
Whole-Genome Sequencing of 502 Individuals from Latvia: The First Step towards a Population-Specific Reference of Genetic Variation

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Characteristics of the Cohort
2.2. Quality of Variant Calling
2.3. Genetic Variation in the Latvian Population
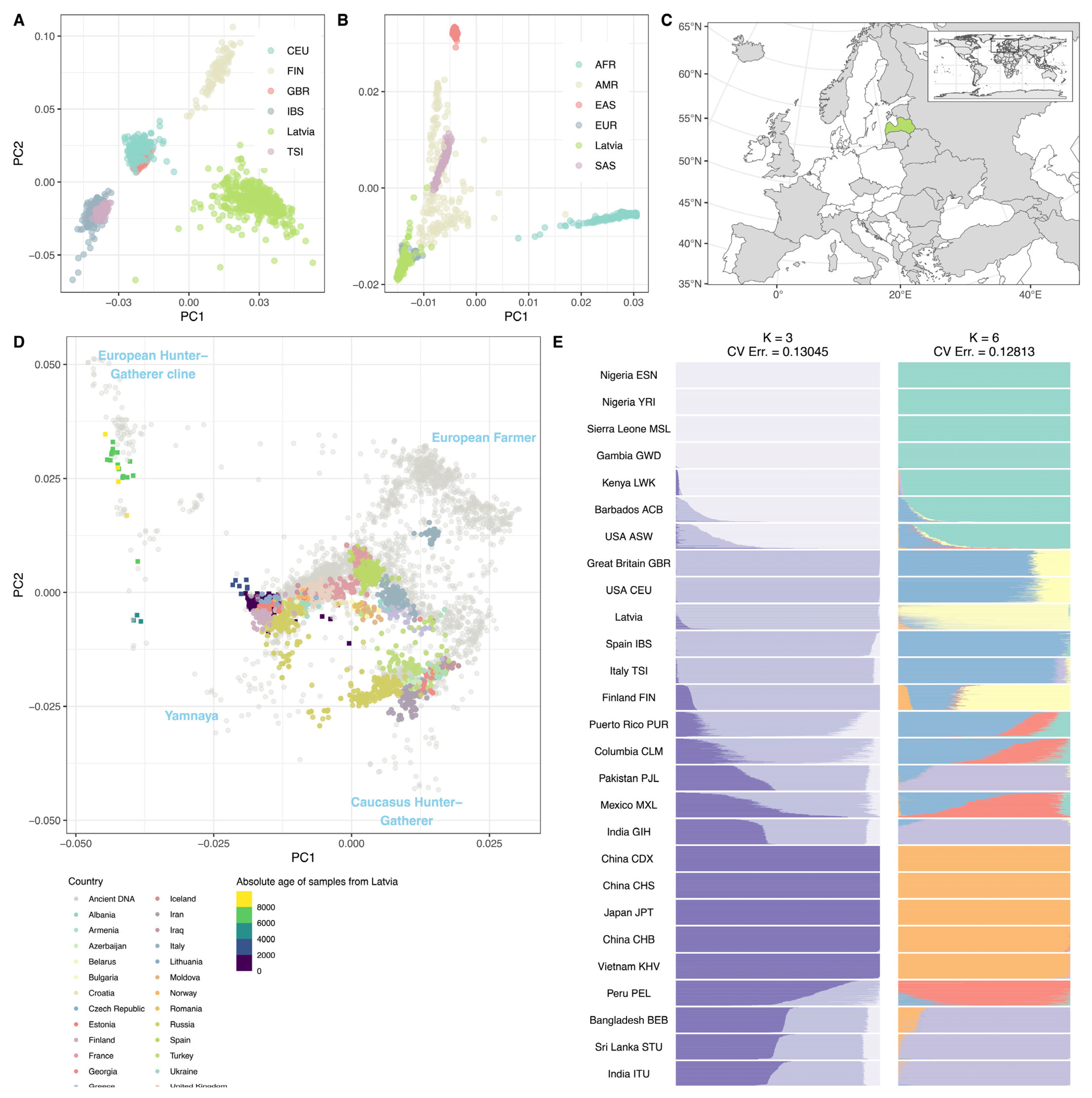
2.4. Relation between Latvian and Global Populations
2.5. The Medical Relevance of Identified Variants
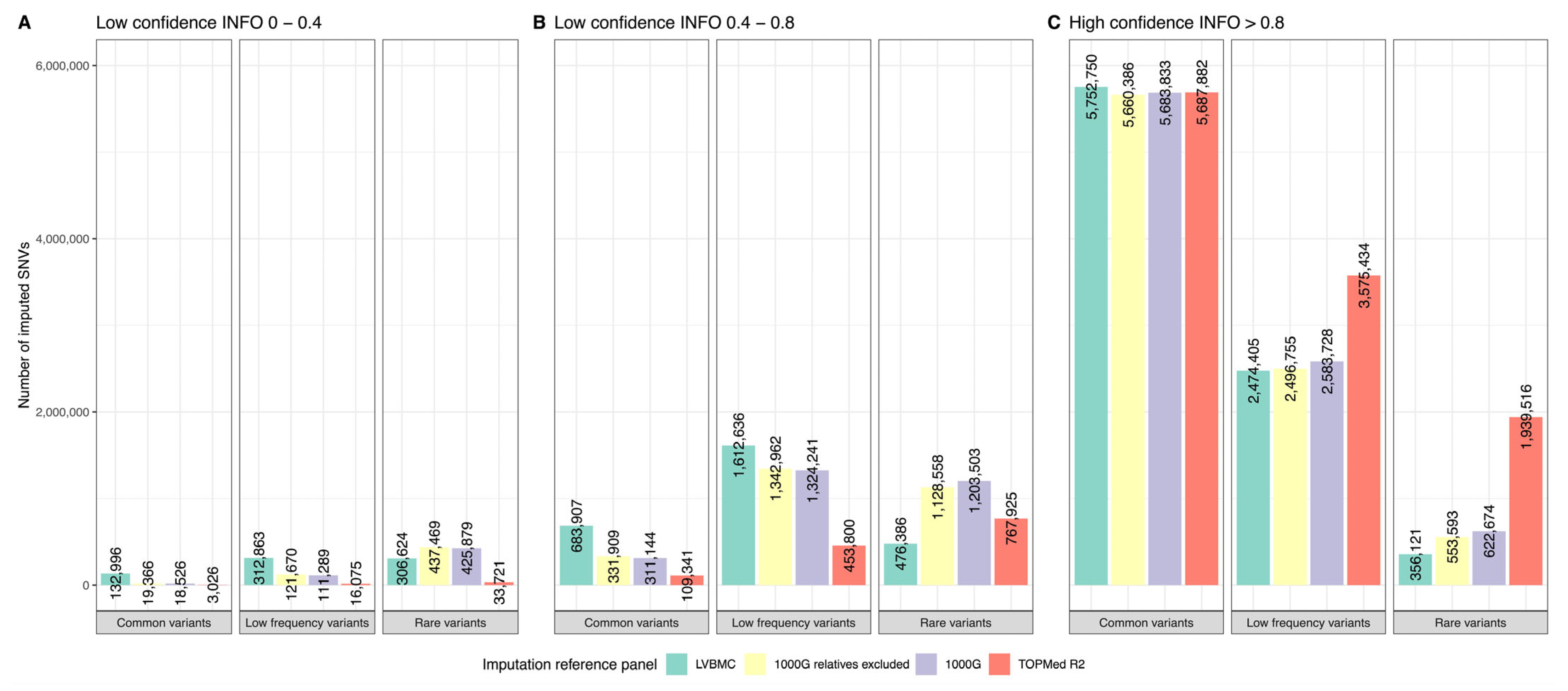
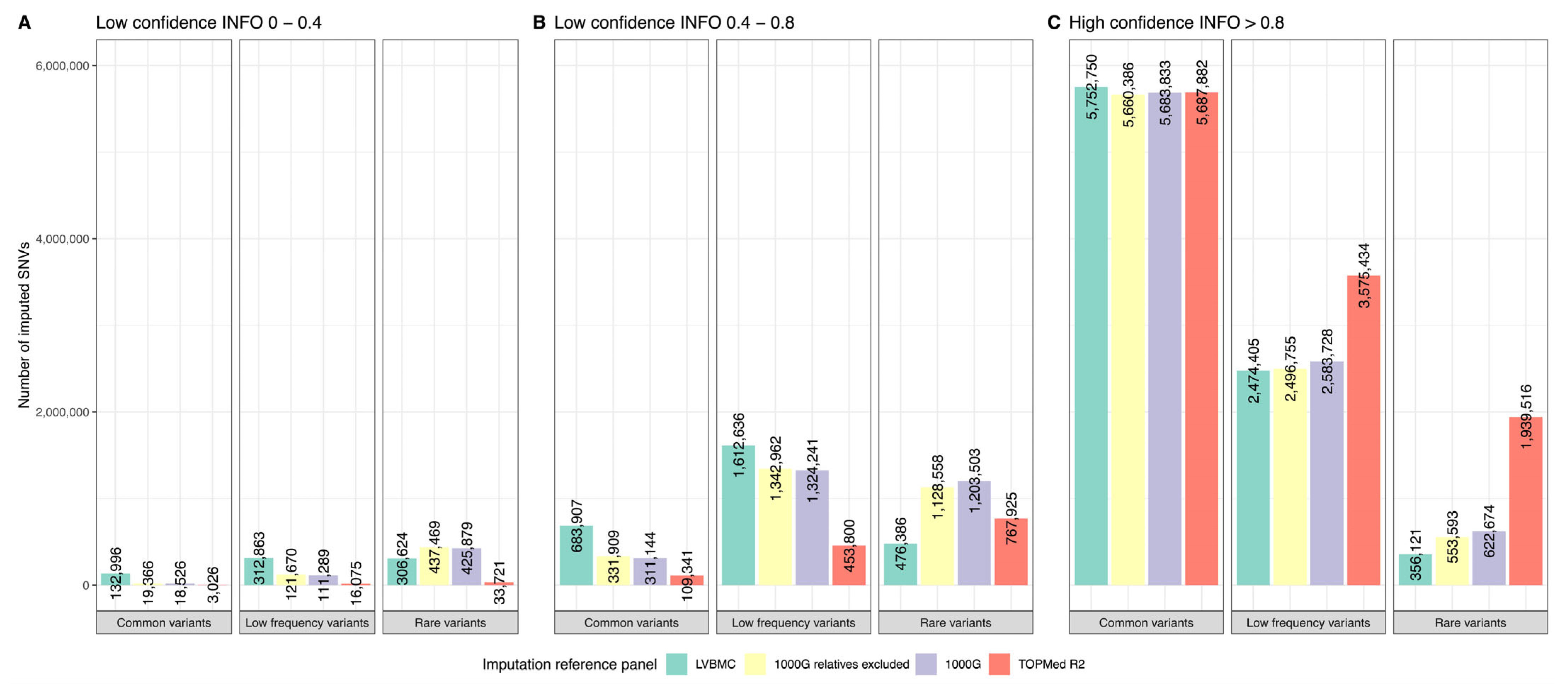
2.6. Imputation Panel Comparison
3. Discussion
4. Materials and Methods
4.1. Cohort
4.2. WGS Sequencing and Genotyping
4.3. Variant Calling
4.4. Quality Control and Annotation
4.5. Phasing, Imputation, and Concordance Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, F.S.; De La Vega, F.M.; Donnelly, P.; Egholm, M.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar]
- Oleksyk, T.K.; Wolfsberger, W.W.; Schubelka, K.; Mangul, S.; O’Brien, S.J. The Pioneer Advantage: Filling the blank spots on the map of genome diversity in Europe. GigaScience 2022, 11, giac081. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, E.; Shanmugam, A.; Cavalleri, G.L. Revealing the recent demographic history of Europe via haplotype sharing in the UK Biobank. Proc. Natl. Acad. Sci. USA 2022, 119, e2119281119. [Google Scholar] [CrossRef]
- Nelis, M.; Esko, T.; Mägi, R.; Zimprich, F.; Zimprich, A.; Toncheva, D.; Karachanak, S.; Piskáčková, T.; Balaščák, I.; Peltonen, L.; et al. Genetic Structure of Europeans: A View from the North–East. PLoS ONE 2009, 4, e5472. [Google Scholar] [CrossRef]
