

Disrupted Neural Regeneration in Dry Eye Secondary to Ankylosing Spondylitis—With a Theoretical Link between Piezo2 Channelopathy and Gateway Reflex, WDR Neurons, and Flare-Ups
 ,
,  ,
,
Abstract
1. Introduction
2. Results
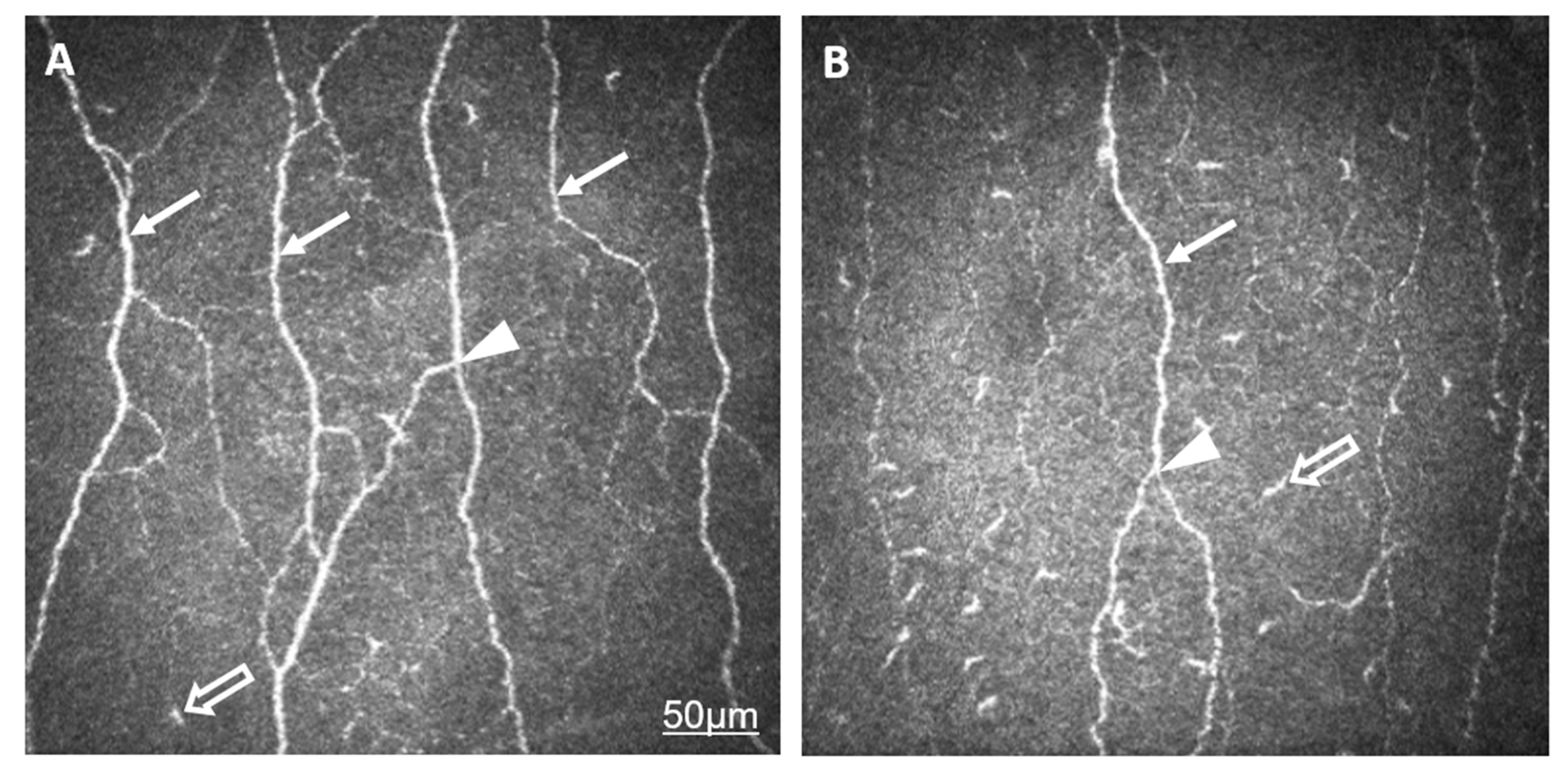
2.1. Comparison of AS and Control Groups
2.2. Subgroup Analysis by Disease Activity
2.3. Correlation Analysis
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| APC | antigen-presenting cell |
| AS | ankylosing spondylitis |
| axSpA | axial spondyloarthritis |
| BASDAI | AS disease activity index |
| CNS | central nervous system |
| CNBD | corneal nerve branch density |
| CNFA | corneal nerve fiber area |
| CNFD | corneal nerve fiber density |
| CNFL | corneal nerve fiber length |
| CTBD | corneal nerve fiber total branch density |
| CRP | C reactive protein |
| DED | dry eye disease |
| DOMS | delayed onset muscle soreness |
| EP4 | prostaglandin EP4 receptor |
| HLA-B27 | human leukocyte antigen B27 |
| IL-6 | Interleukin-6 |
| IL-17 | Interleukin-17 |
| L5 | lumbar 5 |
| LC | Langerhans cells |
| IVCM | In vivo corneal microscopy |
| LCD | Langerhans cell density |
| LIPCOF | lid parallel conjunctival folds |
| LN | lymph node |
| MT1-MMP | membrane type matrix metalloproteinase-1 |
| NKT cell | natural killer-T cell |
| NKT17 | IL17 producing NKT |
| NF-κB | nuclear factor kappa B |
| OSDI | ocular surface disease index |
| PGE2 | prostaglandin E2 |
| PD-L | programmed death-ligand |
| RA | rheumatoid arthritis |
| SBNP | subbasal nerve plexus |
| SLE | systemic lupus erythematosus |
| TBUT | tear film break-up time |
| Treg | T regulatory cell |
| Tγδ17 | γδ17 cell |
| UPR | unfolding protein reaction |
| VIP | vasoactive intestinal peptide |
| WDR | wide dynamic range |
References
- Braun, J.; Sieper, J. Ankylosing spondylitis. Lancet 2007, 369, 1379–1390. [Google Scholar] [CrossRef]
- Sonkodi, B.; Berkes, I.; Koltai, E. Have We Looked in the Wrong Direction for More Than 100 Years? Delayed Onset Muscle Soreness Is, in Fact, Neural Microdamage Rather Than Muscle Damage. Antioxidants 2020, 9, 212. [Google Scholar] [CrossRef]
- Sonkodi, B. Delayed Onset Muscle Soreness (DOMS): The Repeated Bout Effect and Chemotherapy-Induced Axonopathy May Help Explain the Dying-Back Mechanism in Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases. Brain Sci. 2021, 11, 108. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, M.; Zhang, Y.; Huang, Y.; Wu, J.; Xie, Y.; Wei, Q.; Liao, Z.; Gu, J. Dynamics of Adaptive Immune Cell and NK Cell Subsets in Patients with Ankylosing Spondylitis After IL-17A Inhibition by Secukinumab. Front. Pharmacol. 2021, 12, 738316. [Google Scholar] [CrossRef] [PubMed]
- The epidemiology of dry eye disease: Report of the Epidemiology Subcommittee of the International Dry Eye WorkShop (2007). Ocul. Surf. 2007, 5, 93–107. [CrossRef] [PubMed]
- Galor, A.; Moein, H.R.; Lee, C.; Rodriguez, A.; Felix, E.R.; Sarantopoulos, K.D.; Levitt, R.C. Neuropathic pain and dry eye. Ocul. Surf. 2018, 16, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, J. Definition and Diagnostic Criteria of Dry Eye Disease: Historical Overview and Future Directions. Invest. Ophthalmol. Vis. Sci. 2018, 59, DES7–DES12. [Google Scholar] [CrossRef]
- Glover, K.; Mishra, D.; Singh, T.R.R. Epidemiology of Ocular Manifestations in Autoimmune Disease. Front. Immunol. 2021, 12, 744396. [Google Scholar] [CrossRef]
- Lee, C.Y.; Chen, H.C.; Huang, J.Y.; Yen, C.H.; Hwang, Y.S.; Chang, C.K.; Yang, S.F. The Presence of Ankylosing Spondylitis and the Incidence of Subsequent External Eye Diseases: A Population-Based Cohort Study. Int. J. Environ. Res. Public. Health 2022, 19, 16296. [Google Scholar] [CrossRef]
- Sonkodi, B.; Resch, M.D.; Hortobagyi, T. Is the Sex Difference a Clue to the Pathomechanism of Dry Eye Disease? Watch out for the NGF-TrkA-Piezo2 Signaling Axis and the Piezo2 Channelopathy. J. Mol. Neurosci. 2022, 72, 1598–1608. [Google Scholar] [CrossRef]
