
Investigating Antibody Reactivity to the Intestinal Microbiome in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Feasibility Study
 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Recruiting Severe ME/CFS Patients and Same-Household Controls
2.2. Study Population Characteristics
2.3. Stool Consistency Does Not Separate Severe ME/CFS Patients with IBS from Matched Household Controls without IBS
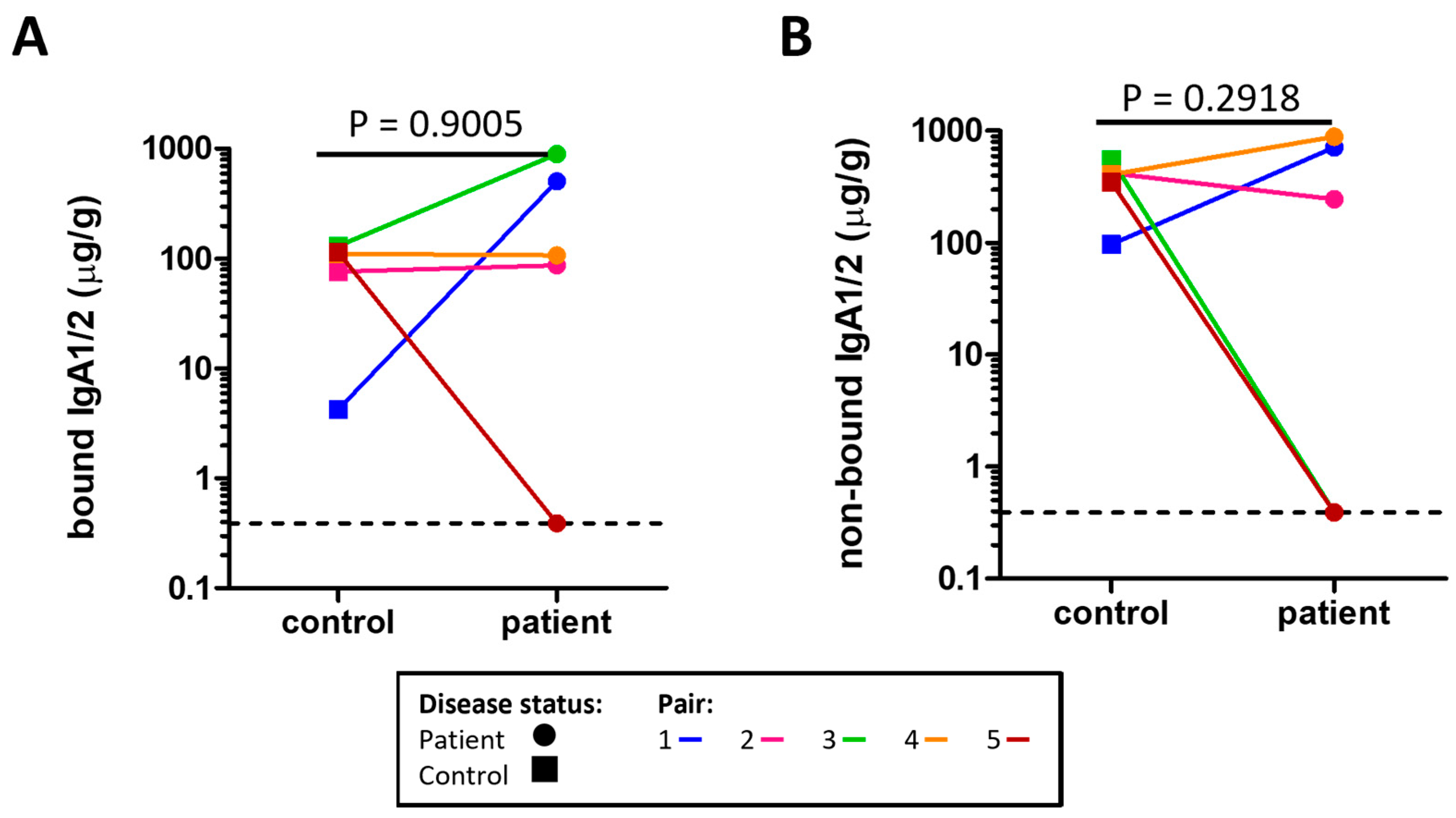
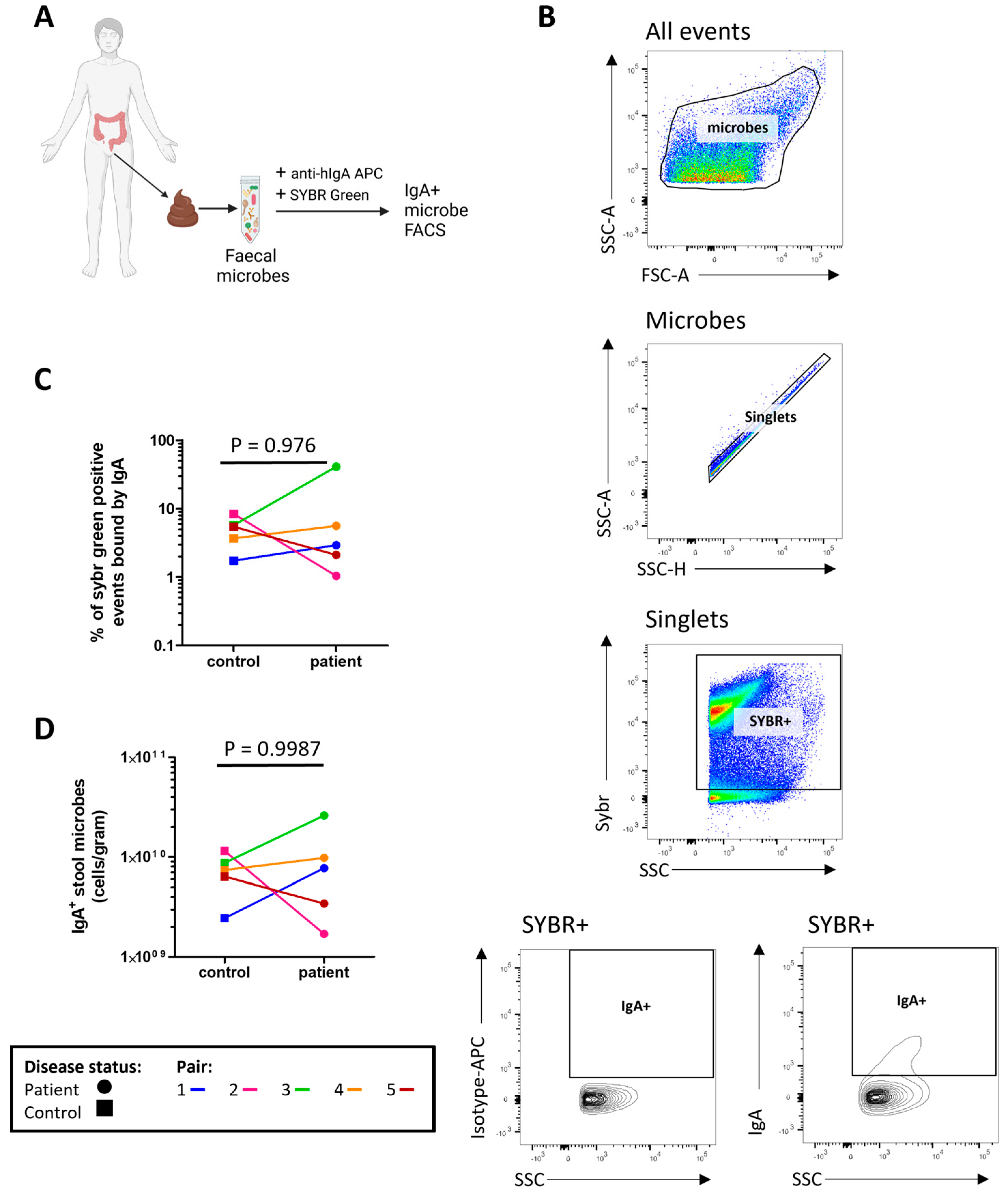
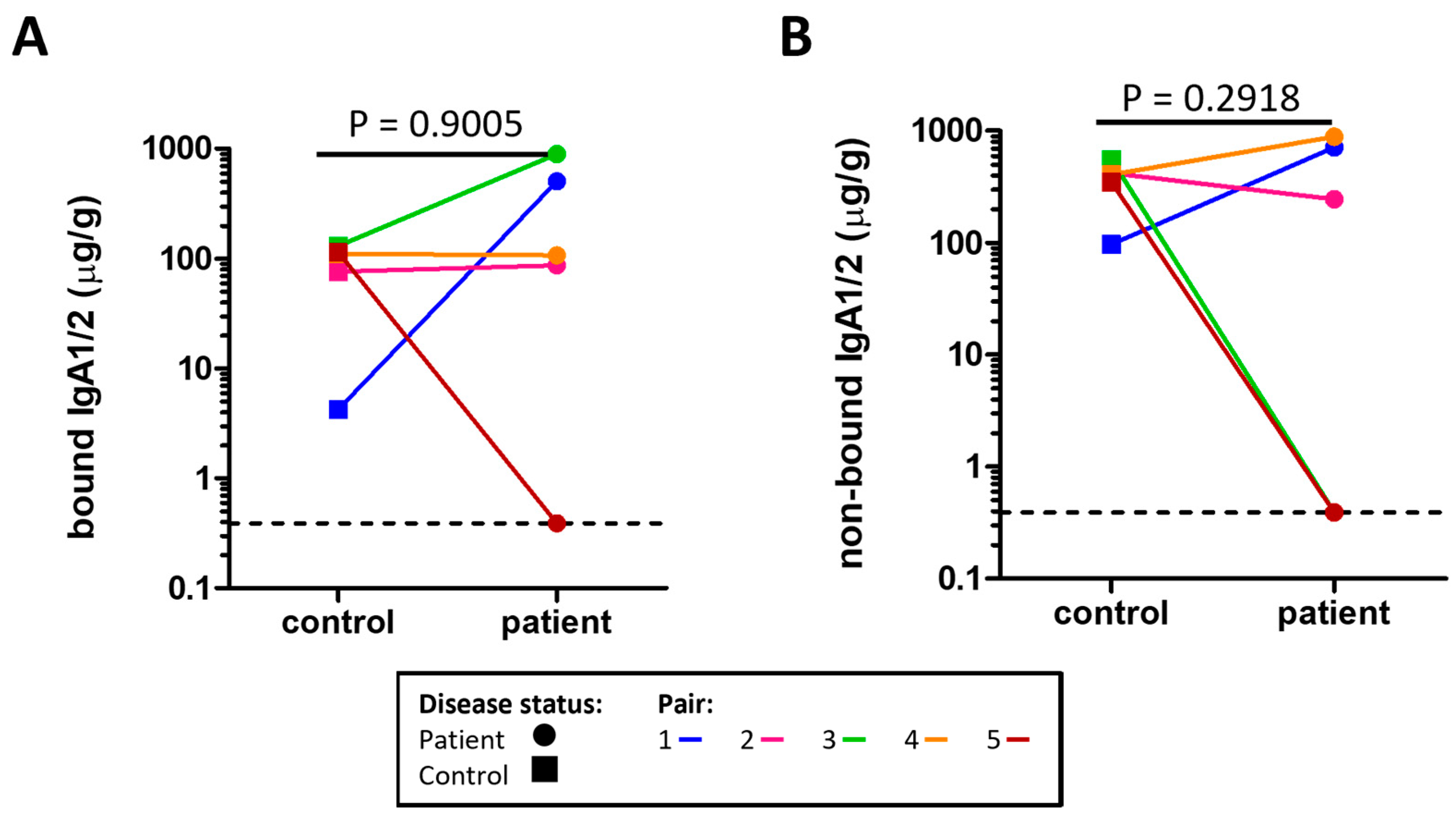
2.4. Assessment of Secretory IgA (sIgA) in Stool
