (0.20) |
| 6A:C6F5I | 2.41 | 6B:C6F5I | 0.72 (0.16) |
| 7A:C6F5I | 2.34 | 7B:C6F5I | 0.72 (0.20) |
| 8A:C6F5I | 3.61 | 8B:C6F5I | 1.38 (0.14) |
| 9A:C6F5I | 3.10 | 9B:C6F5I | 1.53 (0.12) |
| 10A:C6F5I | 2.51 | 10B:C6F5I | 1.40 (0.11) |
| 11A:C6F5I | 2.58 | 11B:C6F5I | 1.68 (0.14) |
| 12A:C6F5I | 4.20 | 12B:C6F5I | 1.60 (0.15) |
| 13A:C6F5I | 4.18 | 13B:C6F5I | 1.61 (0.16) |
| 14A:C6F5I | 4.77 | 14B:C6F5I | 1.38 (0.16) |
| 15A:C6F5I | 8.09 | 15B:C6F5I | 3.30 |
| 16A:C6F5I | 7.11 | 16B:C6F5I | 3.10 |
| 17A:C6F5I | 5.83 | 17B:C6F5I | 2.83 |
| 18A:C6F5I | 5.78 | 18B:C6F5I | 3.09 |
| 19A:C6F5I | 9.25 | 19B:C6F5I | 3.69 |
| 20A:C6F5I | 9.16 | 20B:C6F5I | 3.82 |
| 21A:C6F5I | 9.99 | 21B:C6F5I | 3.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burguera, S.; Bauzá, A.; Frontera, A. Tuning the Nucleophilicity and Electrophilicity of Group 10 Elements through Substituent Effects: A DFT Study. Int. J. Mol. Sci. 2023, 24, 15597. https://doi.org/10.3390/ijms242115597
Burguera S, Bauzá A, Frontera A. Tuning the Nucleophilicity and Electrophilicity of Group 10 Elements through Substituent Effects: A DFT Study. International Journal of Molecular Sciences. 2023; 24(21):15597. https://doi.org/10.3390/ijms242115597
Chicago/Turabian StyleBurguera, Sergi, Antonio Bauzá, and Antonio Frontera. 2023. "Tuning the Nucleophilicity and Electrophilicity of Group 10 Elements through Substituent Effects: A DFT Study" International Journal of Molecular Sciences 24, no. 21: 15597. https://doi.org/10.3390/ijms242115597
APA StyleBurguera, S., Bauzá, A., & Frontera, A. (2023). Tuning the Nucleophilicity and Electrophilicity of Group 10 Elements through Substituent Effects: A DFT Study. International Journal of Molecular Sciences, 24(21), 15597. https://doi.org/10.3390/ijms242115597