- Allentoft, M.E.; Sikora, M.; Refoyo-Martinez, A.; Irving-Pease, E.K.; Fischer, A.; Barrie, W.; Ingason, A.; Stenderup, J.; Sjögren, K.-G.; Pearson, A.; et al. Population Genomics of Stone Age Eurasia. biorXiv 2022. [Google Scholar] [CrossRef]
- Urnikyte, A.; Flores-Bello, A.; Mondal, M.; Molyte, A.; Comas, D.; Calafell, F.; Bosch, E.; Kučinskas, V. Patterns of genetic structure and adaptive positive selection in the Lithuanian population from high-density SNP data. Sci. Rep. 2019, 9, 9163. [Google Scholar] [CrossRef]
- Kushniarevich, A.; Utevska, O.; Chuhryaeva, M.; Agdzhoyan, A.; Dibirova, K.; Uktveryte, I.; Möls, M.; Mulahasanovic, L.; Pshenichnov, A.; Frolova, S.; et al. Genetic Heritage of the Balto-Slavic Speaking Populations: A Synthesis of Autosomal, Mitochondrial and Y-Chromosomal Data. PLoS ONE 2015, 10, e0135820. [Google Scholar] [CrossRef]
- Haak, W.; Lazaridis, I.; Patterson, N.; Rohland, N.; Mallick, S.; Llamas, B.; Brandt, G.; Nordenfelt, S.; Harney, E.; Stewardson, K.; et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 2015, 522, 207–211. [Google Scholar] [CrossRef]
- Mittnik, A.; Wang, C.-C.; Pfrengle, S.; Daubaras, M.; Zarina, G.; Hallgren, F.; Allmäe, R.; Khartanovich, V.; Moiseyev, V.; Tõrv, M.; et al. The genetic prehistory of the Baltic Sea region. Nat. Commun. 2018, 9, 442. [Google Scholar] [CrossRef]