- Woo, S.H.; Lukacs, V.; de Nooij, J.C.; Zaytseva, D.; Criddle, C.R.; Francisco, A.; Jessell, T.M.; Wilkinson, K.A.; Patapoutian, A. Piezo2 is the principal mechanotransduction channel for proprioception. Nat. Neurosci. 2015, 18, 1756–1762. [Google Scholar] [CrossRef]
- Sonkodi, B. Miswired Proprioception in Amyotrophic Lateral Sclerosis in Relation to Pain Sensation (and in Delayed Onset Muscle Soreness)-Is Piezo2 Channelopathy a Principal Transcription Activator in Proprioceptive Terminals Besides Being the Potential Primary Damage? Life 2023, 13, 657. [Google Scholar] [CrossRef]
- Sonkodi, B.; Bardoni, R.; Poór, G. Osteoporosis in Light of a New Mechanism Theory of Delayed Onset Muscle Soreness and Non-Contact Anterior Cruciate Ligament Injury. Int. J. Mol. Sci. 2022, 23, 9046. [Google Scholar] [CrossRef]
- Sonkodi, B. Psoriasis, Is It a Microdamage of Our “Sixth Sense”? A Neurocentric View. Int. J. Mol. Sci. 2022, 23, 11940. [Google Scholar] [CrossRef] [PubMed]
- Bozhkova, E. Vladimir Mikhailovich Bekhterev. Lancet Neurol. 2018, 17, 744. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sonkodi, B.; Hegedus, A.; Kopper, B.; Berkes, I. Significantly Delayed Medium-Latency Response of the Stretch Reflex in Delayed-Onset Muscle Soreness of the Quadriceps Femoris Muscles Is Indicative of Sensory Neuronal Microdamage. J. Funct. Morphol. Kinesiol. 2022, 7, 43. [Google Scholar] [CrossRef]
- Sonkodi, B.; Kopa, Z.; Nyirady, P. Post Orgasmic Illness Syndrome (POIS) and Delayed Onset Muscle Soreness (DOMS): Do They Have Anything in Common? Cells 2021, 10, 1867. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.F.; Sonkodi, B.; Pal, M.; Klivenyi, P.; Szell, M. Likely Pathogenic Variants of Ca(v)1.3 and Na(v)1.1 Encoding Genes in Amyotrophic Lateral Sclerosis Could Elucidate the Dysregulated Pain Pathways. Biomedicines 2023, 11, 933. [Google Scholar] [CrossRef]
- Sonkodi, B.; Bardoni, R.; Hangody, L.; Radak, Z.; Berkes, I. Does Compression Sensory Axonopathy in the Proximal Tibia Contribute to Noncontact Anterior Cruciate Ligament Injury in a Causative Way?—A New Theory for the Injury Mechanism. Life 2021, 11, 443. [Google Scholar] [CrossRef]
- Sonkodi, B. Delayed Onset Muscle Soreness and Critical Neural Microdamage-Derived Neuroinflammation. Biomolecules 2022, 12, 1207. [Google Scholar] [CrossRef]
- Perrotta, F.M.; Lories, R.; Lubrano, E. To move or not to move: The paradoxical effect of physical exercise in axial spondyloarthritis. RMD Open 2021, 7, e001480. [Google Scholar] [CrossRef]
- Klingberg, E.; Lorentzon, M.; Mellstrom, D.; Geijer, M.; Gothlin, J.; Hilme, E.; Hedberg, M.; Carlsten, H.; Forsblad-d’Elia, H. Osteoporosis in ankylosing spondylitis—Prevalence, risk factors and methods of assessment. Arthritis Res. Ther. 2012, 14, R108. [Google Scholar] [CrossRef] [PubMed]
- Ozen, T.; Tonga, E.; Polat, M.G.; Bayraktar, D.; Akar, S. Cervical proprioception accuracy is impaired in patients with axial spondyloarthritis. Musculoskelet. Sci. Pract. 2021, 51, 102304. [Google Scholar] [CrossRef] [PubMed]
- Ulutatar, F.; Unal-Ulutatar, C.; Duruoz, M.T. Cervical proprioceptive impairment in patients with rheumatoid arthritis. Rheumatol. Int. 2019, 39, 2043–2051. [Google Scholar] [CrossRef]
- Ma, S.; Dubin, A.E.; Romero, L.O.; Loud, M.; Salazar, A.; Chu, S.; Klier, N.; Masri, S.; Zhang, Y.; Wang, Y.; et al. Excessive mechanotransduction in sensory neurons causes joint contractures. Science 2023, 379, 201–206. [Google Scholar] [CrossRef]
- Suchyna, T.M. Piezo channels and GsMTx4: Two milestones in our understanding of excitatory mechanosensitive channels and their role in pathology. Prog. Biophys. Mol. Biol. 2017, 130, 244–253. [Google Scholar] [CrossRef]
- Szczot, M.; Liljencrantz, J.; Ghitani, N.; Barik, A.; Lam, R.; Thompson, J.H.; Bharucha-Goebel, D.; Saade, D.; Necaise, A.; Donkervoort, S.; et al. PIEZO2 mediates injury-induced tactile pain in mice and humans. Sci. Transl. Med. 2018, 10, eaat9892. [Google Scholar] [CrossRef] [PubMed]
