
Quantitative Lipid Profiling Reveals Major Differences between Liver Organoids with Normal Pi*M and Deficient Pi*Z Variants of Alpha-1-antitrypsin
 , , , ,
, , , , 
Abstract
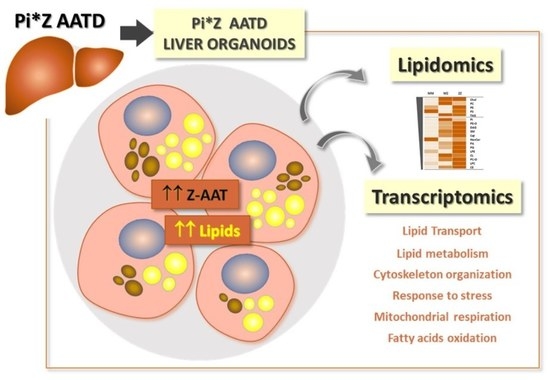
1. Introduction
2. Results
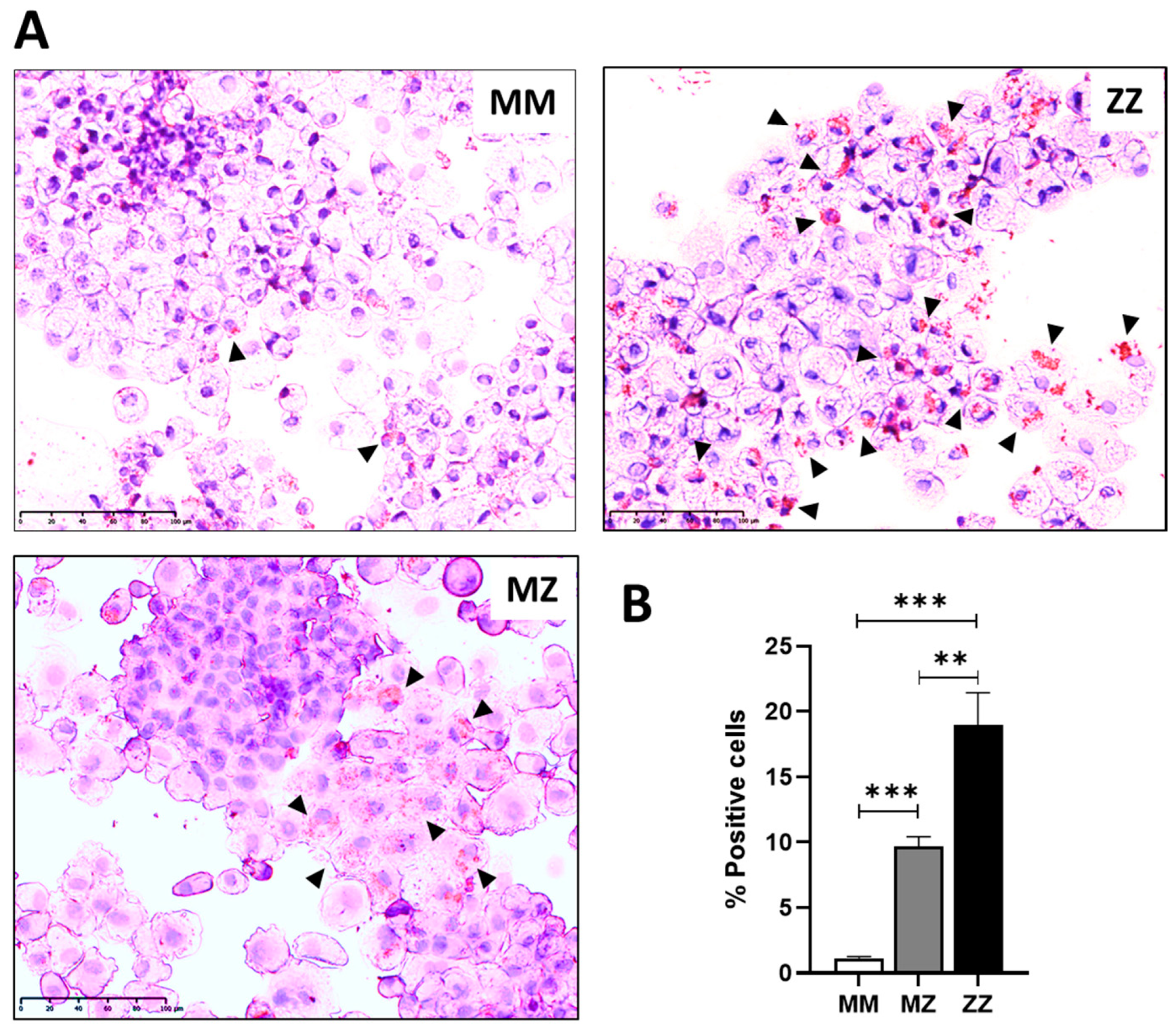
2.1. Intracellular Accumulation of Z-AAT Polymers Parallel with Increased Lipid Content
2.2. Differences in Lipidomic Profiles between MM-, MZ-, and ZZ-ORGs
2.3. Distribution of Specific Lipids within ZZ-, MZ-, and MM-ORG Relative to the Total Lipid Content
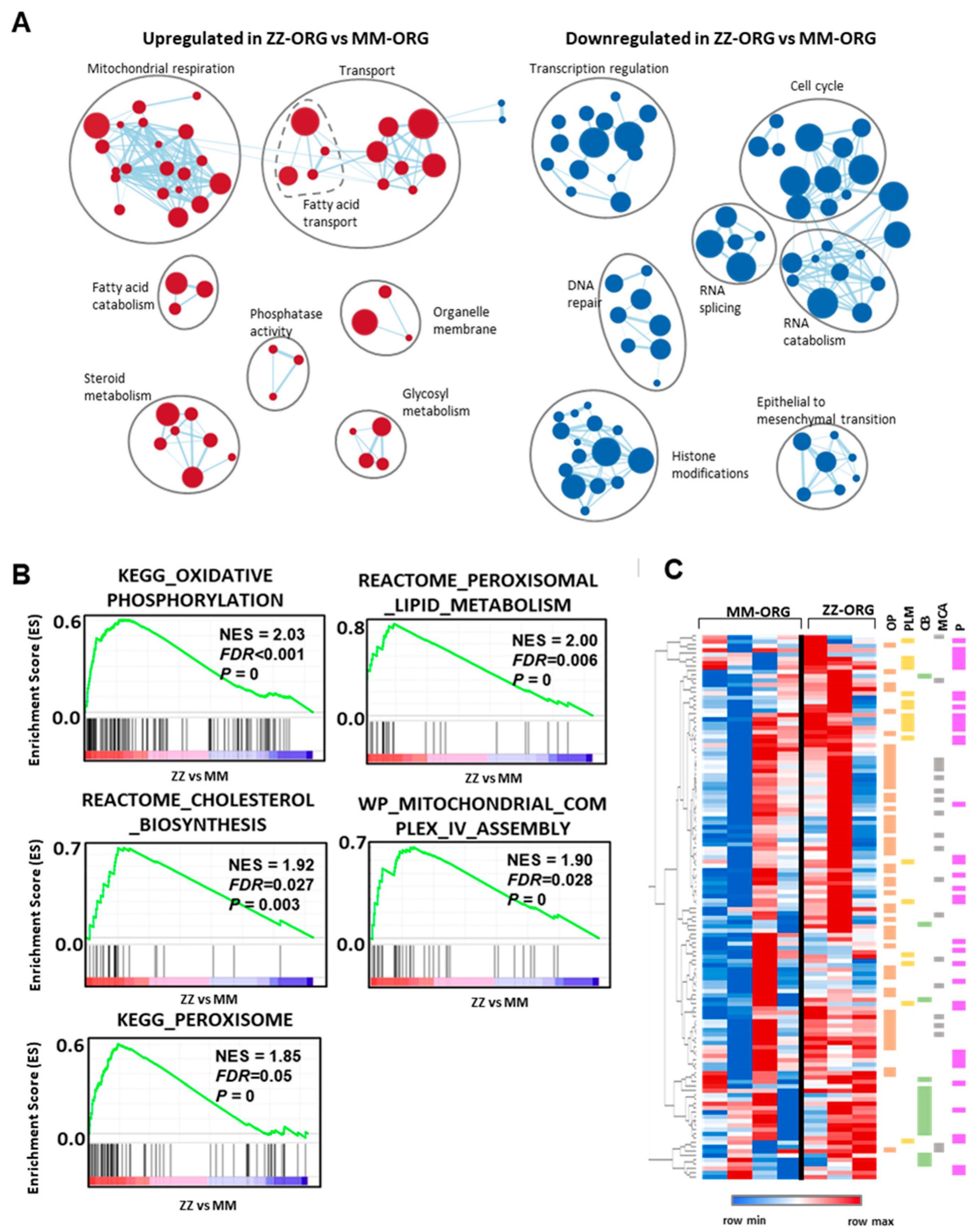
2.4. Functional Annotation Analysis of Differentially Expressed Genes (DEGs) between ZZ- and MM-ORG
2.5. Gene Set Enrichment Analysis (GSEA) in ZZ-ORG Versus MM-ORG
2.6. Expression Levels of Specific DEGs Related to Lipid Transport and Metabolism
3. Discussion
4. Materials and Methods
4.1. Culture of Human Liver Organoids
4.2. Culture and Transfection of HepG2 Cell Line
4.3. Lipid Extraction for Mass Spectrometry Lipidomics
4.4. Mass Spectrometry Data Acquisition, Analysis, and Post-Processing
4.5. Determination of Intracellular Lipid Content
4.6. Western-Blot Analysis of AAT
4.7. Detection of Intracellular AAT by Immunofluoresce
4.8. Transcriptomic Analysis of Liver Organoids (RNA-Seq)
4.9. Data Processing and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The Discovery of A1-Antitrypsin and Its Role in Health and Disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef]
- Foil, K.E. Variants of SERPINA1 and the Increasing Complexity of Testing for Alpha-1 Antitrypsin Deficiency. Ther. Adv. Chronic Dis. 2021, 12, 20406223211015950. [Google Scholar] [CrossRef]
- Sinden, N.J.; Baker, M.J.; Smith, D.J.; Kreft, J.-U.; Dafforn, T.R.; Stockley, R.A. α-1-Antitrypsin Variants and the Proteinase/Antiproteinase Imbalance in Chronic Obstructive Pulmonary Disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L179–L190. [Google Scholar] [CrossRef]
- Miravitlles, M. Alpha-1-Antitrypsin and Other Proteinase Inhibitors. Curr. Opin. Pharmacol. 2012, 12, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.; Lipsker, D.; Lara, B.; Janciauskiene, S. Neutrophilic Panniculitis Associated with Alpha-1-Antitrypsin Deficiency: An Update. Br. J. Dermatol. 2016, 174, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Xu, Z.; Zhu, C.; Chen, T.; Muñoz, L.E.; Dai, L.; Zhao, Y. Alpha-1 Antitrypsin in Autoimmune Diseases: Roles and Therapeutic Prospects. Int. Immunopharmacol. 2022, 110, 109001. [Google Scholar] [CrossRef]
- Seixas, S.; Marques, P.I. Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum. Appl. Clin. Genet. 2021, 14, 173–194. [Google Scholar] [CrossRef]