- Bick, D.; Fraser, P.C.; Gutzeit, M.F.; Harris, J.M.; Hambuch, T.M.; Helbling, D.C.; Jacob, H.J.; Kersten, J.N.; Leuthner, S.R.; May, T.; et al. Successful Application of Whole Genome Sequencing in a Medical Genetics Clinic. J. Pediatr. Genet. 2017, 6, 61–76. [Google Scholar] [CrossRef]
- Godard, B.; Kate, L.T.; Evers-Kiebooms, G.; Aymé, S. Population genetic screening programmes: Principles, techniques, practices, and policies. Eur. J. Hum. Genet. 2003, 11, S49–S87. [Google Scholar] [CrossRef]
- Mitt, M.; Kals, M.; Pärn, K.; Gabriel, S.B.; Lander, E.S.; Palotie, A.; Ripatti, S.; Morris, A.P.; Metspalu, A.; Esko, T.; et al. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur. J. Hum. Genet. 2017, 25, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Micco, A.; Mah, M.; Ringbauer, H.; Lasaridis, I.; Olalde, I.; Patterson, N.; Reich, D. The Allen Ancient DNA Resource (AADR): A curated compendium of ancient human genomes. bioRxiv 2023. [Google Scholar] [CrossRef]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 2022, 185, 3426–3440.e19. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Zagury, J.-F.; Marchini, J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 2013, 10, 5–6. [Google Scholar] [CrossRef]
- Oleksyk, T.K.; Wolfsberger, W.W.; Weber, A.M.; Shchubelka, K.; Oleksyk, O.T.; Levchuk, O.; Patrus, A.; Lazar, N.; Castro-Marquez, S.O.; Hasynets, Y.; et al. Genome diversity in Ukraine. GigaScience 2021, 10, giaa159. [Google Scholar] [CrossRef] [PubMed]