- Eijkelkamp, N.; Linley, J.E.; Torres, J.M.; Bee, L.; Dickenson, A.H.; Gringhuis, M.; Minett, M.S.; Hong, G.S.; Lee, E.; Oh, U.; et al. A role for Piezo2 in EPAC1-dependent mechanical allodynia. Nat. Commun. 2013, 4, 1682. [Google Scholar] [CrossRef]
- Nencini, S.; Morgan, M.; Thai, J.; Jobling, A.I.; Mazzone, S.B.; Ivanusic, J.J. Piezo2 Knockdown Inhibits Noxious Mechanical Stimulation and NGF-Induced Sensitization in A-Delta Bone Afferent Neurons. Front. Physiol. 2021, 12, 644929. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.; Powell, J.; Kozak, C.; Song, Y. Mechanosensitive Ion Channels, Axonal Growth, and Regeneration. Neuroscientist 2023, 29, 421–444. [Google Scholar] [CrossRef] [PubMed]
- De, I.; Sharma, P.; Singh, M. Emerging approaches of neural regeneration using physical stimulations solely or coupled with smart piezoelectric nano-biomaterials. Eur. J. Pharm. Biopharm. 2022, 173, 73–91. [Google Scholar] [CrossRef]
- Marino, A.; Genchi, G.G.; Sinibaldi, E.; Ciofani, G. Piezoelectric Effects of Materials on Bio-Interfaces. ACS Appl. Mater. Interfaces 2017, 9, 17663–17680. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, E.C.; Yagci, I. The structural, functional and electrophysiological assessment of paraspinal musculature of patients with ankylosing spondylitis and non-radiographic axial spondyloarthropathy. Rheumatol. Int. 2021, 41, 595–603. [Google Scholar] [CrossRef]
- Khedr, E.M.; Rashad, S.M.; Hamed, S.A.; El-Zharaa, F.; Abdalla, A.K. Neurological complications of ankylosing spondylitis: Neurophysiological assessment. Rheumatol. Int. 2009, 29, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Sonkodi, B.; Marsovszky, L.; Csorba, A.; Balog, A.; Kopper, B.; Nagy, Z.Z.; Resch, M.D. Neural Regeneration in Dry Eye Secondary to Systemic Lupus Erythematosus Is Also Disrupted like in Rheumatoid Arthritis, but in a Progressive Fashion. Int. J. Mol. Sci. 2023, 24, 10680. [Google Scholar] [CrossRef] [PubMed]
- Sonkodi, B.; Csorba, A.; Marsovszky, L.; Balog, A.; Kopper, B.; Nagy, Z.Z.; Resch, M.D. Evidence of Disruption in Neural Regeneration in Dry Eye Secondary to Rheumatoid Arthritis. Int. J. Mol. Sci. 2023, 24, 7514. [Google Scholar] [CrossRef]
- Sonkodi, B.; Pállinger, É.; Radovits, T.; Csulak, E.; Shenker-Horváth, K.; Kopper, B.; Buzás, E.I.; Sydó, N.; Merkely, B. CD3+/CD56+ NKT-like Cells Show Imbalanced Control Immediately after Exercise in Delayed-Onset Muscle Soreness. Int. J. Mol. Sci. 2022, 23, 11117. [Google Scholar] [CrossRef]
- Kim, T.J.; Lee, S.J.; Cho, Y.N.; Park, S.C.; Jin, H.M.; Kim, M.J.; Park, D.J.; Kee, S.J.; Lee, S.S.; Park, Y.W. Immune cells and bone formation in ankylosing spondylitis. Clin. Exp. Rheumatol. 2012, 30, 469–475. [Google Scholar] [PubMed]
- Jaiswal, A.K.; Sadasivam, M.; Hamad, A.R.A. Unexpected alliance between syndecan-1 and innate-like T cells to protect host from autoimmune effects of interleukin-17. World J. Diabetes 2018, 9, 220–225. [Google Scholar] [CrossRef]
- Marshall, L.J.; Ramdin, L.S.; Brooks, T.; PC, D.P.; Shute, J.K. Plasminogen activator inhibitor-1 supports IL-8-mediated neutrophil transendothelial migration by inhibition of the constitutive shedding of endothelial IL-8/heparan sulfate/syndecan-1 complexes. J. Immunol. 2003, 171, 2057–2065. [Google Scholar] [CrossRef]
- Segaliny, A.I.; Brion, R.; Mortier, E.; Maillasson, M.; Cherel, M.; Jacques, Y.; Le Goff, B.; Heymann, D. Syndecan-1 regulates the biological activities of interleukin-34. Biochim. Biophys. Acta 2015, 1853, 1010–1021. [Google Scholar] [CrossRef]
- Kenna, T.J.; Davidson, S.I.; Duan, R.; Bradbury, L.A.; McFarlane, J.; Smith, M.; Weedon, H.; Street, S.; Thomas, R.; Thomas, G.P.; et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012, 64, 1420–1429. [Google Scholar] [CrossRef]