- de Serres, F.J.; Blanco, I. Prevalence of A1-Antitrypsin Deficiency Alleles PI*S and PI*Z Worldwide and Effective Screening for Each of the Five Phenotypic Classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: A Comprehensive Review. Ther. Adv. Respir. Dis. 2012, 6, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The Mechanism of Z Alpha 1-Antitrypsin Accumulation in the Liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Oton-Gonzalez, L.; Selvatici, R.; Rizzo, P.; Pavasini, R.; Campo, G.C.; Lanzillotti, C.; Mazziotta, C.; De Mattei, M.; Tognon, M.; et al. SERPINA1 Gene Promoter Is Differentially Methylated in Peripheral Blood Mononuclear Cells of Pregnant Women. Front. Cell Dev. Biol. 2020, 8, 550543. [Google Scholar] [CrossRef]
- Qiu, W.; Baccarelli, A.; Carey, V.J.; Boutaoui, N.; Bacherman, H.; Klanderman, B.; Rennard, S.; Agusti, A.; Anderson, W.; Lomas, D.A.; et al. Variable DNA Methylation Is Associated with Chronic Obstructive Pulmonary Disease and Lung Function. Am. J. Respir. Crit. Care Med. 2012, 185, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Hamesch, K.; Mandorfer, M.; Pereira, V.M.; Moeller, L.S.; Pons, M.; Dolman, G.E.; Reichert, M.C.; Schneider, C.V.; Woditsch, V.; Voss, J.; et al. Liver Fibrosis and Metabolic Alterations in Adults With Alpha-1-Antitrypsin Deficiency Caused by the Pi*ZZ Mutation. Gastroenterology 2019, 157, 705–719.e18. [Google Scholar] [CrossRef] [PubMed]
- Winther, S.V.; Ahmed, D.; Al-Shuweli, S.; Landt, E.M.; Nordestgaard, B.G.; Seersholm, N.; Dahl, M. Severe A1-Antitrypsin Deficiency Associated with Lower Blood Pressure and Reduced Risk of Ischemic Heart Disease: A Cohort Study of 91,540 Individuals and a Meta-Analysis. Respir. Res. 2022, 23, 55. [Google Scholar] [CrossRef]
- Khodayari, N.; Wang, R.L.; Oshins, R.; Lu, Y.; Millett, M.; Aranyos, A.M.; Mostofizadeh, S.; Scindia, Y.; Flagg, T.O.; Brantly, M. The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease. Int. J. Mol. Sci. 2021, 22, 13255. [Google Scholar] [CrossRef]
- Anand, P.K. Lipids, Inflammasomes, Metabolism, and Disease. Immunol. Rev. 2020, 297, 108–122. [Google Scholar] [CrossRef]
- Krahmer, N.; Farese, R.V.; Walther, T.C. Balancing the Fat: Lipid Droplets and Human Disease. EMBO Mol. Med. 2013, 5, 905–915. [Google Scholar] [CrossRef]
- Gómez-Mariano, G.; Matamala, N.; Martínez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzón, S.; Cuesta, I.; Garfia, C.; Martínez, M.T.; et al. Liver Organoids Reproduce Alpha-1 Antitrypsin Deficiency-Related Liver Disease. Hepatol. Int. 2020, 14, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, M.; Vallier, L.; Guillot, A. Modeling Nonalcoholic Fatty Liver Disease in the Dish Using Human-Specific Platforms: Strategies and Limitations. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, B.; Mandorfer, M.; Viveiros, A.; Finkenstedt, A.; Ferenci, P.; Schneeberger, S.; Tilg, H.; Zoller, H. Heterozygosity for the Alpha-1-Antitrypsin Z Allele in Cirrhosis Is Associated with More Advanced Disease. Liver Transpl. 2018, 24, 744–751. [Google Scholar] [CrossRef]
- Prior, N.; Inacio, P.; Huch, M. Liver Organoids: From Basic Research to Therapeutic Applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef]