- AlSafar, H.S.; Al-Ali, M.; Elbait, G.D.; Al-Maini, M.H.; Ruta, D.; Peramo, B.; Henschel, A.; Tay, G.K. Introducing the first whole genomes of nationals from the United Arab Emirates. Sci. Rep. 2019, 9, 14725. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Scliar, M.O.; Yamamoto, G.L.; Wang, J.Y.T.; Zverinova, S.; Karp, T.; Nunes, K.; Ricardo, J.; Ceroni, M.; Lima de Carvalho, D.; et al. Whole-genome sequencing of 1171 elderly admixed individuals from Brazil. Nat. Commun. 2022, 13, 1004. [Google Scholar] [CrossRef]
- Kaja, E.; Lejman, A.; Sielski, D.; Sypniewski, M.; Gambin, T.; Dawidziuk, M.; Suchocki, T.; Golik, P.; Wojtaszewska, M.; Mroczek, M.; et al. The Thousand Polish Genomes—A Database of Polish Variant Allele Frequencies. Int. J. Mol. Sci. 2022, 23, 4532. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Kember, R.L.; Roach, J.C.; O’Connell, J.R.; Craig, D.W.; Bucan, M.; Scott, W.K.; Pericak-Vance, M.; Haines, J.L.; Crawford, M.H.; et al. A population-specific reference panel empowers genetic studies of Anabaptist populations. Sci. Rep. 2017, 7, 6079. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, X.; Chen, H.; Tan, X.; Yang, Z.; Deng, L.; Wang, B.; Kong, S.; Li, S.; Cui, Y.; et al. A pangenome reference of 36 Chinese populations. Nature 2023, 619, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J.K.; Monlong, J.; Abel, H.J.; et al. A draft human pangenome reference. Nature 2023, 617, 312–324. [Google Scholar] [CrossRef]
