
Flexible Quality Control for Protein Turnover Rates Using d2ome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
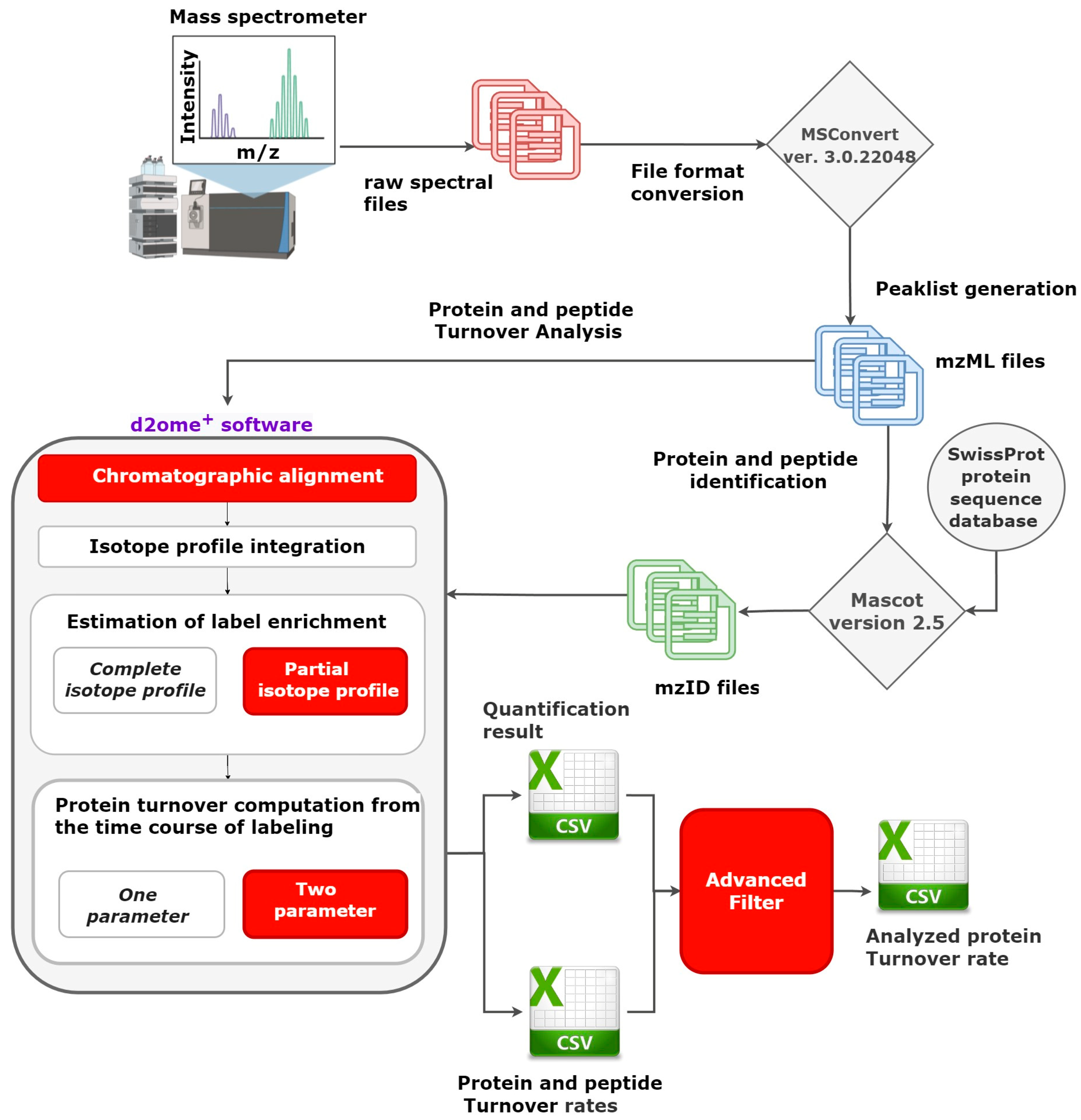
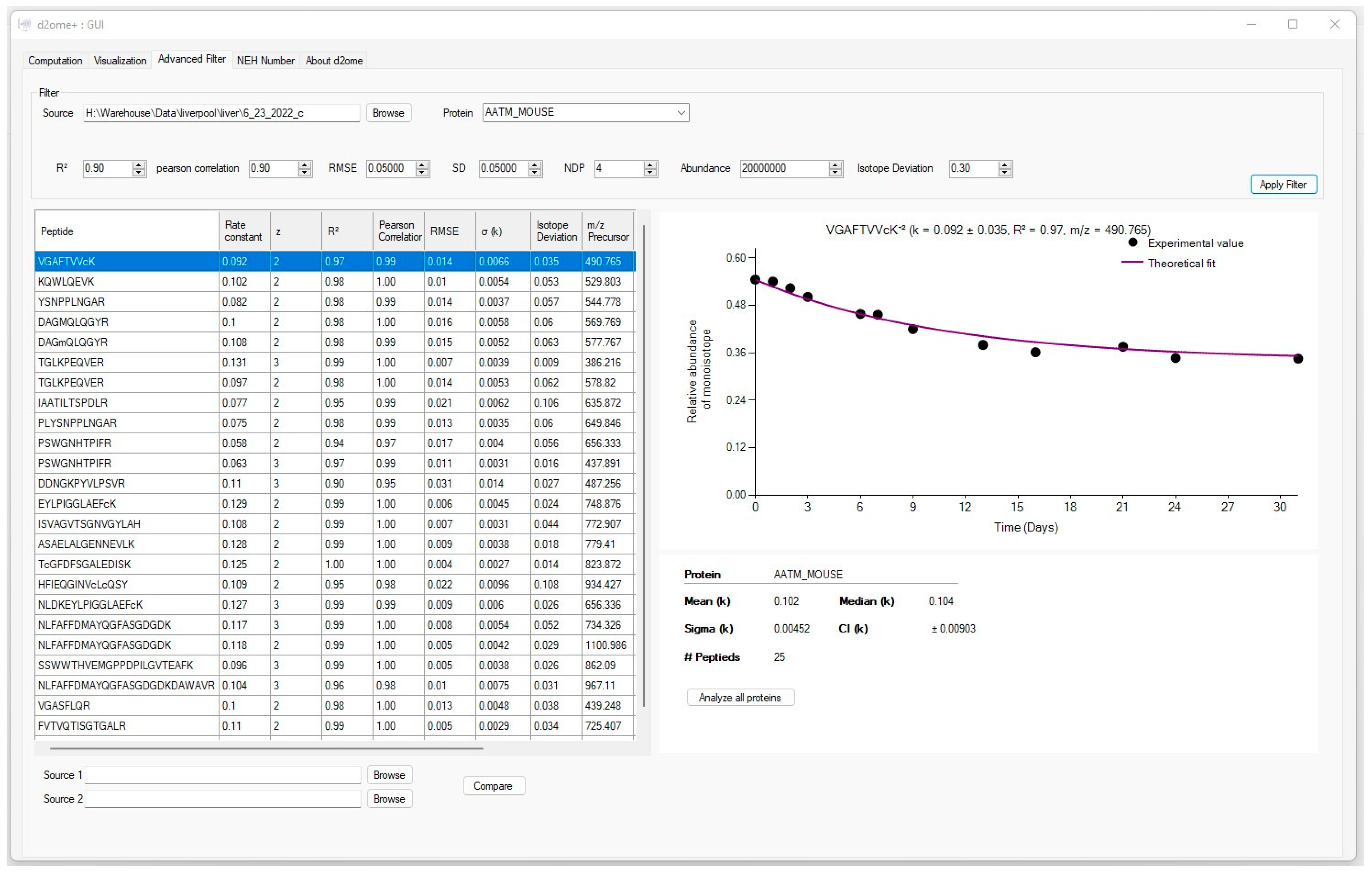
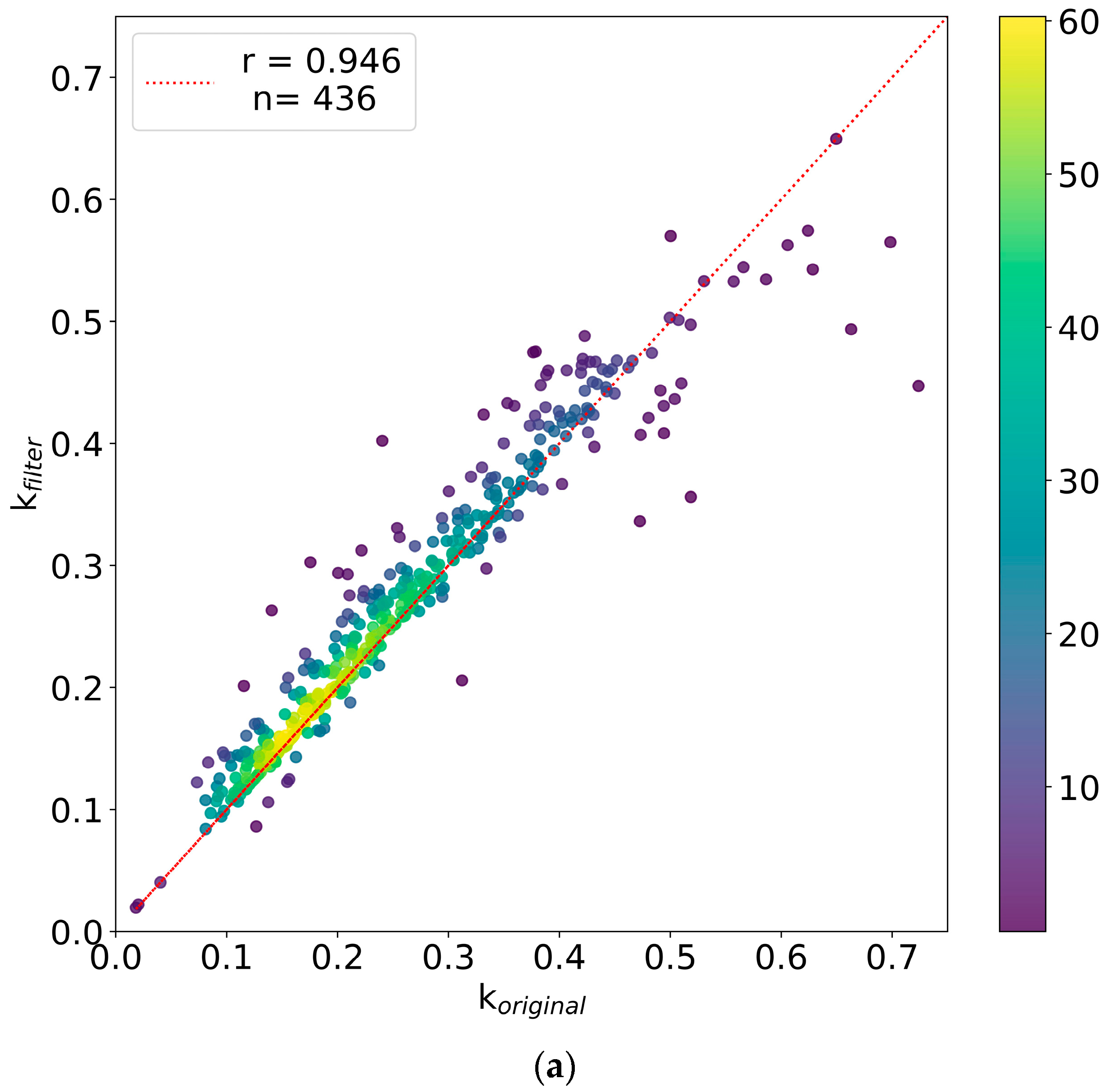
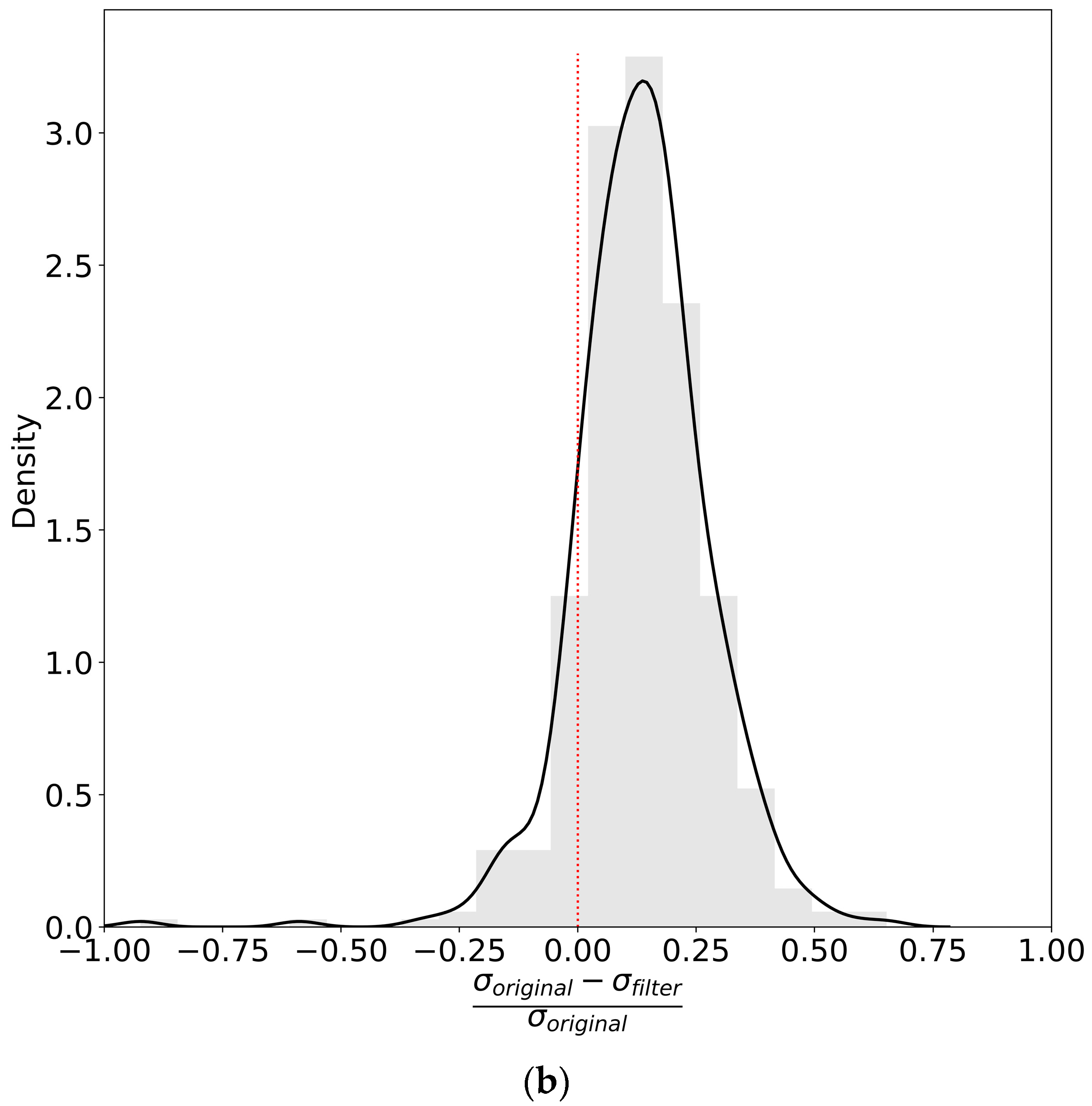
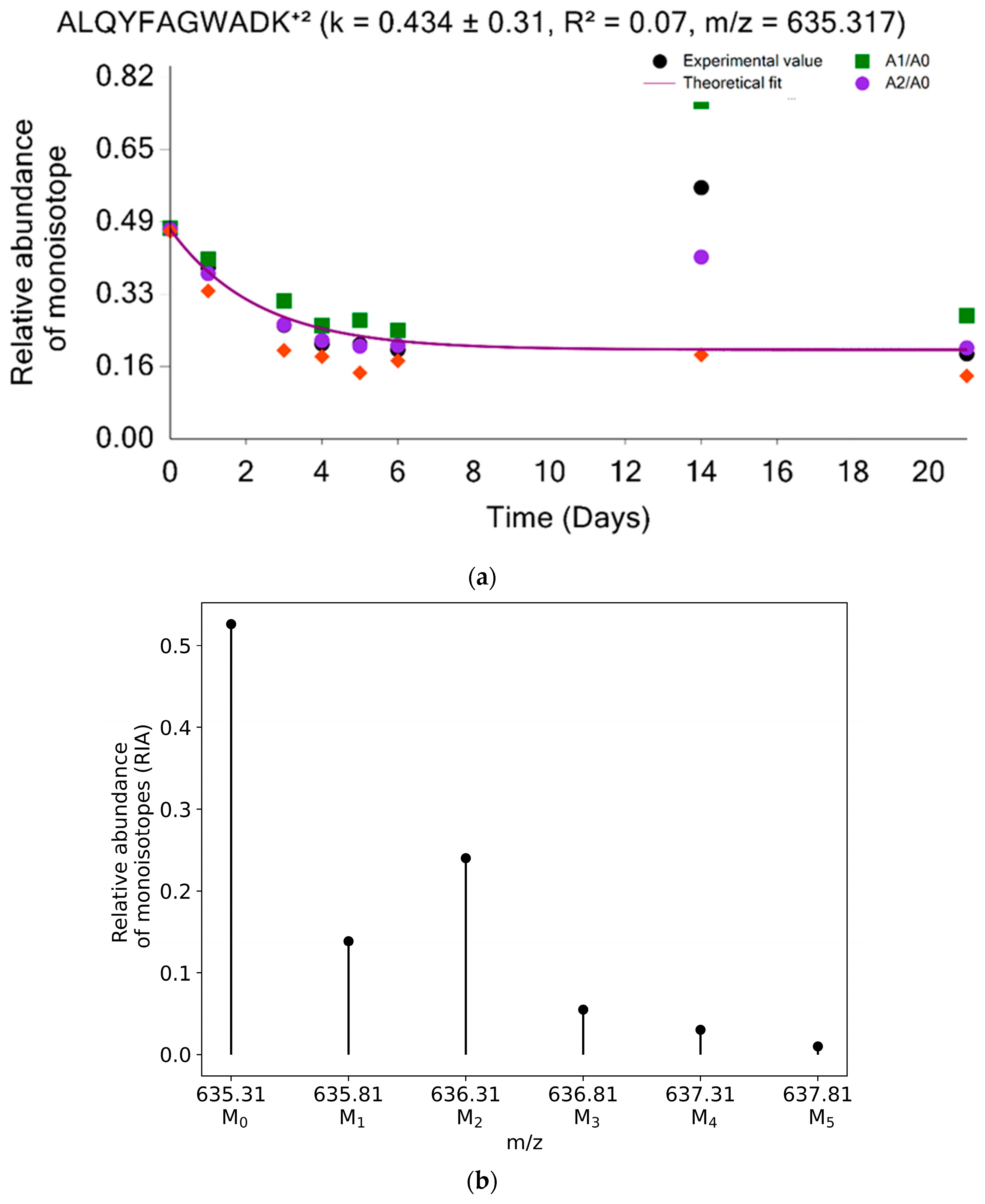
2.1. Advanced Filters to Facilitate Protein Turnover Rate Analysis
2.2. Recent Developments in d2ome Software
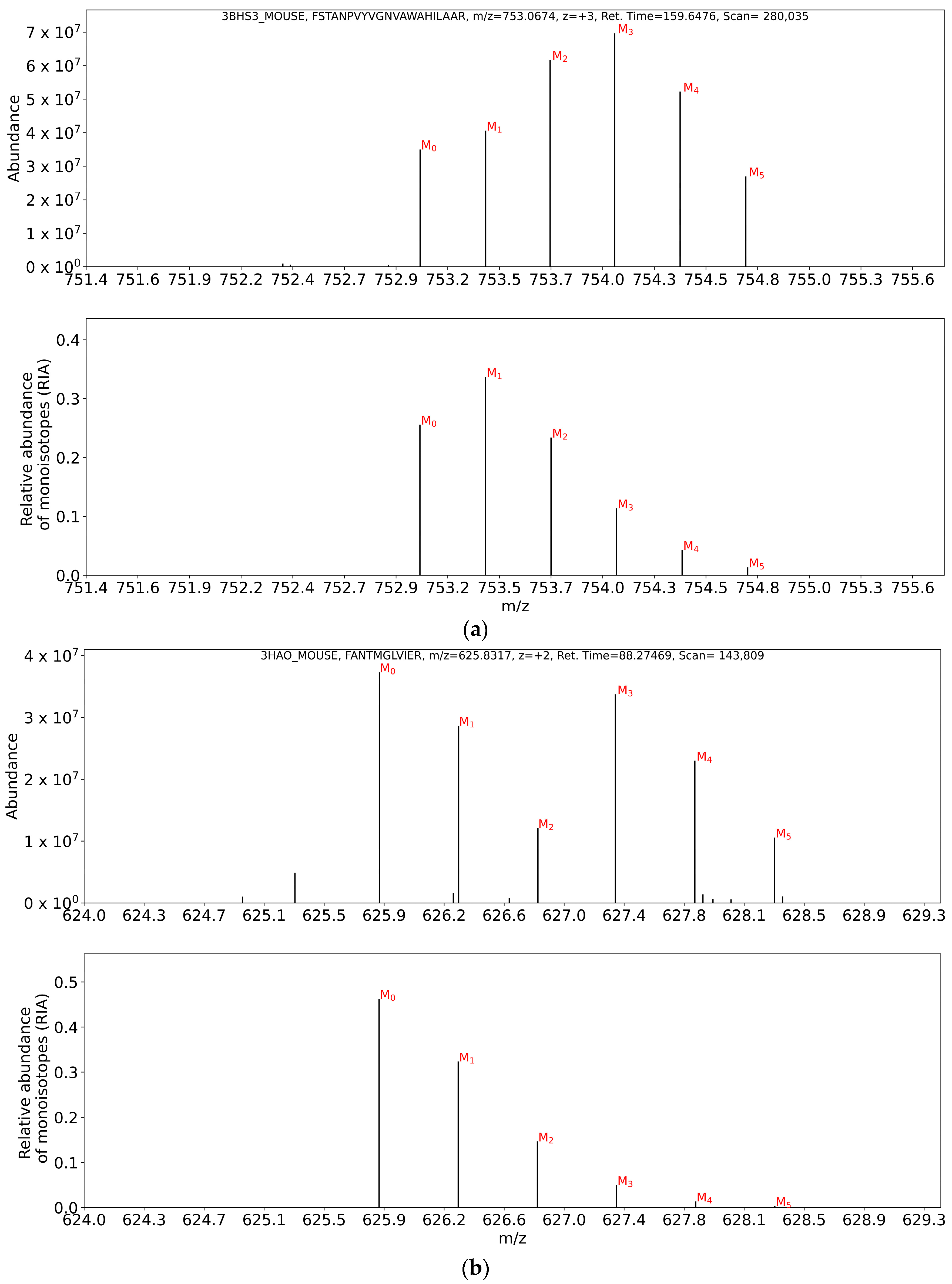
2.2.1. Quantification of Label Enrichment from Partial Isotope Profiles
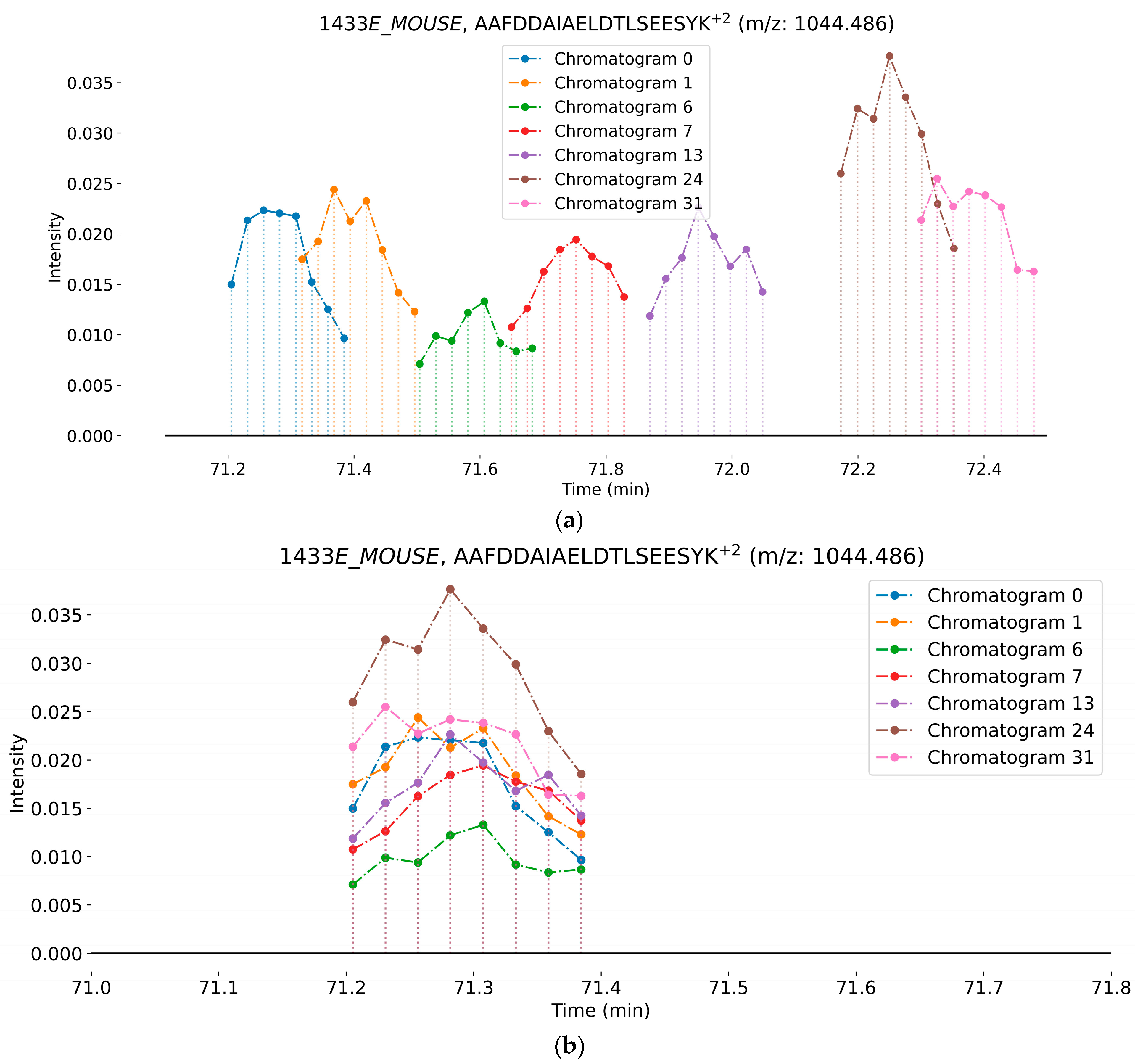
2.2.2. Retention Time Alignment
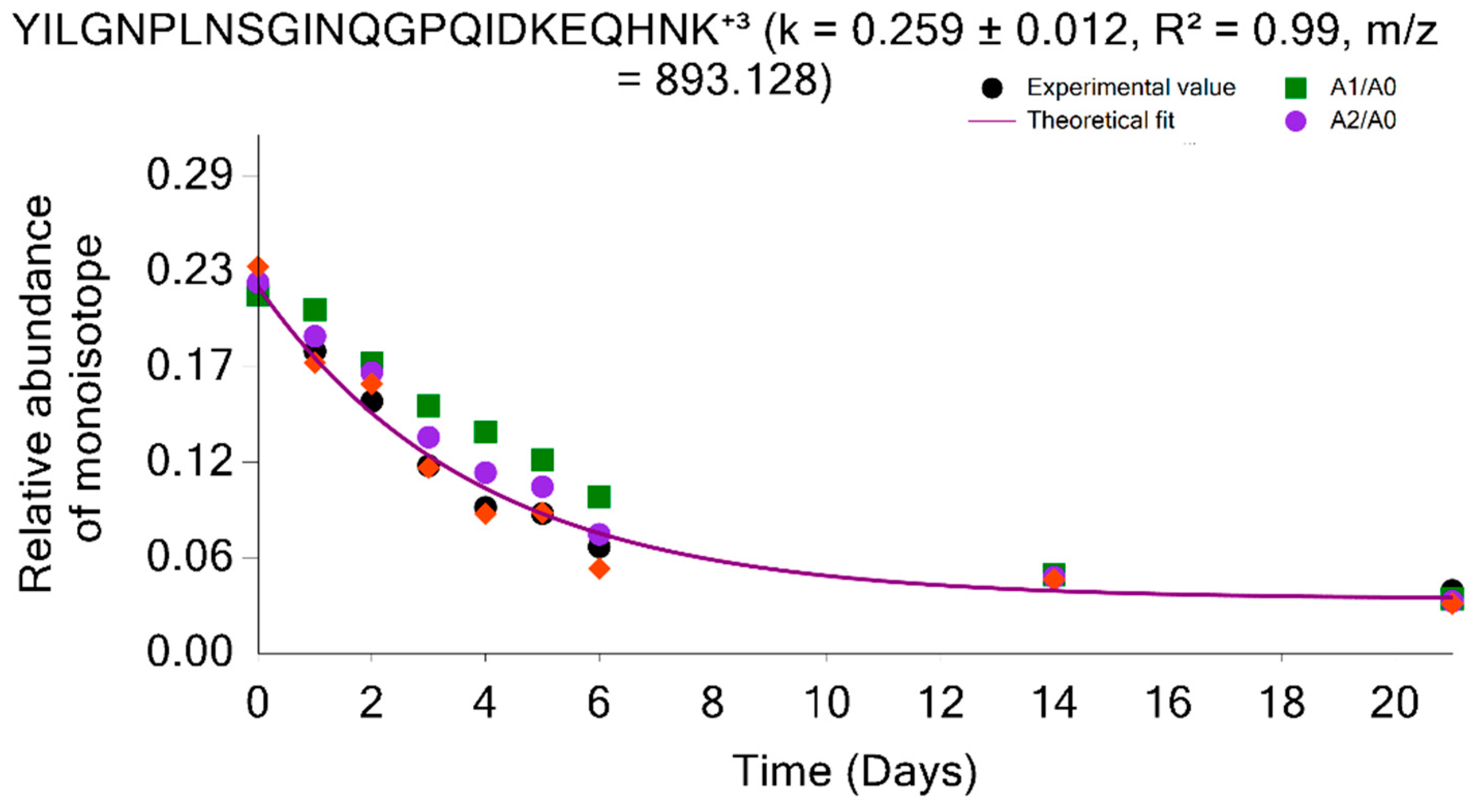
2.2.3. Two-Parameter Modeling
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hammond, D.E.; Simpson, D.M.; Franco, C.; Wright Muelas, M.; Waters, J.; Ludwig, R.W.; Prescott, M.C.; Hurst, J.L.; Beynon, R.J.; Lau, E. Harmonizing Labeling and Analytical Strategies to Obtain Protein Turnover Rates in Intact Adult Animals. Mol. Cell Proteom. 2022, 21, 100252. [Google Scholar] [CrossRef]