- Shi, S.; Verstegen, M.M.A.; Roest, H.P.; Ardisasmita, A.I.; Cao, W.; Roos, F.J.M.; de Ruiter, P.E.; Niemeijer, M.; Pan, Q.; IJzermans, J.N.M.; et al. Recapitulating Cholangiopathy-Associated Necroptotic Cell Death In Vitro Using Human Cholangiocyte Organoids. Cell Mol. Gastroenterol. Hepatol. 2021, 13, 541–564. [Google Scholar] [CrossRef]
- Schneider, C.V.; Hamesch, K.; Gross, A.; Mandorfer, M.; Moeller, L.S.; Pereira, V.; Pons, M.; Kuca, P.; Reichert, M.C.; Benini, F.; et al. Liver Phenotypes of European Adults Heterozygous or Homozygous for Pi∗Z Variant of AAT (Pi∗MZ vs Pi∗ZZ Genotype) and Noncarriers. Gastroenterology 2020, 159, 534–548.e11. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic Lipids Stored by Kupffer Cells Correlates with Their Pro-Inflammatory Phenotype at an Early Stage of Steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and Its Management. Nutr. Clin. Pract. 2020, 35, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, A.N.; Heinecke, J.W. Lipoproteomics: Using Mass Spectrometry-Based Proteomics to Explore the Assembly, Structure, and Function of Lipoproteins. J. Lipid Res. 2009, 50, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Krahmer, N.; Guo, Y.; Wilfling, F.; Hilger, M.; Lingrell, S.; Heger, K.; Newman, H.W.; Schmidt-Supprian, M.; Vance, D.E.; Mann, M.; et al. Phosphatidylcholine Synthesis for Lipid Droplet Expansion Is Mediated by Localized Activation of CTP:Phosphocholine Cytidylyltransferase. Cell Metab. 2011, 14, 504–515. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Georgakilas, A.; Ravanat, J.-L. Targeted and Off-Target (Bystander and Abscopal) Effects of Radiation Therapy: Redox Mechanisms and Risk/Benefit Analysis. Antioxid. Redox Signal. 2018, 29, 1447–1487. [Google Scholar] [CrossRef]
- Kinoshita, M.; Matsumori, N. Inimitable Impacts of Ceramides on Lipid Rafts Formed in Artificial and Natural Cell Membranes. Membranes 2022, 12, 727. [Google Scholar] [CrossRef]
- Idowu, J.Y.; Hagenbuch, B. Free Cholesterol Affects the Function and Localization of Human Na+/Taurocholate Cotransporting Polypeptide (NTCP) and Organic Cation Transporter 1 (OCT1). Int. J. Mol. Sci. 2022, 23, 8457. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Shen, T.; Huynh, H.; Fang, X.; Han, Z.; Ouyang, K. Cardiolipin Regulates Mitochondrial Ultrastructure and Function in Mammalian Cells. Genes 2022, 13, 1889. [Google Scholar] [CrossRef]
- Prola, A.; Pilot-Storck, F. Cardiolipin Alterations during Obesity: Exploring Therapeutic Opportunities. Biology 2022, 11, 1638. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent Insights into the Biological Functions of Liver Fatty Acid Binding Protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef]