- Rovite, V.; Wolff-Sagi, Y.; Zaharenko, L.; Nikitina-Zake, L.; Grens, E.; Klovins, J. Genome Database of the Latvian Population (LGDB): Design, Goals, and Primary Results. J. Epidemiol. 2018, 28, 353–360. [Google Scholar] [CrossRef]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Fuchsberger, C.; Abecasis, G.R.; Hinds, D.A. minimac2: Faster genotype imputation. Bioinformatics 2014, 31, 782–784. [Google Scholar] [CrossRef]
- Kim, H.-M.; Jeon, S.; Chung, O.; Jun, J.H.; Kim, H.-S.; Blazyte, A.; Lee, H.-Y.; Yu, Y.; Cho, Y.S.; Bolser, D.M.; et al. Comparative analysis of 7 short-read sequencing platforms using the Korean Reference Genome: MGI and Illumina sequencing benchmark for whole-genome sequencing. GigaScience 2021, 10, giab014. [Google Scholar] [CrossRef]
- Meslier, V.; Quinquis, B.; Da Silva, K.; Oñate, F.P.; Pons, N.; Roume, H.; Podar, M.; Almeida, M. Benchmarking second and third-generation sequencing platforms for microbial metagenomics. Sci. Data 2022, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bähler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14061. [Google Scholar] [CrossRef]
- Gardner, E.J.; Lam, V.K.; Harris, D.N.; Chuang, N.T.; Scott, E.C.; Pittard, W.S.; Mills, R.E.; Devine, S.E. The Mobile Element Locator Tool (MELT): Population-scale mobile element discovery and biology. Genome Res. 2017, 27, 1916–1929. [Google Scholar] [CrossRef]
- Tange, O. GNU Parallel—The Command-Line Power Tool. Login USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Purcell, S.; Chang, C. PLINK 2.0. Available online: https://www.cog-genomics.org/plink/2.0/ (accessed on 1 February 2021).