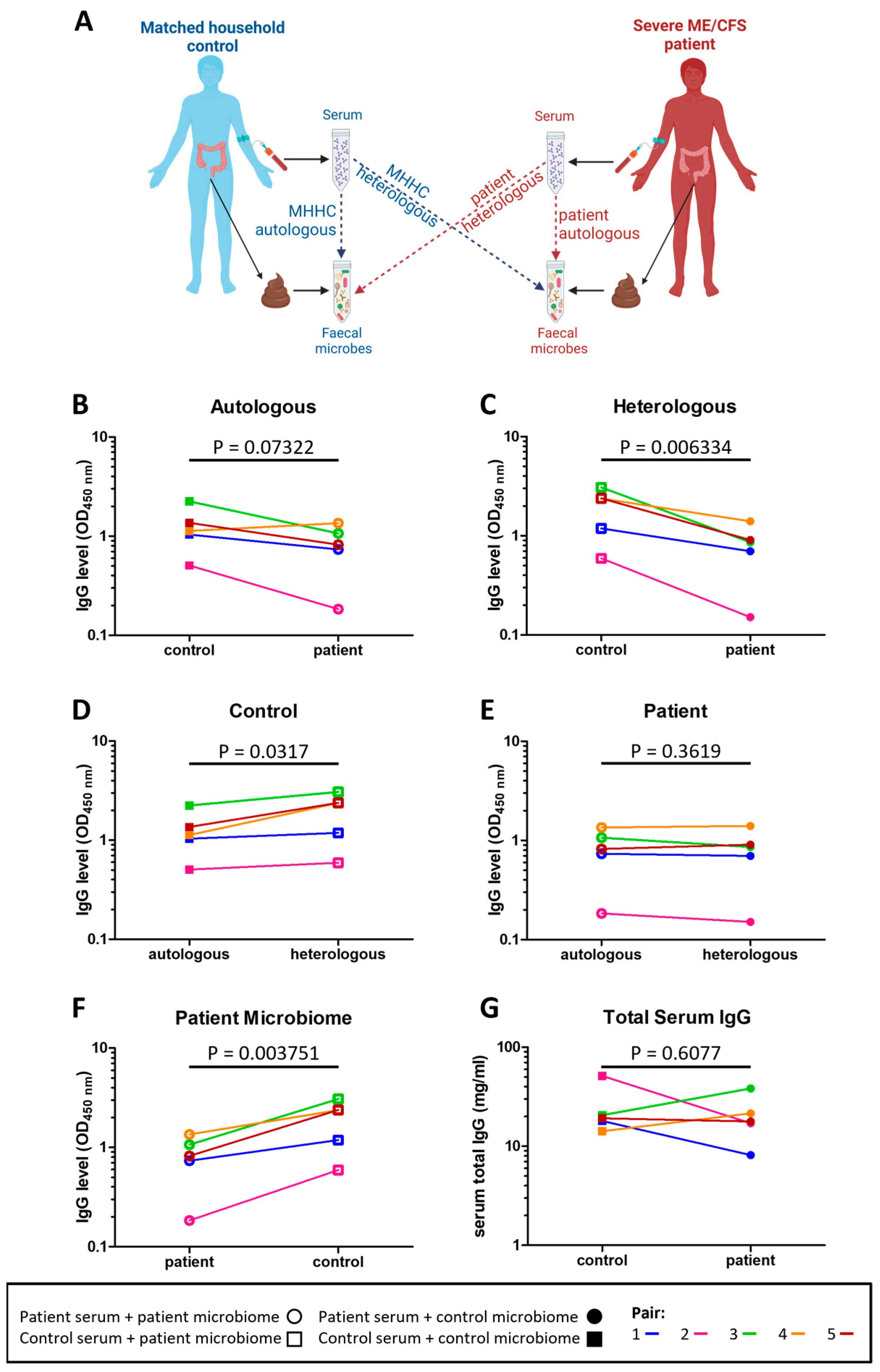
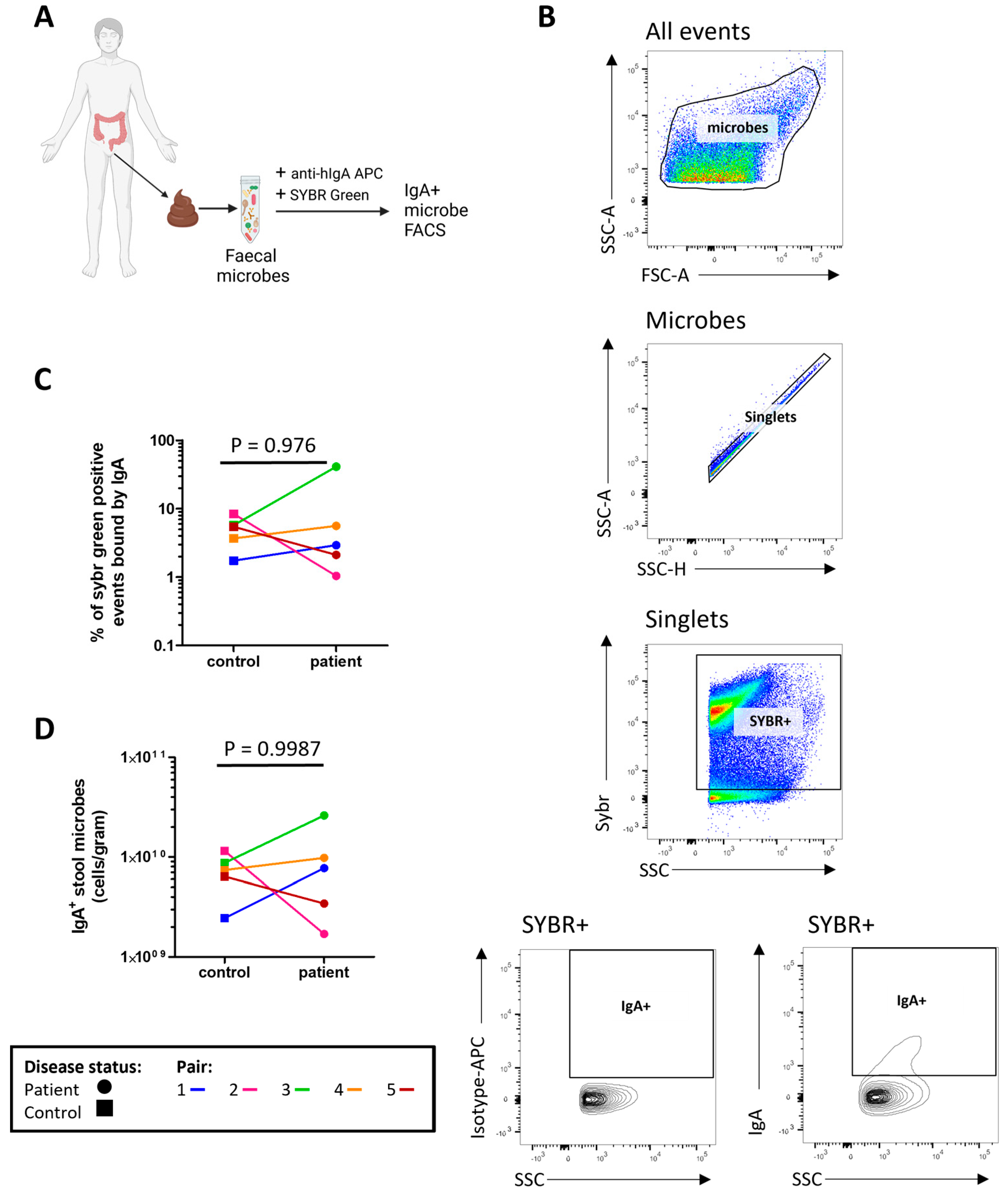
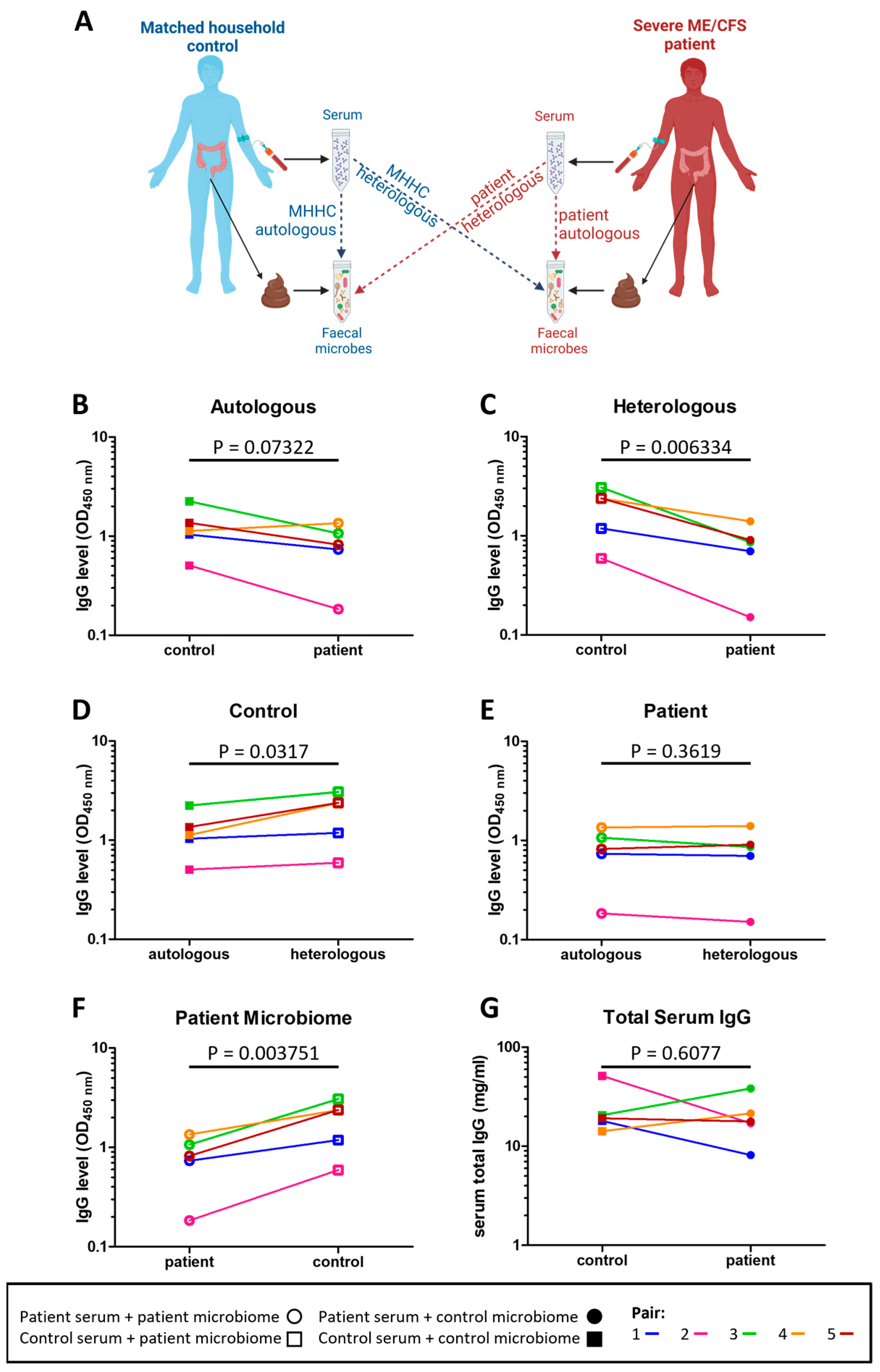
2.5. Severe ME/CFS Patients Have a Reduced Serum IgG Immune Response to Stool Microbes
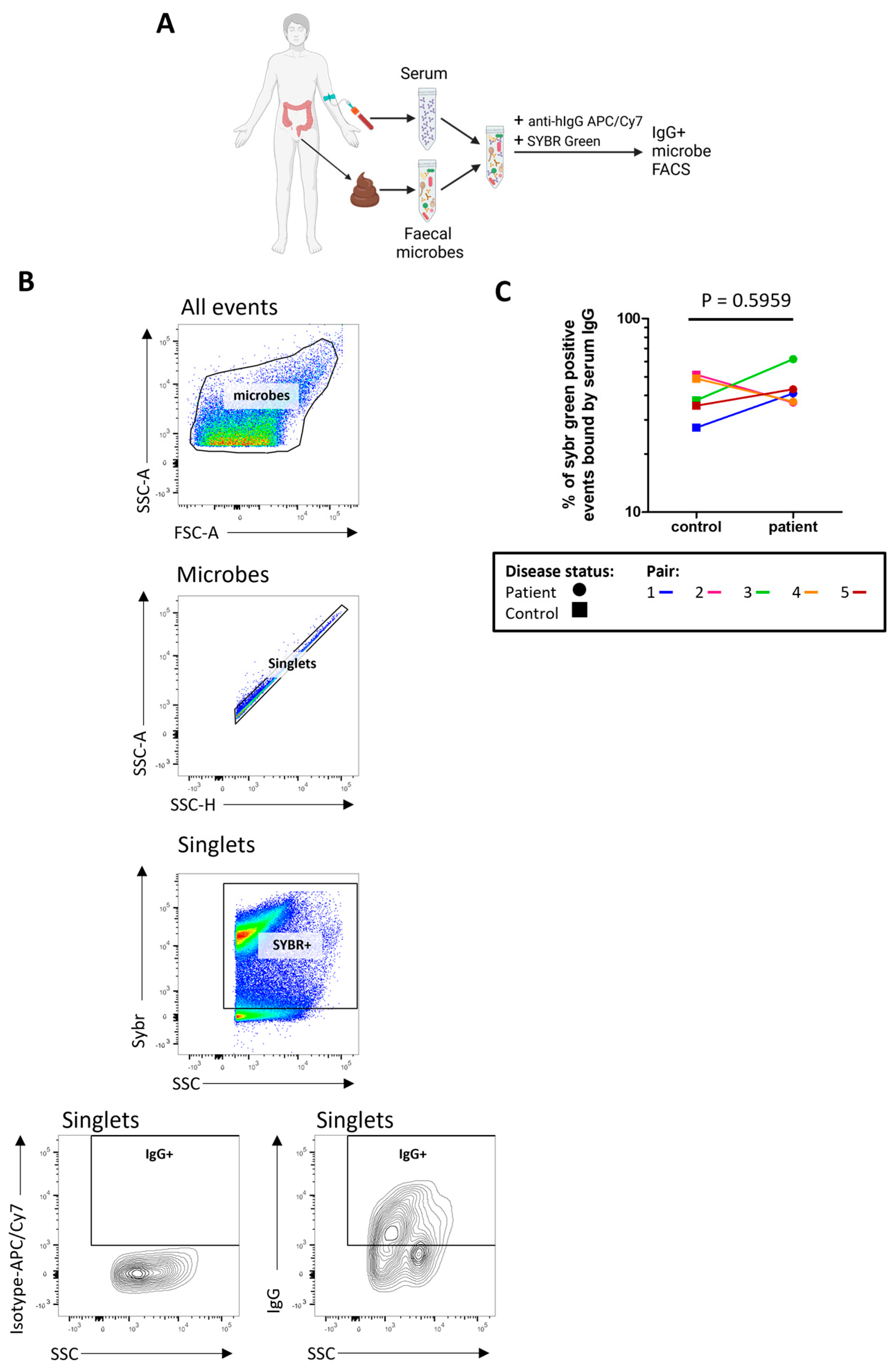
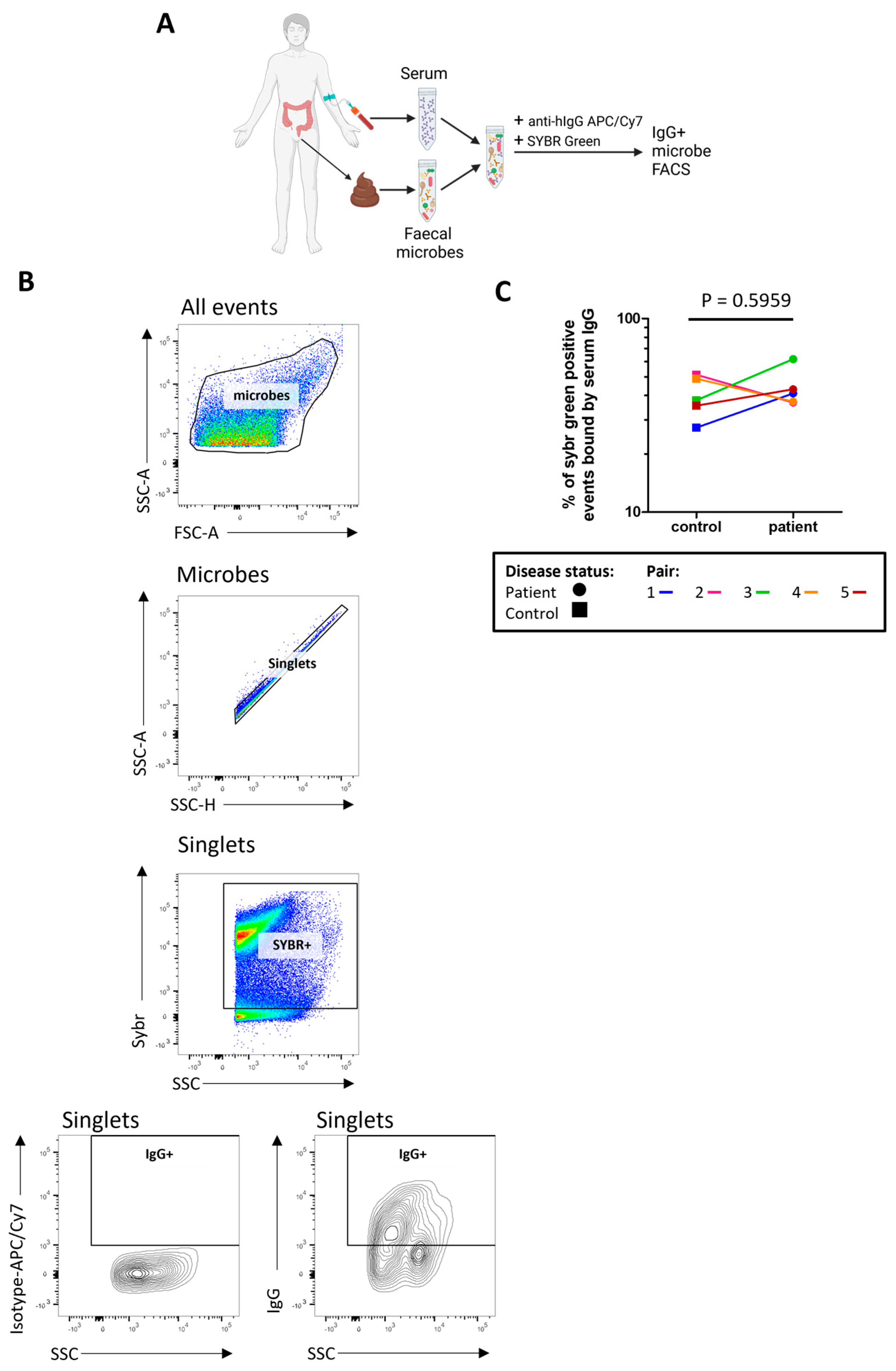
2.6. The Proportion of Microbes Bound by Serum IgG Is High in Both Patients and Controls
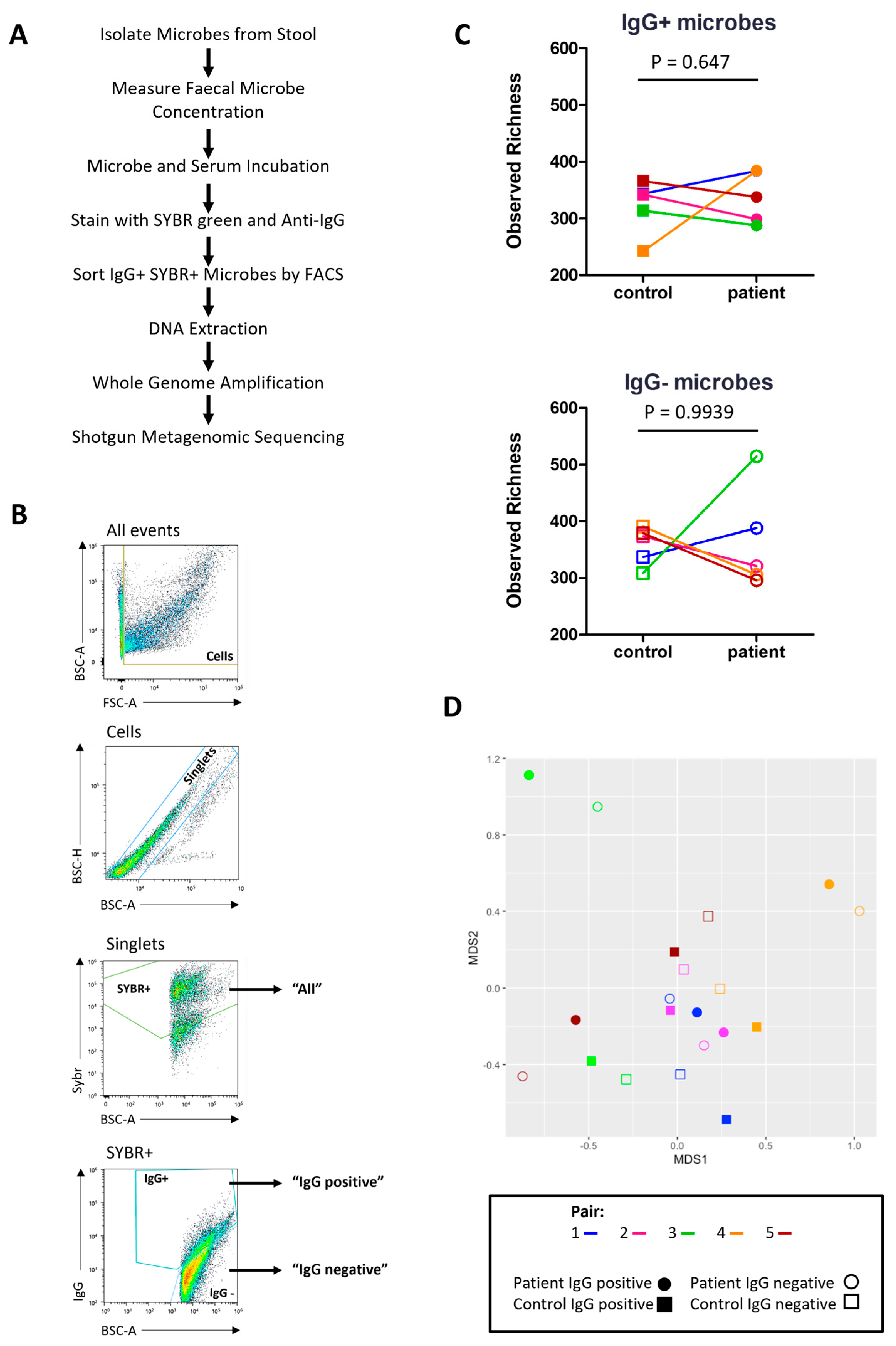
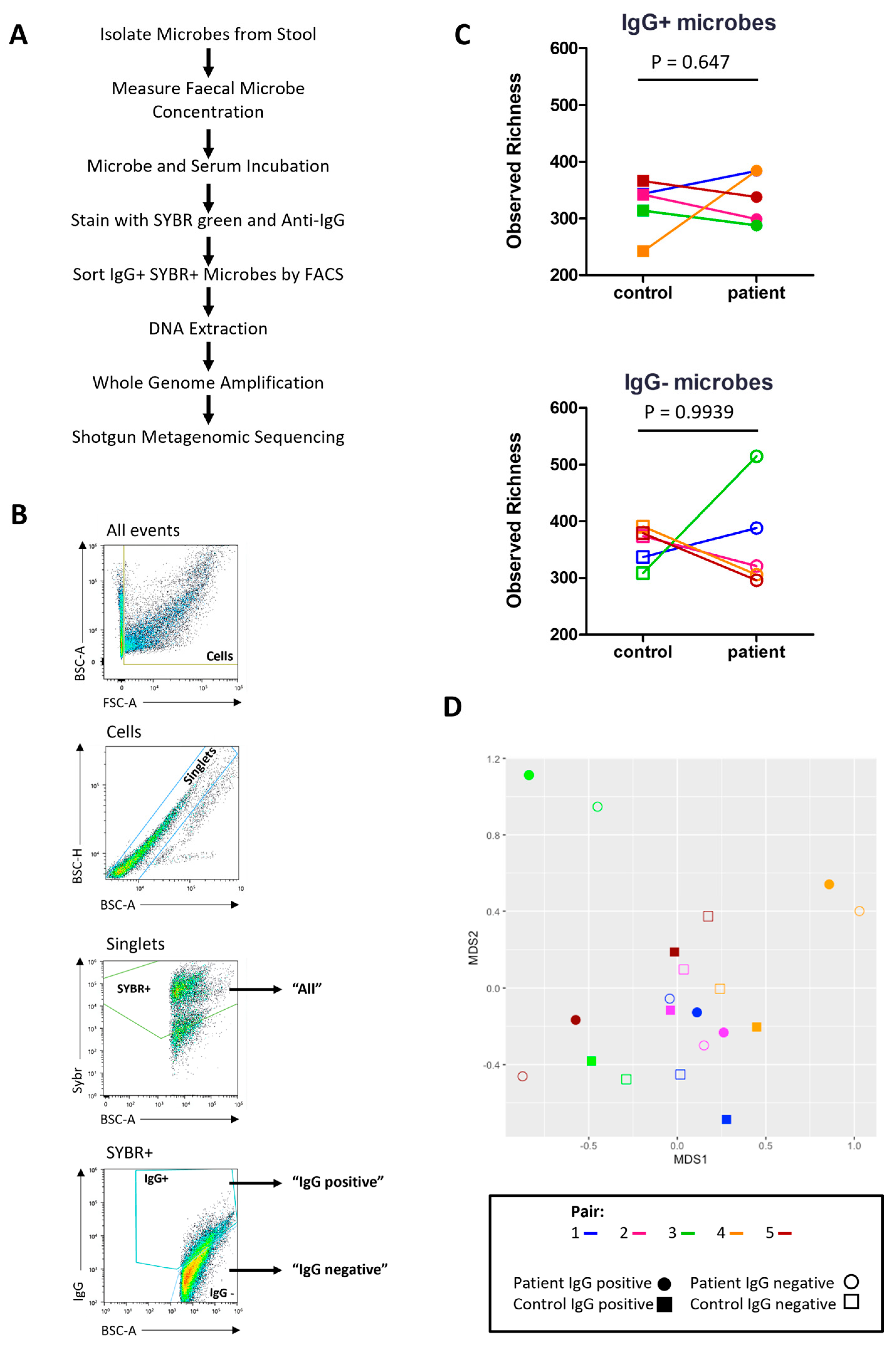
2.7. Characterising the Stool Microbiome
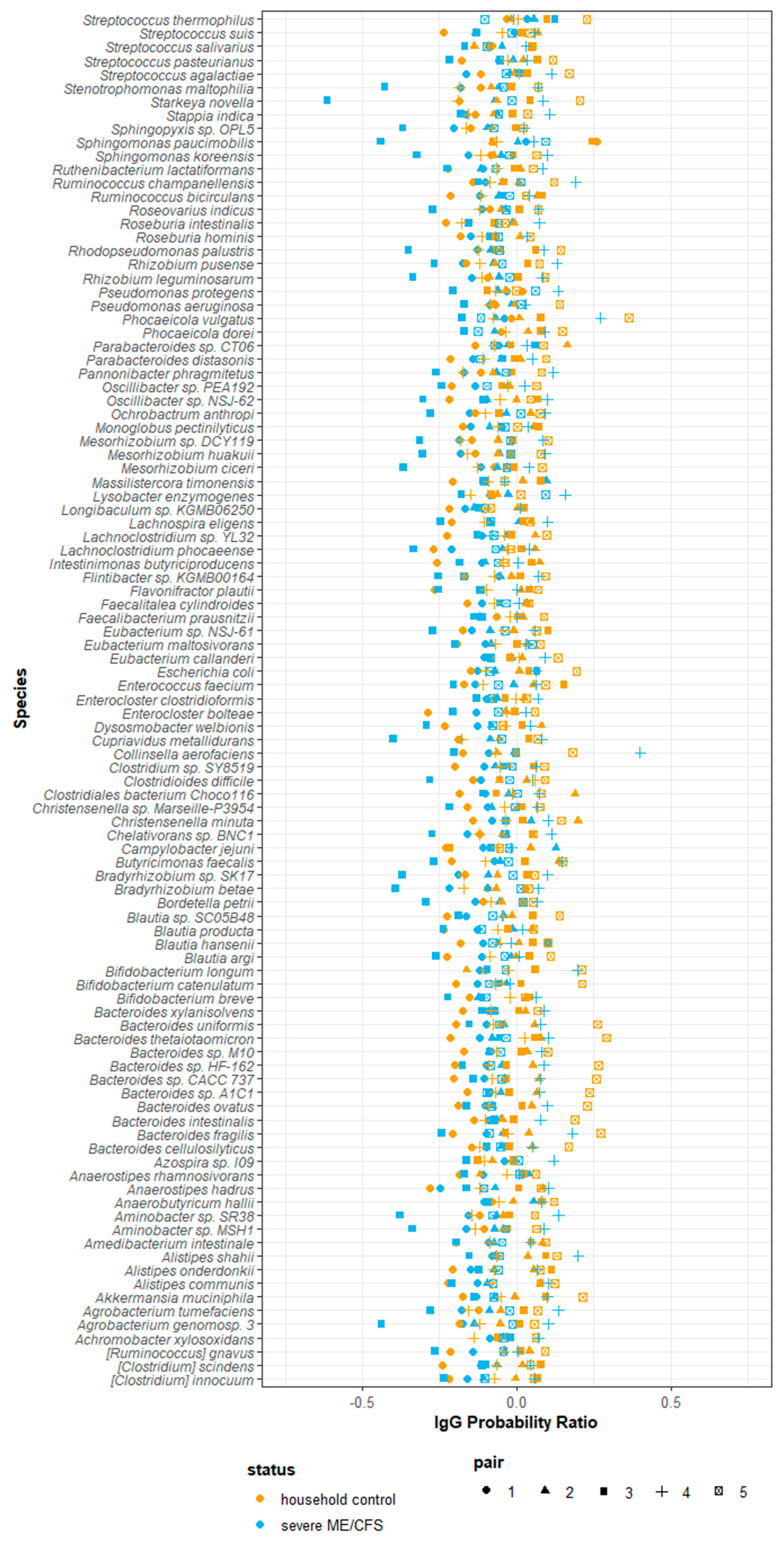