- Smathers, R.L.; Galligan, J.J.; Shearn, C.T.; Fritz, K.S.; Mercer, K.; Ronis, M.; Orlicky, D.J.; Davidson, N.O.; Petersen, D.R. Susceptibility of L-FABP-/- Mice to Oxidative Stress in Early-Stage Alcoholic Liver. J. Lipid Res. 2013, 54, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Petrescu, A.D.; Huang, H.; Martin, G.G.; McIntosh, A.L.; Storey, S.M.; Landrock, D.; Kier, A.B.; Schroeder, F. Impact of L-FABP and Glucose on Polyunsaturated Fatty Acid Induction of PPARα-Regulated β-Oxidative Enzymes. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G241–G256. [Google Scholar] [CrossRef]
- Diraison, F.; Moulin, P.; Beylot, M. Contribution of Hepatic de Novo Lipogenesis and Reesterification of Plasma Non Esterified Fatty Acids to Plasma Triglyceride Synthesis during Non-Alcoholic Fatty Liver Disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Adiels, M.; Olofsson, S.-O.; Taskinen, M.-R.; Borén, J. Overproduction of Very Low-Density Lipoproteins Is the Hallmark of the Dyslipidemia in the Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1225–1236. [Google Scholar] [CrossRef]
- Aggarwal, D.J.; Kathariya, M.G.; Verma, D.P.K. LDL-C, NON-HDL-C and APO-B for Cardiovascular Risk Assessment: Looking for the Ideal Marker. Indian. Heart J. 2021, 73, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Gayet, C.; Ginsberg, H.N. Inhibition of Apolipoprotein B100 Secretion by Lipid-Induced Hepatic Endoplasmic Reticulum Stress in Rodents. J. Clin. Investig. 2008, 118, 316–332. [Google Scholar] [CrossRef]
- Chen, C.; Li, H.; Song, J.; Zhang, C.; Li, M.; Mao, Y.; Liu, A.; Du, J. Role of Apolipoprotein A1 in PPAR Signaling Pathway for Nonalcoholic Fatty Liver Disease. PPAR Res. 2022, 2022, 4709300. [Google Scholar] [CrossRef] [PubMed]
- Deprince, A.; Hennuyer, N.; Kooijman, S.; Pronk, A.C.M.; Baugé, E.; Lienard, V.; Verrijken, A.; Dirinck, E.; Vonghia, L.; Woitrain, E.; et al. Apolipoprotein F Is Reduced in Humans with Steatosis and Controls Plasma Triglyceride-Rich Lipoprotein Metabolism. Hepatology 2023, 77, 1278–1302. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Yoda, E.; Sasaki, Y.; Nakatani, Y.; Kuwata, H. Calcium-Independent Phospholipase A2γ (IPLA2γ) and Its Roles in Cellular Functions and Diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 861–868. [Google Scholar] [CrossRef]
- Teckman, J.H.; An, J.-K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial Autophagy and Injury in the Liver in Alpha 1-Antitrypsin Deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G851–G862. [Google Scholar] [CrossRef]
- Papp, E.; Száraz, P.; Korcsmáros, T.; Csermely, P. Changes of Endoplasmic Reticulum Chaperone Complexes, Redox State, and Impaired Protein Disulfide Reductase Activity in Misfolding Alpha1-Antitrypsin Transgenic Mice. FASEB J. 2006, 20, 1018–1020. [Google Scholar] [CrossRef]
- Kaserman, J.E.; Werder, R.B.; Wang, F.; Matte, T.; Higgins, M.I.; Dodge, M.; Lindstrom-Vautrin, J.; Bawa, P.; Hinds, A.; Bullitt, E.; et al. Human IPSC-Hepatocyte Modeling of Alpha-1 Antitrypsin Heterozygosity Reveals Metabolic Dysregulation and Cellular Heterogeneity. Cell Rep. 2022, 41, 111775. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.; Haemmerle, G. Fat in the Heart: The Enzymatic Machinery Regulating Cardiac Triacylglycerol Metabolism. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 1500–1512. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Faust, P.L.; Wanders, R.J.A. Bile Acids: The Role of Peroxisomes. J. Lipid Res. 2009, 50, 2139–2147. [Google Scholar] [CrossRef]