- Bernal-Alferes, B.; Gomez-Mosqueira, R.; Ortega-Tapia, G.T.; Burgos-Vargas, R.; Garcia-Latorre, E.; Dominguez-Lopez, M.L.; Romero-Lopez, J.P. The role of gammadelta T cells in the immunopathogenesis of inflammatory diseases: From basic biology to therapeutic targeting. J. Leukoc. Biol. 2023, qiad046. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, Z.; Chen, D.; Cui, H.; Wang, J.; Li, Z.; Li, X.; Zheng, Z.; Zhan, Z.; Liu, H. Piezo1-mediated mechanotransduction promotes entheseal pathological new bone formation in ankylosing spondylitis. Ann. Rheum. Dis. 2022, 82, 533–545. [Google Scholar] [CrossRef]
- Xueyi, L.; Lina, C.; Zhenbiao, W.; Qing, H.; Qiang, L.; Zhu, P. Levels of circulating Th17 cells and regulatory T cells in ankylosing spondylitis patients with an inadequate response to anti-TNF-alpha therapy. J. Clin. Immunol. 2013, 33, 151–161. [Google Scholar] [CrossRef]
- Fasching, P.; Stradner, M.; Graninger, W.; Dejaco, C.; Fessler, J. Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules 2017, 22, 134. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dana, R. Autoimmunity in dry eye disease—An updated review of evidence on effector and memory Th17 cells in disease pathogenicity. Autoimmun. Rev. 2021, 20, 102933. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, M.; Toumi, H.; Suzuki, D.; Redman, S.; Emery, P.; McGonagle, D. Microdamage and altered vascularity at the enthesis-bone interface provides an anatomic explanation for bone involvement in the HLA-B27-associated spondylarthritides and allied disorders. Arthritis Rheum. 2007, 56, 224–233. [Google Scholar] [CrossRef]
- Sun, W.; Chi, S.; Li, Y.; Ling, S.; Tan, Y.; Xu, Y.; Jiang, F.; Li, J.; Liu, C.; Zhong, G.; et al. The mechanosensitive Piezo1 channel is required for bone formation. Elife 2019, 8, e47454. [Google Scholar] [CrossRef]
- Sugimoto, A.; Miyazaki, A.; Kawarabayashi, K.; Shono, M.; Akazawa, Y.; Hasegawa, T.; Ueda-Yamaguchi, K.; Kitamura, T.; Yoshizaki, K.; Fukumoto, S.; et al. Piezo type mechanosensitive ion channel component 1 functions as a regulator of the cell fate determination of mesenchymal stem cells. Sci. Rep. 2017, 7, 17696. [Google Scholar] [CrossRef]
- Marsovszky, L.; Resch, M.D.; Nemeth, J.; Toldi, G.; Medgyesi, E.; Kovacs, L.; Balog, A. In vivo confocal microscopic evaluation of corneal Langerhans cell density, and distribution and evaluation of dry eye in rheumatoid arthritis. Innate Immun. 2013, 19, 348–354. [Google Scholar] [CrossRef]
- Resch, M.D.; Marsovszky, L.; Nemeth, J.; Bocskai, M.; Kovacs, L.; Balog, A. Dry eye and corneal langerhans cells in systemic lupus erythematosus. J. Ophthalmol. 2015, 2015, 543835. [Google Scholar] [CrossRef]
- Marsovszky, L.; Nemeth, J.; Resch, M.D.; Toldi, G.; Legany, N.; Kovacs, L.; Balog, A. Corneal Langerhans cell and dry eye examinations in ankylosing spondylitis. Innate Immun. 2014, 20, 471–477. [Google Scholar] [CrossRef]
- Song, Y.; Li, D.; Farrelly, O.; Miles, L.; Li, F.; Kim, S.E.; Lo, T.Y.; Wang, F.; Li, T.; Thompson-Peer, K.L.; et al. The Mechanosensitive Ion Channel Piezo Inhibits Axon Regeneration. Neuron 2019, 102, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Motterle, L.; Diebold, Y.; Enriquez de Salamanca, A.; Saez, V.; Garcia-Vazquez, C.; Stern, M.E.; Calonge, M.; Leonardi, A. Altered expression of neurotransmitter receptors and neuromediators in vernal keratoconjunctivitis. Arch. Ophthalmol. 2006, 124, 462–468. [Google Scholar] [CrossRef]
- Barabino, S.; Chen, Y.; Chauhan, S.; Dana, R. Ocular surface immunity: Homeostatic mechanisms and their disruption in dry eye disease. Prog. Retin. Eye Res. 2012, 31, 271–285. [Google Scholar] [CrossRef]
- Akita, K.; Toda, M.; Hosoki, Y.; Inoue, M.; Fushiki, S.; Oohira, A.; Okayama, M.; Yamashina, I.; Nakada, H. Heparan sulphate proteoglycans interact with neurocan and promote neurite outgrowth from cerebellar granule cells. Biochem. J. 2004, 383, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.J.; Hammarlund, M. Syndecan promotes axon regeneration by stabilizing growth cone migration. Cell Rep. 2014, 8, 272–283. [Google Scholar] [CrossRef][Green Version]