- Peinado-Izaguerri, J.; Zarzuela, E.; McLaughlin, M.; Small, A.C.; Riva, F.; McKeegan, D.E.F.; Bain, M.; Munoz, J.; Bhide, M.; Preston, T. A novel dynamic proteomics approach for the measurement of broiler chicken protein fractional synthesis rate. Rapid Commun. Mass. Spectrom. 2023, 37, e9497. [Google Scholar] [CrossRef]
- Deberneh, H.M.; Sadygov, R.G. Retention Time Alignment for Protein Turnover Studies Using Heavy Water Metabolic Labeling. J. Proteome Res. 2023, 22, 410–419. [Google Scholar] [CrossRef]
- Rauniyar, N.; McClatchy, D.B.; Yates, J.R. 3rd. Stable isotope labeling of mammals (SILAM) for in vivo quantitative proteomic analysis. Methods 2013, 61, 260–268. [Google Scholar] [CrossRef]
- Lee, K.; Haddad, A.; Osme, A.; Kim, C.; Borzou, A.; Ilchenko, S.; Allende, D.; Dasarathy, S.; McCullough, A.; Sadygov, R.G.; et al. Hepatic Mitochondrial Defects in a Nonalcoholic Fatty Liver Disease Mouse Model Are Associated with Increased Degradation of Oxidative Phosphorylation Subunits. Mol. Cell Proteom. 2018, 17, 2371–2386. [Google Scholar] [CrossRef]