- Kleiboeker, B.; Lodhi, I.J. Peroxisomal Regulation of Energy Homeostasis: Effect on Obesity and Related Metabolic Disorders. Mol. Metab. 2022, 65, 101577. [Google Scholar] [CrossRef]
- Hultcrantz, R.; Mengarelli, S. Ultrastructural Liver Pathology in Patients with Minimal Liver Disease and Alpha 1-Antitrypsin Deficiency: A Comparison between Heterozygous and Homozygous Patients. Hepatology 1984, 4, 937–945. [Google Scholar] [CrossRef]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.-K.; Huch, M. Culture and Establishment of Self-Renewing Human and Mouse Adult Liver and Pancreas 3D Organoids and Their Genetic Manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef]
- Matamala, N.; Lara, B.; Gomez-Mariano, G.; Martínez, S.; Retana, D.; Fernandez, T.; Silvestre, R.A.; Belmonte, I.; Rodriguez-Frias, F.; Vilar, M.; et al. Characterization of Novel Missense Variants of SERPINA1 Gene Causing Alpha-1 Antitrypsin Deficiency. Am. J. Respir. Cell Mol. Biol. 2018, 58, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global Analysis of the Yeast Lipidome by Quantitative Shotgun Mass Spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.; Dominaitiene, R.; Sternby, N.H.; Piitulainen, E.; Eriksson, S. Detection of Circulating and Endothelial Cell Polymers of Z and Wild Type Alpha 1-Antitrypsin by a Monoclonal Antibody. J. Biol. Chem. 2002, 277, 26540–26546. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class 1 | MM | MZ | ZZ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Media (pmol) | SD * | Media (pmol) | SD | FC ** | p-Value *** | Media (pmol) | SD | FC | p-Value *** | ||
| Cholesterol | Chol | 5070.61 | 488.56 | 5776.7 | 357.55 | 1.14 | 0.0352 | 5793.16 | 678.17 | 1.14 | 0.1075 |
| Phosphatidylcholine | PC | 3814.2 | 391.59 | 4848.09 | 426.64 | 1.27 | 0.0746 | 4116.96 | 398.06 | 1.08 | 0.3082 |
| Phosphatidylethanolamine | PE | 2116.96 | 207.09 | 2001.29 | 144.14 | 0.95 | 0.3366 | 2257.92 | 284.74 | 1.07 | 0.3556 |
| Phosphatidylserine | PS | 1076.2 | 145.32 | 1025.35 | 95.45 | 0.95 | 0.3933 | 1071.83 | 101.36 | 1 | 0.4908 |
| Triacylglycerol | TAG | 1065.28 | 60.73 | 3325.34 | 312.73 | 3.12 | 0.0079 | 3112.48 | 399.46 | 2.92 | 0.0168 |
| Phosphatidylinositol | PI | 525.25 | 26.77 | 775.52 | 41.67 | 1.48 | 0.0054 | 601.58 | 53.74 | 1.15 | 0.1475 |
| Phosphatidylethanolamine (-ether) | PE-O | 460.33 | 43.84 | 641.14 | 49.71 | 1.39 | 0.0267 | 868.37 | 108.36 | 1.89 | 0.0243 |
| Diacylglycerol | DAG | 376.61 | 25.09 | 417.04 | 39.62 | 1.11 | 0.2227 | 466.18 | 65.62 | 1.24 | 0.1527 |
| Sphingomyelin | SM | 290.89 | 31.92 | 350.47 | 22.77 | 1.2 | 0.1053 | 367.73 | 43.07 | 1.26 | 0.1154 |