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 2020, 367, eaay5012. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; Alföldi, J.; Watts, N.A.; Vittal, C.; Gauthier, L.D.; et al. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020; pp. 275–286. [Google Scholar]
- Parn, K.; Isokallio, M.A.; Nunez-Fontarnau, J.; Palotie, A.; Ripatti, S.; Palta, P. Genotype imputation workflow v3.0 V.2. Protocols.io 2019. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Eberle, M.; Fritzilas, E.; Peter, K.; Kallberg, M.; Moore, B.L.; Bekritsky, M.A.; Iqbal, Z.; Chuang, H.-Y.; Humphray, S.J.; Halpern, A.L.; et al. A reference data aset of 5.4 million phased human varianats validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017, 27, 157–164. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Female | Male | Total | |
|---|---|---|---|
| (N = 318) | (N = 184) | (N = 502) | |
| Age | |||
| Mean (SD) | 54.4 (13.6) | 50.0 (14.4) | 52.8 (14.0) |
| Median [Min; Max] | 55.0 [21.0; 91.0] | 50.5 [18.0; 87.0] | 54.0 [18.0; 91.0] |
| Region | |||
| Courland | 47 (14.8%) | 32 (17.4%) | 79 (15.7%) |
| Latgale | 63 (19.8%) | 34 (18.5%) | 97 (19.3%) |
| Riga | 53 (16.7%) | 41 (22.3%) | 94 (18.7%) |
| Semigallia | 28 (8.8%) | 8 (4.3%) | 36 (7.2%) |
| Vidzeme | 74 (23.3%) | 49 (26.6%) | 123 (24.5%) |
| Missing | 53 (16.7%) | 20 (10.9%) | 73 (14.5%) |
| Ethnicity | |||
| Belarussian | 9 (2.8%) | 4 (2.2%) | 13 (2.6%) |
| Latvian | 192 (60.4%) | 124 (67.4%) | 316 (62.9%) |
| Polish | 4 (1.3%) | 3 (1.6%) | 7 (1.4%) |
| Russian | 90 (28.3%) | 43 (23.4%) | 133 (26.5%) |
| Ukrainian | 13 (4.1%) | 3 (1.6%) | 16 (3.2%) |
| Missing | 10 (3.1%) | 7 (3.8%) | 17 (3.4%) |
| Variant Type | Total Count | Novel gnomAD | Mean (SD [Min; Max]) | Mean Novel (SD [Min; Max]) |
|---|---|---|---|---|
| Small variants * | ||||
| SNV | 15,158,133 | 466,749 | 3,822,725 (74,939 [3,643,058; 4,022,661]) | 9788 (4938 [4386; 23,979]) |
| Deletion | 1,608,579 | 49,249 | 326,497 (7337 [304,340; 349,310]) | 1444 (278 [895; 2478]) |
| Inframe deletion | 1398 | 76 | 203 (14 [160; 292]) | 4 (3 [1; 56]) |
| Insertion | 1,435,886 | 59,499 | 327,355 (7352 [305,045; 349,737]) | 2399 (327 [1686; 5622]) |
| Inframe insertion | 1083 | 61 | 176 (13 [138; 301]) | 3 (4 [1;67]) |
| Structural variation ** | ||||
| DEL | 28,191 | 10,337 | 3465 (244 [2568; 4179]) | 794 (88 [514; 1067]) |
| DUP | 6866 | 3139 | 456 (71 [256; 668]) | 192 (29 [104; 277]) |
| INS | 10,405 | 5791 | 798 (345 [327; 1993]) | 454 (198 [196; 1135]) |
| INV | 3881 | 1534 | 297 (36 [179; 396]) | 134 (17 [84; 184]) |
| TRA | 7431 | 3980 | 514 (99 [379; 796]) | 292 (56 [208; 438]) |
| Mobile element insertions ** | ||||
| ALU | 4074 | 760 | 183 (108 [30; 504]) | 19 (11 [1; 61]) |
| LINE1 | 528 | 141 | 19 (15 [1; 74]) | 7 (4 [1; 24]) |