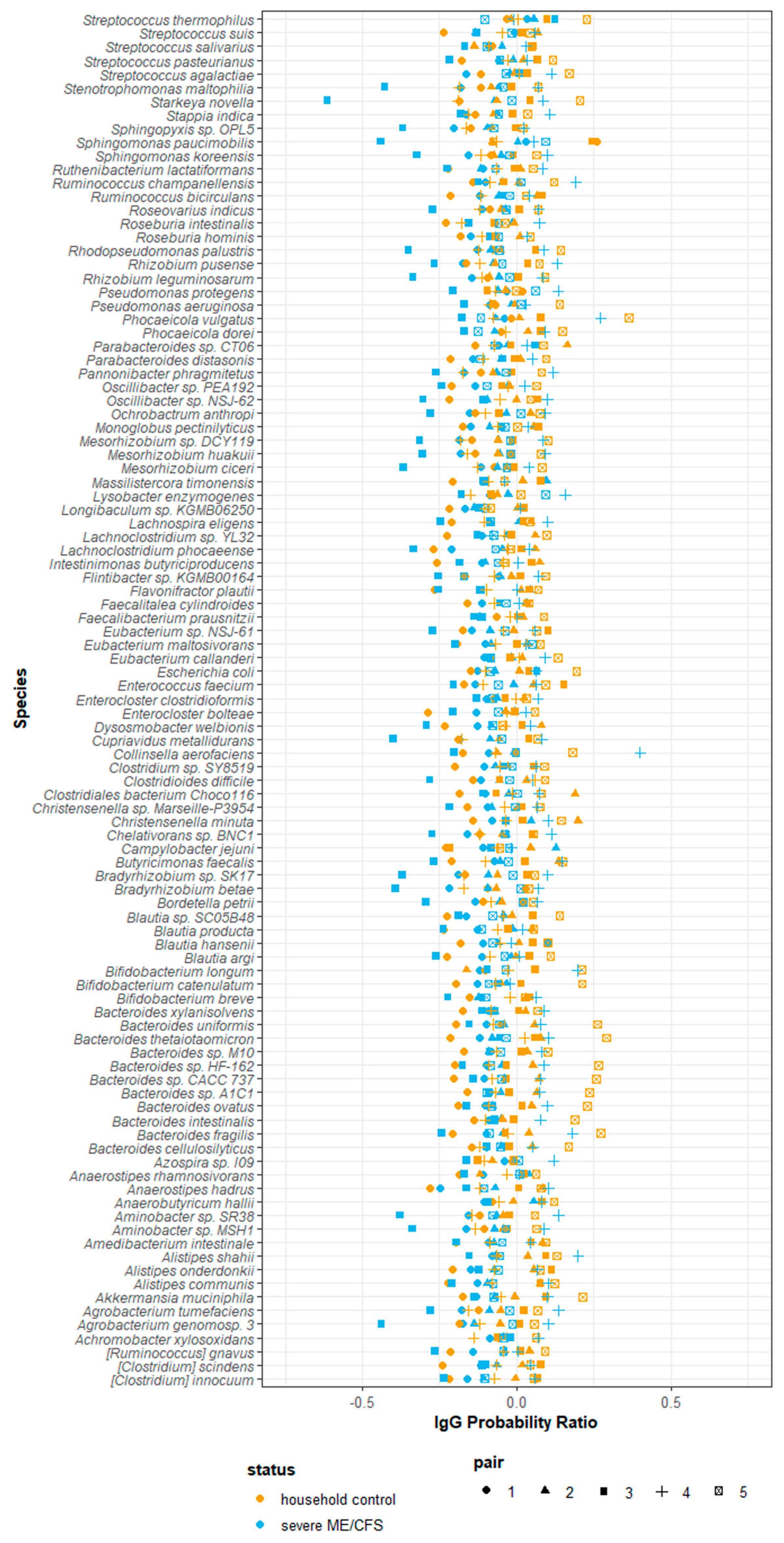
2.8. IgG-Seq Identifies Antimicrobial Signatures Unique to Each Participant
3. Discussion
3.1. Lessons Learnt for the Inclusion of Severe ME/CFS Patients in Biomedical Research
3.2. Severe ME/CFS Patients Have Serum IgG Hyporeactivity to Stool Microbes
3.3. Limitations of this Pilot Study in Addressing the Study Hypothesis
3.4. Conclusions
4. Materials and Methods
4.1. Participant Recruitment
4.2. Sample Collection and Processing
4.3. Stool Water Content
4.4. Stool Microbial Load
4.5. Stool IgA Concentration