| Ceramide | Cer | 231.32 | 18.2 | 363.72 | 18.65 | 1.57 | 0.0035 | 434.46 | 57.11 | 1.88 | 0.0297 |
| Hexosylceramide | HexCer | 87.76 | 9 | 41.95 | 1.72 | 0.48 | 0.0163 | 65.46 | 6.45 | 0.75 | 0.0608 |
| Phosphatidate | PA | 69.81 | 5.12 | 76.21 | 5.81 | 1.09 | 0.2279 | 83.79 | 10.23 | 1.2 | 0.1552 |
| Phosphatidylglycerol | PG | 47.58 | 4.89 | 60.24 | 4.36 | 1.27 | 0.0631 | 81.52 | 10.36 | 1.71 | 0.0317 |
| lyso-Phasphatidylethanolamine | LPE | 25.94 | 2.46 | 16.82 | 0.57 | 0.65 | 0.0295 | 44.62 | 5.14 | 1.72 | 0.0247 |
| Cardiolipins | CL | 25.06 | 1.6 | 54 | 2.69 | 2.15 | 0.001 | 47.67 | 7.27 | 1.9 | 0.0417 |
| Phosphatidylcholine (-ether) | PC-O | 24.71 | 2.34 | 50.62 | 4.23 | 2.05 | 0.0058 | 59.8 | 8.12 | 2.42 | 0.0204 |
| lyso-Phasphatidylcholine | LPC | 22.38 | 2.17 | 17.96 | 2.68 | 0.8 | 0.1359 | 49.03 | 4.96 | 2.19 | 0.0099 |
| Cholesterol esters | CE | 13.47 | 0.59 | 120.21 | 7.92 | 8.92 | 0.0026 | 173.25 | 24.02 | 12.86 | 0.0109 |
| lyso-Phasphatidate | LPA | 4.61 | 0.24 | 0 | 0 | 0 | 0.0014 | 7.48 | 1.04 | 1.62 | 0.0519 |
| lyso-Phosphatidylserine | LPS | 3.12 | 0.53 | 0.71 | 0.71 | 0.23 | 0.0287 | 4.28 | 0.49 | 1.37 | 0.092 |
| lyso-Phosphatidylinositol | LPI | 2.7 | 0.41 | 1.51 | 1.51 | 0.56 | 0.2583 | 2.54 | 0.32 | 0.94 | 0.3845 |
| lyso-Phosphatidylethanolamine (-ether) | LPE-O | 2.34 | 0.22 | 0 | 0 | 0 | 0.0043 | 6.6 | 0.72 | 2.81 | 0.01 |
| lyso-Phosphatidylglycerol | LPG | 1.59 | 0.1 | 0 | 0 | 0 | 0.0019 | 3.66 | 0.5 | 2.31 | 0.0246 |
| lyso-Phosphatidylcholine (-ether) | LPC-O | 0.86 | 0.18 | 0 | 0 | 0 | 0.0209 | 3.04 | 0.28 | 3.53 | 0.0025 |
| Class | MM | MZ | ZZ | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Media (%) | SD * | Media (%) | SD * | p-Value MM | Media (%) | SD * | p-Value MM | p-Value MZ | ||
| Cholesterol | Chol | 33 | 0.0801 | 28.99 | 0.5554 | 0.017 | 29.35 | 0.0865 | 6.87 × 10-6 | 0.5837 |
| Phosphatidylcholine | PC | 24.8 | 0.4197 | 24.24 | 0.4711 | 0.4215 | 20.95 | 0.41 | 0.0028 | 0.0066 |
| Phosphatidylethanolamine | PE | 13.78 | 0.1345 | 10.03 | 0.1567 | 0.0001 | 11.42 | 0.1663 | 0.0005 | 0.0005 |
| Triacylglycerol | TAG | 6.98 | 0.2678 | 16.61 | 0.3737 | 0.0001 | 15.74 | 0.3171 | 3.70 × 10-5 | 0.1512 |
| Phosphatidylserine | PS | 6.96 | 0.2644 | 5.14 | 0.288 | 0.0098 | 5.46 | 0.2001 | 0.0126 | 0.4155 |
| Phosphatidylinositol | PI | 3.45 | 0.1424 | 3.9 | 0.087 | 0.0663 | 3.07 | 0.0792 | 0.0991 | 0.0022 |
| Phosphatidylethanolamine (-ether) | PE-O | 3 | 0.042 | 3.21 | 0.0287 | 0.0179 | 4.39 | 0.0482 | 3.01 × 10-5 | 0.0001 |
| Diacylglycerol | DAG | 2.47 | 0.0944 | 2.08 | 0.0472 | 0.037 | 2.35 | 0.074 | 0.3865 | 0.0479 |
| Sphingomyelin | SM | 1.89 | 0.0695 | 1.76 | 0.0171 | 0.1905 | 1.87 | 0.0507 | 0.7852 | 0.1581 |
| Ceramide | Cer | 1.51 | 0.0316 | 1.83 | 0.0668 | 0.0254 | 2.19 | 0.0876 | 0.0093 | 0.0328 |
| Hexosylceramide | HexCer | 0.57 | 0.0343 | 0.21 | 0.0071 | 0.007 | 0.33 | 0.0062 | 0.0174 | 0.0002 |