| SVA | 207 | 49 | 7 (6 [1; 27]) | 2 (2 [1; 9]) |
| HERVK | 22 | 9 | 1 (0 [1; 4]) | 1 (0 [1; 2]) |
| Variant Type | Total Count | Novel gnomAD | Mean (SD [Min; Max]) | Mean Novel (SD [Min; Max]) |
|---|---|---|---|---|
| Variants by location | ||||
| Upstream gene variant | 2,972,390 | 76,822 | 713,153 (25,203 [663,145; 775,844]) | 1924 (1428 [736; 6062]) |
| 5 prime UTR variant | 58,753 | 4215 | 12,434 (432 [11,331; 14,018]) | 90 (117 [9; 531]) |
| Noncoding transcript exon variant | 504,846 | 13,166 | 123,426 (5194 [113,905; 134,625]) | 316 (309 [82; 1210]) |
| 3 prime UTR variant | 237,749 | 5596 | 57,344 (1625 [54,020; 61,640]) | 127 (120 [28; 472]) |
| Downstream gene variant | 3,112,542 | 71,227 | 757,570 (26,960 [704,675; 821,758]) | 1818 (1259 [722; 5243]) |
| Intergenic variant | 7,374,606 | 362,260 | 1,651,845 (25,220 [1,564,267; 1,736,263]) | 9031 (1648 [5484; 14,930]) |
| Intron variant | 10,367,399 | 194,729 | 2,634,492 (48,769 [2,512,167; 2,764,304]) | 4368 (3055 [1751; 12,885]) |
| Functional variants by type | ||||
| Splice acceptor variant | 1319 | 66 | 269 (13 [232; 307]) | 3 (1 [1; 13]) |
| Splice donor variant | 1855 | 70 | 393 (16 [351; 449]) | 6 (2 [2; 16]) |
| Splice-donor-region variant | 3471 | 123 | 839 (45 [754; 953]) | 3 (3 [1; 15]) |
| Splice donor fifth-base variant | 1499 | 73 | 341 (17 [299; 389]) | 5 (2 [1; 12]) |
| Splice-region variant | 19,629 | 254 | 4862 (179 [4603; 5262]) | 5 (5 [1; 36]) |
| Transcript ablation | 4 | 0 | 2 (1 [1; 3]) | 0 |
| Frameshift variant | 2453 | 269 | 351 (66 [273; 980]) | 11 (21 [1; 335]) |
| Missense variant | 67,397 | 3617 | 12,257 (1176 [10,682; 14,876]) | 75 (112 [1; 421]) |
| Start lost | 234 | 10 | 44 (5 [29; 60]) | 1 (1 [1; 6]) |
| Stop gained | 1087 | 28 | 152 (18 [122; 203]) | 1 (2 [1; 28]) |
| Stop lost | 264 | 9 | 77 (7 [58; 98]) | 1 (0 [1; 3]) |
| Synonymous variant | 51,160 | 1180 | 12,225 (706 [11,127; 13,741]) | 27 (35 [1; 140]) |
| Protein-altering variant | 67 | 16 | 4 (5 [1; 90]) | 2 (5 [1; 44]) |
| Medically relevant variants in the Latvian population | ||||
| Pathogenic | 344 | - | 40 (5 [26; 56]) | - |
| Likely pathogenic | 177 | - | 19 (3 [11; 30]) | - |
| Benign | 93,468 | - | 35,285 (575 [33,879; 36,871]) | - |
| Likely benign | 19,519 | - | 1412 (58 [1253; 1617]) | - |
| Protective | 43 | - | 18 (3 [9; 26]) | - |
| Drug response | 203 | - | 76 (9 [53; 106]) | - |
| Association | 183 | - | 81 (8 [58; 101]) | - |
| WGS Genotype Overlap (% [Mean; SD (Min; Max)]) | ||||
|---|---|---|---|---|
| Compared Set | REF | ALT_1 | ALT_2 | |
| Array (n = 316) | REF | 98.5 [318,418; 2491 (309,942; 324,671)] | 0.2 [219; 25 (167; 424)] | 0.2 [135; 162 (1; 642)] |
| Imputed with 1000G panel (n = 200) | REF | 96.4 [4,976,995; 31,027 (4,839,014; 5,030,586)] | 10.8 [211,324; 9452 (193,120; 277,490)] | 1.0 [13,574; 1339 (10,398; 20,400)] |