- Papageorgopoulos, C.; Caldwell, K.; Shackleton, C.; Schweingrubber, H.; Hellerstein, M.K. Measuring protein synthesis by mass isotopomer distribution analysis (MIDA). Anal. Biochem. 1999, 267, 1–16. [Google Scholar] [CrossRef]
- Kim, T.Y.; Wang, D.; Kim, A.K.; Lau, E.; Lin, A.J.; Liem, D.A.; Zhang, J.; Zong, N.C.; Lam, M.P.; Ping, P. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol. Cell Proteom. 2012, 11, 1586–1594. [Google Scholar] [CrossRef]
- Naylor, B.C.; Porter, M.T.; Wilson, E.; Herring, A.; Lofthouse, S.; Hannemann, A.; Piccolo, S.R.; Rockwood, A.L.; Price, J.C. DeuteRater: A tool for quantifying peptide isotope precision and kinetic proteomics. Bioinformatics 2017, 33, 1514–1520. [Google Scholar] [CrossRef]
- Sadygov, R.G.; Avva, J.; Rahman, M.; Lee, K.; Ilchenko, S.; Kasumov, T.; Borzou, A. d2ome, Software for in Vivo Protein Turnover Analysis Using Heavy Water Labeling and LC-MS, Reveals Alterations of Hepatic Proteome Dynamics in a Mouse Model of NAFLD. J. Proteome Res. 2018, 17, 3740–3748. [Google Scholar] [CrossRef]