| Phasphatidate | PA | 0.46 | 0.0108 | 0.38 | 0.0072 | 0.007 | 0.42 | 0.0037 | 0.0843 | 0.0135 |
| Phosphatidylglycerol | PG | 0.31 | 0.0044 | 0.3 | 0.0073 | 0.4399 | 0.41 | 0.0057 | 0.0002 | 0.0004 |
| lyso-Phosphatidylethanolamine | LPE | 0.17 | 0.008 | 0.08 | 0.0035 | 0.0035 | 0.23 | 0.0048 | 0.0068 | 0 |
| Cardiolipins | CL | 0.16 | 0.0052 | 0.27 | 0.0079 | 0.0007 | 0.24 | 0.0117 | 0.0121 | 0.0946 |
| Phosphatidylcholine (-ether) | PC-O | 0.16 | 0.0031 | 0.26 | 0.0264 | 0.0675 | 0.3 | 0.0098 | 0.0025 | 0.2206 |
| lyso-Phosphatidylcholine | LPC | 0.15 | 0.0022 | 0.09 | 0.0068 | 0.0091 | 0.25 | 0.0033 | 4.18 × 10-5 | 0.0003 |
| Cholesterol esters | CE | 0.09 | 0.0103 | 0.6 | 0.0103 | 3.88 × 10-6 | 0.87 | 0.022 | 0.0001 | 0.002 |
| lyso-Phosphatidate | LPA | 0.03 | 0.0032 | 0 | 0 | 0.0106 | 0.04 | 0.0028 | 0.1618 | 0.0055 |
| lyso-Phosphatidylserine | LPS | 0.02 | 0.0039 | 0 | 0.004 | 0.0425 | 0.02 | 0.0002 | 0.7866 | 0.0483 |
| lyso-Phosphatidylinositol | LPI | 0.02 | 0.001 | 0.01 | 0.0067 | 0.2457 | 0.01 | 0.0011 | 0.0409 | 0.4465 |
| lyso-Phosphatidylethanolamine (-ether) | LPE-O | 0.02 | 0.0011 | 0 | 0 | 0.0052 | 0.03 | 0.0004 | 0.0014 | 0.0014 |
| lyso-Phosphatidylglycerol | LPG | 0.01 | 0.0005 | 0 | 0 | 0.0023 | 0.02 | 0.0007 | 0.0009 | 0.0013 |
| lyso-Phosphatidylcholine (-ether) | LPC-O | 0.01 | 0.0007 | 0 | 0 | 0.017 | 0.02 | 0.0015 | 0.01 | 0.0091 |
| Cluster | Term ID | Term Description | Observed Gene Count | Background Gene Count | Strength | FDR * |
|---|---|---|---|---|---|---|
| (1) | GO:0003779 | Actin binding | 15 | 438 | 0.93 | 8.25 × 10−7 |
| Actin Cytoskeleton | GO:0007010 | Cytoskeleton organization | 18 | 1126 | 0.6 | 0.0017 |
| GO:0031982 | Vesicle | 29 | 3879 | 0.27 | 0.0393 | |
| (2) | GO:0019222 | Regulation of metabolic process | 62 | 6948 | 0.32 | 1.45 × 10−9 |
| Regulation of metabolic processes | GO:0060255 | Regulation of macromolecule metabolic process | 60 | 6407 | 0.34 | 1.45 × 10−9 |
| GO:0010468 | Regulation of gene expression | 51 | 4813 | 0.4 | 3.64 × 10−9 | |
| GO:0006950 | Response to stress | 37 | 3485 | 0.4 | 4.68 × 10−6 | |
| GO:0030162 | Regulation of proteolysis | 15 | 747 | 0.68 | 8.76 × 10−5 | |
| GO:0001817 | Regulation of cytokine production | 16 | 742 | 0.71 | 1.82 × 10−5 | |
| GO:0042981 | Regulation of apoptotic process | 21 | 1550 | 0.5 | 0.00019 | |
| GO:0006954 | Inflammatory response | 11 | 515 | 0.7 | 0.0011 | |
| GO:0006955 | Immune response | 18 | 1588 | 0.43 | 0.0061 | |
| (3) | GO:0032787 | Monocarboxylic acid metabolic process | 13 | 515 | 0.73 | 0.0119 |
| Fatty acid metabolic processes | GO:0019752 | Carboxylic acid metabolic process | 16 | 853 | 0.61 | 0.0138 |
| GO:0006631 | Fatty acid metabolic process | 9 | 311 | 0.79 | 0.0448 | |
| KEGG-hsa00982 | Drug metabolism - cytochrome P450 | 7 | 64 | 1.37 | 4.22 × 10−6 | |
| KEGG-hsa00140 | Steroid hormone biosynthesis | 5 | 59 | 1.26 | 0.00056 | |
| KEGG-hsa04976 | Bile secretion | 5 | 89 | 1.08 | 0.0025 | |