- Mouthon, M.A.; Morizur, L.; Dutour, L.; Pineau, D.; Kortulewski, T.; Boussin, F.D. Syndecan-1 Stimulates Adult Neurogenesis in the Mouse Ventricular-Subventricular Zone after Injury. iScience 2020, 23, 101784. [Google Scholar] [CrossRef]
- Yilmaz, P.D.; Kadiyoran, C.; Goktepe, M.H.; Akkubak, Y.; Icli, A.; Kucuk, A. Syndecan 1 may slow the progression of subclinical atherosclerosis in patients with ankylosing spondylitis. Clin. Exp. Hypertens. 2023, 45, 2156529. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef]
- Kang, H.; Hong, Z.; Zhong, M.; Klomp, J.; Bayless, K.J.; Mehta, D.; Karginov, A.V.; Hu, G.; Malik, A.B. Piezo1 mediates angiogenesis through activation of MT1-MMP signaling. Am. J. Physiol. Cell Physiol. 2019, 316, C92–C103. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Usui, Y.; Horie, S.; Futagami, Y.; Yamada, Y.; Ma, J.; Kezuka, T.; Hamada, H.; Usui, T.; Mochizuki, M.; et al. Human corneal endothelial cells expressing programmed death-ligand 1 (PD-L1) suppress PD-1+ T helper 1 cells by a contact-dependent mechanism. Invest. Ophthalmol. Vis. Sci. 2009, 50, 263–272. [Google Scholar] [CrossRef]
- De Paiva, C.S.; Corrales, R.M.; Villarreal, A.L.; Farley, W.; Li, D.Q.; Stern, M.E.; Pflugfelder, S.C. Apical corneal barrier disruption in experimental murine dry eye is abrogated by methylprednisolone and doxycycline. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2847–2856. [Google Scholar] [CrossRef] [PubMed]
- Canadas, P.; Alberquilla Garcia-Velasco, M.; Hernandez Verdejo, J.L.; Teus, M.A. Update on Corneal Confocal Microscopy Imaging. Diagnostics 2022, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.; Fadavi, H.; Ishibashi, F.; Howard, S.; Boulton, A.J.M.; Shore, A.C.; Tavakoli, M. Implementation of corneal confocal microscopy for screening and early detection of diabetic neuropathy in primary care alongside retinopathy screening: Results from a feasibility study. Front. Endocrinol. 2022, 13, 891575. [Google Scholar] [CrossRef] [PubMed]
- Merola, A.; Rosso, M.; Romagnolo, A.; Comi, C.; Fasano, A.; Zibetti, M.; Lopez-Castellanos, J.R.; Cocito, D.; Lopiano, L.; Espay, A.J. Peripheral neuropathy as marker of severe Parkinson’s disease phenotype. Mov. Disord. 2017, 32, 1256–1258. [Google Scholar] [CrossRef]
- Kolkedi, Z.; Csutak, A.; Szalai, E. Corneal Cellular and Neuroinflammatory Changes After SARS-CoV-2 Infection. Cornea 2022, 41, 879–885. [Google Scholar] [CrossRef]
- Liu, Y.; Chou, Y.; Dong, X.; Liu, Z.; Jiang, X.; Hao, R.; Li, X. Corneal Subbasal Nerve Analysis Using In Vivo Confocal Microscopy in Patients with Dry Eye: Analysis and Clinical Correlations. Cornea 2019, 38, 1253–1258. [Google Scholar] [CrossRef]
- Ferdousi, M.; Petropoulos, I.N.; Kalteniece, A.; Azmi, S.; Ponirakis, G.; Efron, N.; Soran, H.; Malik, R.A. No Relation Between the Severity of Corneal Nerve, Epithelial, and Keratocyte Cell Morphology with Measures of Dry Eye Disease in Type 1 Diabetes. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5525–5530. [Google Scholar] [CrossRef] [PubMed]
- Sivaskandarajah, G.A.; Halpern, E.M.; Lovblom, L.E.; Weisman, A.; Orlov, S.; Bril, V.; Perkins, B.A. Structure-function relationship between corneal nerves and conventional small-fiber tests in type 1 diabetes. Diabetes Care 2013, 36, 2748–2755. [Google Scholar] [CrossRef]
- Petropoulos, I.N.; Ponirakis, G.; Ferdousi, M.; Azmi, S.; Kalteniece, A.; Khan, A.; Gad, H.; Bashir, B.; Marshall, A.; Boulton, A.J.M.; et al. Corneal Confocal Microscopy: A Biomarker for Diabetic Peripheral Neuropathy. Clin. Ther. 2021, 43, 1457–1475. [Google Scholar] [CrossRef] [PubMed]
- Cashman, C.R.; Hoke, A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 2015, 596, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Luna, C.; Mizerska, K.; Quirce, S.; Belmonte, C.; Gallar, J.; Acosta, M.D.C.; Meseguer, V. Sodium Channel Blockers Modulate Abnormal Activity of Regenerating Nociceptive Corneal Nerves After Surgical Lesion. Investig. Ophthalmol. Vis. Sci. 2021, 62, 2. [Google Scholar] [CrossRef]