- Rolfs, Z.; Frey, B.L.; Shi, X.; Kawai, Y.; Smith, L.M.; Welham, N.V. An atlas of protein turnover rates in mouse tissues. Nat. Commun. 2021, 12, 6778. [Google Scholar] [CrossRef]
- Basisty, N.; Shulman, N.; Wehrfritz, C.; Marsh, A.N.; Shah, S.; Rose, J.; Ebert, S.; Miller, M.; Dai, D.F.; Rabinovitch, P.S.; et al. TurnoveR: A Skyline External Tool for Analysis of Protein Turnover in Metabolic Labeling Studies. J. Proteome Res. 2022, 22, 311–322. [Google Scholar] [CrossRef]
- Deberneh, H.M.; Sadygov, R.G. Software Tool for Visualization and Validation of Protein Turnover Rates Using Heavy Water Metabolic Labeling and LC-MS. Int. J. Mol. Sci. 2022, 23, 14620. [Google Scholar] [CrossRef]
- Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W.H.; Rompp, A.; Neumann, S.; Pizarro, A.D.; et al. mzML--a community standard for mass spectrometry data. Mol. Cell Proteom. 2011, 10, R110 000133. [Google Scholar] [CrossRef]
- Jones, A.R.; Eisenacher, M.; Mayer, G.; Kohlbacher, O.; Siepen, J.; Hubbard, S.J.; Selley, J.N.; Searle, B.C.; Shofstahl, J.; Seymour, S.L.; et al. The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell Proteom. 2012, 11, M111 014381. [Google Scholar] [CrossRef]
- Deberneh, H.M.; Abdelrahman, D.R.; Verma, S.K.; Linares, J.J.; Murton, A.J.; Russell, W.K.; Kuyumcu-Martinez, M.N.; Miller, B.F.; Sadygov, R.G. Quantifying label enrichment from two mass isotopomers increases proteome coverage for in vivo protein turnover using heavy water metabolic labeling. Commun. Chem. 2023, 6, 72. [Google Scholar] [CrossRef]
- Sadygov, R.G.; Zhao, Y.; Haidacher, S.J.; Starkey, J.M.; Tilton, R.G.; Denner, L. Using power spectrum analysis to evaluate (18)O-water labeling data acquired from low resolution mass spectrometers. J. Proteome Res. 2010, 9, 4306–4312. [Google Scholar] [CrossRef] [PubMed]
- Sadygov, R.G. Protein turnover models for LC-MS data of heavy water metabolic labeling. Brief. Bioinform. 2022, 23, bbab598. [Google Scholar] [CrossRef] [PubMed]