4.6. Serum IgG Quantification
4.7. Serum IgG Levels to Autologous and Heterologous Stool Microbes
4.8. Microbial Flow Cytometry
4.9. Metagenomic Shotgun Sequencing Analysis of Total Gut Microbiota and IgG-Coated Microbes (IgG-Seq)
4.9.1. Microbial Cell Sorting
4.9.2. DNA Extraction, Processing and Sequencing
4.9.3. Relative and Quantitative Microbiome Profiling
4.9.4. Analysing IgG Binding of Taxa
4.9.5. Alpha Diversity
4.9.6. Beta Diversity
4.9.7. Functional Analysis
4.10. Statistical Analysis
4.11. Common Data Elements for ME Research
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lim, E.J.; Son, C.G. Review of case definitions for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J. Transl. Med. 2020, 18, 289. [Google Scholar] [CrossRef] [PubMed]
- Pendergrast, T.; Brown, A.; Sunnquist, M.; Jantke, R.; Newton, J.L.; Strand, E.B.; Jason, L.A. Housebound versus nonhousebound patients with myalgic encephalomyelitis and chronic fatigue syndrome. Chronic Illn. 2016, 12, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Valencia, I.J.; Garvert, D.W.; Montoya, J.G. Onset Patterns and Course of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Pediatr. 