| (4) | GO:0030054 | Cell junction | 28 | 2075 | 0.43 | 0.0011 |
| Plasma membrane | GO:0031226 | Intrinsic component of plasma membrane | 22 | 1703 | 0.42 | 0.0229 |
| (5) | GO:0031090 | Organelle membrane | 27 | 3548 | 0.39 | 0.002 |
| Endomembrane system | GO:0005794 | Golgi apparatus | 17 | 1584 | 0.54 | 0.0027 |
| GO:0034703 | Cation channel complex | 7 | 214 | 1.02 | 0.0027 | |
| GO:0012505 | Endomembrane system | 30 | 4542 | 0.33 | 0.003 | |
| GO:0098797 | Plasma membrane protein complex | 9 | 547 | 0.72 | 0.0079 | |
| GO:0008194 | UDP-glycosyltransferase activity | 6 | 143 | 1.13 | 0.0092 |
| Organoids | Sex | Age | Reason of Surgery | Serum AAT | GPT | GOT | GGT | Platelets | Glucose | Albumin | Bilirubin |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | (Years) | (g/L) | (U/L) | (U/L) | (U/L) | (×1000/uL) | (mg/dL) | (g/dL) | (mg/dL) | ||
| (5–45) * | (5–33) * | (8–61) * | (140–450) * | (70–110) * | (3.5–5) * | (0.2–1) * | |||||
| MM | F | 78 | Hepatocellular carcinoma | ND | 31 | 31 | 49 | 246 | 113 | 4.5 | 0.3 |
| MZ | M | 82 | Cholecystectomy | ND | 64 | 36 | 33 | 126 | 110 | 3.9 | 0.3 |
| ZZ | M | 1 | Hepatic failure. Liver transplant | 0.41 | 199 | 360 | 551 | 92 | 71 | 3 | 3.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Luz, S.; Lalchandani, J.; Matamala, N.; Barrero, M.J.; Gil-Martín, S.; Saz, S.R.-D.; Varona, S.; Monzón, S.; Cuesta, I.; Justo, I.; et al. Quantitative Lipid Profiling Reveals Major Differences between Liver Organoids with Normal Pi*M and Deficient Pi*Z Variants of Alpha-1-antitrypsin. Int. J. Mol. Sci. 2023, 24, 12472. https://doi.org/10.3390/ijms241512472
Pérez-Luz S, Lalchandani J, Matamala N, Barrero MJ, Gil-Martín S, Saz SR-D, Varona S, Monzón S, Cuesta I, Justo I, et al. Quantitative Lipid Profiling Reveals Major Differences between Liver Organoids with Normal Pi*M and Deficient Pi*Z Variants of Alpha-1-antitrypsin. International Journal of Molecular Sciences. 2023; 24(15):12472. https://doi.org/10.3390/ijms241512472
Chicago/Turabian StylePérez-Luz, Sara, Jaanam Lalchandani, Nerea Matamala, Maria Jose Barrero, Sara Gil-Martín, Sheila Ramos-Del Saz, Sarai Varona, Sara Monzón, Isabel Cuesta, Iago Justo, and et al. 2023. "Quantitative Lipid Profiling Reveals Major Differences between Liver Organoids with Normal Pi*M and Deficient Pi*Z Variants of Alpha-1-antitrypsin" International Journal of Molecular Sciences 24, no. 15: 12472. https://doi.org/10.3390/ijms241512472
APA StylePérez-Luz, S., Lalchandani, J., Matamala, N., Barrero, M. J., Gil-Martín, S., Saz, S. R.-D., Varona, S., Monzón, S., Cuesta, I., Justo, I., Marcacuzco, A., Hierro, L., Garfia, C., Gomez-Mariano, G., Janciauskiene, S., & Martínez-Delgado, B. (2023). Quantitative Lipid Profiling Reveals Major Differences between Liver Organoids with Normal Pi*M and Deficient Pi*Z Variants of Alpha-1-antitrypsin. International Journal of Molecular Sciences, 24(15), 12472. https://doi.org/10.3390/ijms241512472