- Ilchenko, S.; Haddad, A.; Sadana, P.; Recchia, F.A.; Sadygov, R.G.; Kasumov, T. Calculation of the Protein Turnover Rate Using the Number of Incorporated (2)H Atoms and Proteomics Analysis of a Single Labeled Sample. Anal. Chem. 2019, 91, 14340–14351. [Google Scholar] [CrossRef] [PubMed]
- Sadygov, R.G. Poisson Model To Generate Isotope Distribution for Biomolecules. J. Proteome Res. 2018, 17, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Jensen, D.R.; Ramirez, D.E. Outlier Detection Under Star-Contoured Errors. J. Stat. Theory Pract. 2015, 9, 850–867. [Google Scholar] [CrossRef]
- Deberneh, H.M.; Abdelrahman, D.R.; Verma, S.K.; Linares, J.J.; Murton, A.J.; Russell, W.K.; Kuyumcu-Martinez, M.N.; Miller, B.F.; Sadygov, R.G. A large-scale LC-MS dataset of murine liver proteome from time course of heavy water metabolic labeling. Sci. Data 2023, 10, 635. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Lau, E.; Cao, Q.; Ng, D.C.; Bleakley, B.J.; Dincer, T.U.; Bot, B.M.; Wang, D.; Liem, D.A.; Lam, M.P.; Ge, J.; et al. A large dataset of protein dynamics in the mammalian heart proteome. Sci. Data 2016, 3, 160015. [Google Scholar] [CrossRef] [PubMed]