- Fernandez-Trillo, J.; Florez-Paz, D.; Inigo-Portugues, A.; Gonzalez-Gonzalez, O.; Del Campo, A.G.; Gonzalez, A.; Viana, F.; Belmonte, C.; Gomis, A. Piezo2 Mediates Low-Threshold Mechanically Evoked Pain in the Cornea. J. Neurosci. 2020, 40, 8976–8993. [Google Scholar] [CrossRef]
- Cox, S.M.; Kheirkhah, A.; Aggarwal, S.; Abedi, F.; Cavalcanti, B.M.; Cruzat, A.; Hamrah, P. Alterations in corneal nerves in different subtypes of dry eye disease: An in vivo confocal microscopy study. Ocul. Surf. 2021, 22, 135–142. [Google Scholar] [CrossRef]
- Talotta, R.; Rucci, F.; Scaglione, F. Calcium physiology, metabolism and supplementation: A glance at patients with ankylosing spondylitis. Reumatologia 2020, 58, 297–311. [Google Scholar] [CrossRef]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef]
- Uchida, M.; Yamamoto, R.; Matsuyama, S.; Murakami, K.; Hasebe, R.; Hojyo, S.; Tanaka, Y.; Murakami, M. Gateway reflexes, neuronal circuits that regulate the autoreactive T cells in organs having blood barriers. Int. Immunol. 2022, 34, 59–65. [Google Scholar] [CrossRef]
- Arima, Y.; Harada, M.; Kamimura, D.; Park, J.H.; Kawano, F.; Yull, F.E.; Kawamoto, T.; Iwakura, Y.; Betz, U.A.; Marquez, G.; et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell 2012, 148, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Sonkodi, B. Should We Void Lactate in the Pathophysiology of Delayed Onset Muscle Soreness? Not So Fast! Let’s See a Neurocentric View! Metabolites 2022, 12, 857. [Google Scholar] [CrossRef] [PubMed]
- Labarrade, F.; Perrin, A.; Ferreira, Y.; Botto, J.M.; Imbert, I. Modulation of Piezo1 influences human skin architecture and oxytocin expression. Int. J. Cosmet. Sci. 2023, 45, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Espino, C.M.; Lewis, C.M.; Ortiz, S.; Dalal, M.S.; Garlapalli, S.; Wells, K.M.; O’Neil, D.A.; Wilkinson, K.A.; Griffith, T.N. Na(V)1.1 is essential for proprioceptive signaling and motor behaviors. Elife 2022, 11, e79917. [Google Scholar] [CrossRef]
- Sonkodi, B.; Varga, E.; Hangody, L.; Poor, G.; Berkes, I. Finishing stationary cycling too early after anterior cruciate ligament reconstruction is likely to lead to higher failure. BMC Sports Sci. Med. Rehabil. 2021, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Brewerton, D.A.; Hart, F.D.; Nicholls, A.; Caffrey, M.; James, D.C.; Sturrock, R.D. Ankylosing spondylitis and HL-A 27. Lancet 1973, 1, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Voruganti, A.; Bowness, P. New developments in our understanding of ankylosing spondylitis pathogenesis. Immunology 2020, 161, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Zechini, L.; Camilleri-Brennan, J.; Walsh, J.; Beaven, R.; Moran, O.; Hartley, P.S.; Diaz, M.; Denholm, B. Piezo buffers mechanical stress via modulation of intracellular Ca(2+) handling in the Drosophila heart. Front. Physiol. 2022, 13, 1003999. [Google Scholar] [CrossRef]
- Evans, D.M.; Spencer, C.C.; Pointon, J.J.; Su, Z.; Harvey, D.; Kochan, G.; Oppermann, U.; Dilthey, A.; Pirinen, M.; Stone, M.A.; et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat. Genet. 2011, 43, 761–767. [Google Scholar] [CrossRef]
- Choi, J.H.; Lee, S.H.; Kim, H.R.; Lee, K.A. Association of neuropathic-like pain characteristics with clinical and radiographic features in patients with ankylosing spondylitis. Clin. Rheumatol. 2018, 37, 3077–3086. [Google Scholar] [CrossRef]
- Brazenor, G.A.; Malham, G.M.; Teddy, P.J. Can Central Sensitization After Injury Persist as an Autonomous Pain Generator? A Comprehensive Search for Evidence. Pain. Med. 2022, 23, 1283–1298. [Google Scholar] [CrossRef]
- Treede, R.D. Pain research in 2022: Nociceptors and central sensitisation. Lancet Neurol. 2023, 22, 24–25. [Google Scholar] [CrossRef]