| Imputed with reduced LVBMC panel (n = 200) * | REF | 96.6 [5,724,931; 59,377 (5,532,919; 5,811,414)] | 12.8 [275,223; 20,029 (238,691; 354,592)] | 1.8 [25,167; 2280 (20,140; 34,645)] |
| Array (n = 316) | ALT_1 | 0.9 [3067; 1901 (702; 6486)] | 99.5 [105,725; 3716 (99,028; 113,350)] | 0.5 [281; 180 (46; 955)] |
| Imputed with 1000G panel (n = 200) | ALT_1 | 3.2 [165,345; 11,462 (140,740; 219,026)] | 85.1 [1,671,734; 28,554 (1,583,691; 1,739,678)] | 6.0 [78,923; 3714 (72,727; 104,440)] |
| Imputed with reduced LVBMC panel (n = 200) * | ALT_1 | 3.2 [188,676; 16,716 (158,304; 280,839)] | 83.0 [1,784,775; 37,177 (1,669,064; 1,868,066)] | 6.0 [82,413; 5783 (73,026; 117,744)] |
| Array (n = 316) | ALT_2 | 0.6 [1781; 654 (430; 3442)] | 0.3 [347; 38 (273; 672)] | 99.3 [59,945; 1873 (56,492; 65,564)] |
| Imputed with 1000G panel (n = 200) | ALT_2 | 0.4 [21,463; 5702 (9101; 33,309)] | 4.2 [81,625; 4195 (73,042; 111,549)] | 92.9 [1,214,036; 19,902 (1,150,271; 1,261,170)] |
| Imputed with reduced LVBMC panel (n = 200) * | ALT_2 | 0.2 [13,949; 2369 (9743; 23,925)] | 4.2 [90,491; 6953 (77,116; 133,932)] | 92.2 [1,268,448; 21,304 (1,189,978; 1,317,623)] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reščenko, R.; Brīvība, M.; Atava, I.; Rovīte, V.; Pečulis, R.; Silamiķelis, I.; Ansone, L.; Megnis, K.; Birzniece, L.; Leja, M.; et al. Whole-Genome Sequencing of 502 Individuals from Latvia: The First Step towards a Population-Specific Reference of Genetic Variation. Int. J. Mol. Sci. 2023, 24, 15345. https://doi.org/10.3390/ijms242015345
Reščenko R, Brīvība M, Atava I, Rovīte V, Pečulis R, Silamiķelis I, Ansone L, Megnis K, Birzniece L, Leja M, et al. Whole-Genome Sequencing of 502 Individuals from Latvia: The First Step towards a Population-Specific Reference of Genetic Variation. International Journal of Molecular Sciences. 2023; 24(20):15345. https://doi.org/10.3390/ijms242015345
Chicago/Turabian StyleReščenko, Raimonds, Monta Brīvība, Ivanna Atava, Vita Rovīte, Raitis Pečulis, Ivars Silamiķelis, Laura Ansone, Kaspars Megnis, Līga Birzniece, Mārcis Leja, and et al. 2023. "Whole-Genome Sequencing of 502 Individuals from Latvia: The First Step towards a Population-Specific Reference of Genetic Variation" International Journal of Molecular Sciences 24, no. 20: 15345. https://doi.org/10.3390/ijms242015345
APA StyleReščenko, R., Brīvība, M., Atava, I., Rovīte, V., Pečulis, R., Silamiķelis, I., Ansone, L., Megnis, K., Birzniece, L., Leja, M., Xu, L., Shi, X., Zhou, Y., Slaitas, A., Hou, Y., & Kloviņš, J. (2023). Whole-Genome Sequencing of 502 Individuals from Latvia: The First Step towards a Population-Specific Reference of Genetic Variation. International Journal of Molecular Sciences, 24(20), 15345. https://doi.org/10.3390/ijms242015345