- Sadygov, R.G. Partial Isotope Profiles Are Sufficient for Protein Turnover Analysis Using Closed-Form Equations of Mass Isotopomer Dynamics. Anal. Chem. 2020, 92, 14747–14753. [Google Scholar] [CrossRef]
- Yu, F.; Haynes, S.E.; Nesvizhskii, A.I. IonQuant Enables Accurate and Sensitive Label-Free Quantification With FDR-Controlled Match-Between-Runs. Mol. Cell Proteom. 2021, 20, 100077. [Google Scholar] [CrossRef]
- Lim, M.Y.; Paulo, J.A.; Gygi, S.P. Evaluating False Transfer Rates from the Match-between-Runs Algorithm with a Two-Proteome Model. J. Proteome Res. 2019, 18, 4020–4026. [Google Scholar] [CrossRef]
- Finney, G.L.; Blackler, A.R.; Hoopmann, M.R.; Canterbury, J.D.; Wu, C.C.; MacCoss, M.J. Label-free comparative analysis of proteomics mixtures using chromatographic alignment of high-resolution muLC-MS data. Anal. Chem. 2008, 80, 961–971. [Google Scholar] [CrossRef]
- Byrd, R.H.; Lu, P.H.; Nocedal, J.; Zhu, C.Y. A Limited Memory Algorithm for Bound Constrained Optimization. Siam J. Sci. Comput. 1995, 16, 1190–1208. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deberneh, H.M.; Sadygov, R.G. Flexible Quality Control for Protein Turnover Rates Using d2ome. Int. J. Mol. Sci. 2023, 24, 15553. https://doi.org/10.3390/ijms242115553
Deberneh HM, Sadygov RG. Flexible Quality Control for Protein Turnover Rates Using d2ome. International Journal of Molecular Sciences. 2023; 24(21):15553. https://doi.org/10.3390/ijms242115553
Chicago/Turabian StyleDeberneh, Henock M., and Rovshan G. Sadygov. 2023. "Flexible Quality Control for Protein Turnover Rates Using d2ome" International Journal of Molecular Sciences 24, no. 21: 15553. https://doi.org/10.3390/ijms242115553
APA StyleDeberneh, H. M., & Sadygov, R. G. (2023). Flexible Quality Control for Protein Turnover Rates Using d2ome. International Journal of Molecular Sciences, 24(21), 15553. https://doi.org/10.3390/ijms242115553