- Obeidat, A.M.; Wood, M.J.; Adamczyk, N.S.; Ishihara, S.; Li, J.; Wang, L.; Ren, D.; Bennett, D.A.; Miller, R.J.; Malfait, A.M.; et al. Piezo2 expressing nociceptors mediate mechanical sensitization in experimental osteoarthritis. Nat. Commun. 2023, 14, 2479. [Google Scholar] [CrossRef]
- Borbiro, I.; Badheka, D.; Rohacs, T. Activation of TRPV1 channels inhibits mechanosensitive Piezo channel activity by depleting membrane phosphoinositides. Sci. Signal 2015, 8, ra15. [Google Scholar] [CrossRef] [PubMed]
- Puja, G.; Sonkodi, B.; Bardoni, R. Mechanisms of Peripheral and Central Pain Sensitization: Focus on Ocular Pain. Front. Pharmacol. 2021, 12, 764396. [Google Scholar] [CrossRef]
- Melzack, R.; Wall, P.D. Pain mechanisms: A new theory. Science 1965, 150, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Sengupta, S.; Ghosh, K.; Mukhopadhyay, S.; Ghosh, B. A Curious Case of Ankylosing Spondylosis and Motor Neuron Disease: A Mere Coincidence or Correlation? Int. J. Appl. Basic. Med. Res. 2018, 8, 266–268. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, S.; Valkenburg, H.A.; Cats, A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984, 27, 361–368. [Google Scholar] [CrossRef]
- Garrett, S.; Jenkinson, T.; Kennedy, L.G.; Whitelock, H.; Gaisford, P.; Calin, A. A new approach to defining disease status in ankylosing spondylitis: The Bath Ankylosing Spondylitis Disease Activity Index. J. Rheumatol. 1994, 21, 2286–2291. [Google Scholar]
- Vitali, C.; Bombardieri, S.; Jonsson, R.; Moutsopoulos, H.M.; Alexander, E.L.; Carsons, S.E.; Daniels, T.E.; Fox, P.C.; Fox, R.I.; Kassan, S.S.; et al. Classification criteria for Sjogren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum. Dis. 2002, 61, 554–558. [Google Scholar] [CrossRef]
- Jing, D.; Liu, Y.; Chou, Y.; Jiang, X.; Ren, X.; Yang, L.; Su, J.; Li, X. Change patterns in the corneal sub-basal nerve and corneal aberrations in patients with dry eye disease: An artificial intelligence analysis. Exp. Eye Res. 2022, 215, 108851. [Google Scholar] [CrossRef]
- Schiffman, R.M.; Christianson, M.D.; Jacobsen, G.; Hirsch, J.D.; Reis, B.L. Reliability and validity of the Ocular Surface Disease Index. Arch. Ophthalmol. 2000, 118, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Pult, H.; Purslow, C.; Murphy, P.J. The relationship between clinical signs and dry eye symptoms. Eye 2011, 25, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Zhivov, A.; Stave, J.; Vollmar, B.; Guthoff, R. In vivo confocal microscopic evaluation of Langerhans cell density and distribution in the normal human corneal epithelium. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 1056–1061. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Control | AS | p | |
|---|---|---|---|
| No. of patients | 35 | 24 | NA |
| No. of eyes | 35 | 24 | NA |
| Age (years) | 44.2 ± 19.94 | 41.9 ± 9.8 | 0.907 |
| Gender (male/female) | 11/18 | 1/28 | NA |
| AS duration (years) | - | 9.9 ± 4.5 | NA |
| BASDAI | - | 3.2 ± 2.11 | NA |
| LIPCOF | 1.02 ± 0.7 | 0.95 ± 0.62 | 0.0 |
| TBUT (s) | 11.28 ± 3.0 | 11.47 ± 4.63 | 0.85 |
| Schirmer test (mm/5 min) | 12.1 ± 2.9 | 8.4 ± 8.11 | 0.016 * |
| OSDI | 8.8 ± 6.56 | 21.52 ± 15.44 | ≥0.001 * |
| Control | AS | p | |
|---|---|---|---|
| CNFD | 19.32 ± 7.87 | 13.59 ± 6.58 | 0.005 * |
| CNBD | 25.77 ± 17.2 | 13.38 ± 7.82 | 0.002 * |
| CNFL | 13.4 ± 3.82 | 10.43 ± 3.77 | 0.005 * |
| CTBD | 44.2 ± 24.1 | 28.06 ± 12.4 | 0.005 * |
| CNFA | 0.006 ± 0.002 | 0.005 ± 0.001 | 0.03 * |
| Central LCD | 23.56 ± 25.37 | 77.57 ± 41.94 | ≥0.001 * |
| Peripheral LCD | 75.52 ± 33.03 | 128.25 ± 56.33 | ≥0.001 * |
| Central LCM | 1.0 ± 0.68 | 1.71 ± 0.69 | ≥0.001 * |
| Peripheral LCM | 2.34 ± 0.54 | 2.53 ± 0.59 | 0.185 |
| HLA-B27 + | HLA-B27 − | p | |
|---|---|---|---|
| No. of patients | 12 | 12 | NA |
| No. of eyes | 12 | 12 | NA |
| Age (years) | 42.7 ± 7.6 | 43.1 ± 8.7 | |
| AS duration (years) | 8.4 ± 3.3 | 11.4 ± 5.1 | 0.104 |
| BASDAI | 3.0 ± 1.2 | 3.4 ± 2.7 | 0.670 |
| ESR (We) | 23.4 ± 17.9 | 23.5 ± 20.7 | 0.983 |
| CRP | 15.9 ± 18.7 | 18.6 ± 16.6 | 0.714 |
| LIPCOF | 0.92 ± 0.52 | 1.00 ± 0.74 | 0.752 |
| TBUT (s) | 11.5 ± 5.0 | 11.4 ± 4.5 | 0.949 |
| Schirmer test (mm/5 min) | 9.2 ± 8.2 | 7.6 ± 8.4 | 0.644 |
| OSDI | 23.9 ± 11.7 | 19.5 ± 18.2 | 0.509 |
| CNFD | 14.4 ± 5.4 | 12.8 ± 5.4 | 0.960 |
| CNBD | 13.3 ± 7.3 | 13.5 ± 8.7 | 0.857 |
| CNFL | 10.6 ± 3.2 | 10.3 ± 4.4 | 0.879 |
| CTBD | 2.76 ± 11.7 | 28.5 ± 13.7 | 0.312 |
| CNFA | 0.005 ± 0.002 | 0.006 ± 0.001 | 0.618 |
| Central LCD | 79.3 ± 36.7 | 75.8 ± 48.2 | 0.843 |
| Peripheral LCD | 141.4 ± 53.5 | 115.1 ± 58.2 | 0.261 |
| Central LCM | 1.8 ± 0.7 | 1.6 ± 0.7 | 0.387 |
| Peripheral LCM | 2.7 ± 0.5 | 2.4 ± 0.7 | 0.308 |
| Endothelium | 2645 ± 566 | 2627 ± 472 | 0.935 |
| BASDAI ≤ 4 | BASDAI > 4 | p | |
|---|---|---|---|
| No. of patients | 17 | 7 | NA |
| No. of eyes | 17 | 7 | NA |
| Age (years) | 41.5 ± 9.4 | 45.4 ± 10.3 | 0.408 |
| AS duration (years) | 8.7 ± 3.9 | 13.5 ± 4.7 | 0.026 * |
| BASDAI | 2.2 ± 1.2 | 5.9 ± 1.6 | <0.001 * |
| ESR (We) | 18.0 ± 13.7 | 32.2 ± 22.7 | 0.082 |
| CRP | 12.3 ± 13.5 | 23.5 ± 15.2 | 0.105 |
| LIPCOF | 0.82 ± 0.64 | 1.33 ± 0.52 | 0.093 |
| TBUT (s) | 12.5 ± 4.5 | 9.0 ± 4.1 | 0.133 |
| Schirmer test (mm/5 min) | 10.2 ± 8.5 | 4.3 ± 5.8 | 0.137 |
| OSDI | 21.2 ± 14.6 | 17.6 ± 17.2 | 0.644 |
| CNFD | 15.2 ± 6.2 | 8.1 ± 6.1 | 0.025 * |
| CNBD | 14.5 ± 7.5 | 10.5 ± 8.6 | 0.305 |
| CNFL | 10.8 ± 3.6 | 8.6 ± 3.7 | 0.213 |
| CTBD | 27.5 ± 12.3 | 29.4 ± 13.6 | 0.756 |
| CNFA | 0.005 ± 0.001 | 0.005 ± 0.002 | 0.755 |
| Central LCD | 73.8 ± 45.1 | 97.0 ± 23.2 | 0.246 |
| Peripheral LCD | 109.1 ± 44.2 | 175.6 ± 63.6 | 0.010 * |
| Central LCM | 1.8 ± 0.6 | 1.5 ± 0.8 | 0.334 |
| Peripheral LCM | 2.4 ± 0.6 | 3.0 ± 0.0 | 0.321 |
| Endothelium | 2559 ± 510 | 2720 ± 451 | 0.503 |
| AS Duration | BASDAI | ESR (We) | CRP | LIPCOF | TBUT | Schirmer Test | CNFD | CNBD | CNFL | CTBD | CNFA | Peripheral LCD | Peripheral LCM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AS duration | + | − | ||||||||||||
| BASDAI | + | + | − | |||||||||||
| ESR (We) | + | − | ||||||||||||
| CRP | + | − | ||||||||||||
| LIPCOF | + | |||||||||||||
| TBUT | − | |||||||||||||
| Schirmer test | − | − | − | + | + | − | ||||||||
| CNFD | − | + | − | − | ||||||||||
| CNBD | − | − | ||||||||||||
| CNFL | + | − | − | |||||||||||
| CTBD | − | |||||||||||||
| Peripheral LCD | − | − | − | |||||||||||
| Peripheral LCM | − | − | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonkodi, B.; Marsovszky, L.; Csorba, A.; Balog, A.; Kopper, B.; Keller-Pintér, A.; Nagy, Z.Z.; Resch, M.D. Disrupted Neural Regeneration in Dry Eye Secondary to Ankylosing Spondylitis—With a Theoretical Link between Piezo2 Channelopathy and Gateway Reflex, WDR Neurons, and Flare-Ups. Int. J. Mol. Sci. 2023, 24, 15455. https://doi.org/10.3390/ijms242015455
Sonkodi B, Marsovszky L, Csorba A, Balog A, Kopper B, Keller-Pintér A, Nagy ZZ, Resch MD. Disrupted Neural Regeneration in Dry Eye Secondary to Ankylosing Spondylitis—With a Theoretical Link between Piezo2 Channelopathy and Gateway Reflex, WDR Neurons, and Flare-Ups. International Journal of Molecular Sciences. 2023; 24(20):15455. https://doi.org/10.3390/ijms242015455
Chicago/Turabian StyleSonkodi, Balázs, László Marsovszky, Anita Csorba, Attila Balog, Bence Kopper, Anikó Keller-Pintér, Zoltán Zsolt Nagy, and Miklós D. Resch. 2023. "Disrupted Neural Regeneration in Dry Eye Secondary to Ankylosing Spondylitis—With a Theoretical Link between Piezo2 Channelopathy and Gateway Reflex, WDR Neurons, and Flare-Ups" International Journal of Molecular Sciences 24, no. 20: 15455. https://doi.org/10.3390/ijms242015455
APA StyleSonkodi, B., Marsovszky, L., Csorba, A., Balog, A., Kopper, B., Keller-Pintér, A., Nagy, Z. Z., & Resch, M. D. (2023). Disrupted Neural Regeneration in Dry Eye Secondary to Ankylosing Spondylitis—With a Theoretical Link between Piezo2 Channelopathy and Gateway Reflex, WDR Neurons, and Flare-Ups. International Journal of Molecular Sciences, 24(20), 15455. https://doi.org/10.3390/ijms242015455