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Ahn, Y.C.; Jang, E.S.; Lee, S.W.; Lee, S.H.; Son, C.G. Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). J. Transl. Med. 2020, 18, 100. [Google Scholar] [CrossRef]
- Salari, N.; Khodayari, Y.; Hosseinian-Far, A.; Zarei, H.; Rasoulpoor, S.; Akbari, H.; Mohammadi, M. Global prevalence of chronic fatigue syndrome among long COVID-19 patients: A systematic review and meta-analysis. Biopsychosoc. Med. 2022, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Missailidis, D.; Annesley, S.J.; Fisher, P.R. Pathological Mechanisms Underlying Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Diagnostics 2019, 9, 80. [Google Scholar] [CrossRef]
- Aaron, L.A.; Burke, M.M.; Buchwald, D. Overlapping conditions among patients with chronic fatigue syndrome, fibromyalgia, and temporomandibular disorder. Arch. Intern. Med. 2000, 160, 221–227. [Google Scholar] [CrossRef]
- Giloteaux, L.; Goodrich, J.K.; Walters, W.A.; Levine, S.M.; Ley, R.E.; Hanson, M.R. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 2016, 4, 30. [Google Scholar] [CrossRef]
- Nagy-Szakal, D.; Williams, B.L.; Mishra, N.; Che, X.; Lee, B.; Bateman, L.; Klimas, N.G.; Komaroff, A.L.; Levine, S.; Montoya, J.G.; et al. Fecal metagenomic profiles in subgroups of patients with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 2017, 5, 44. [Google Scholar] [CrossRef]
- Armstrong, C.W.; McGregor, N.R.; Lewis, D.P.; Butt, H.L.; Gooley, P.R. The association of fecal microbiota and fecal, blood serum and urine metabolites in myalgic encephalomyelitis/chronic fatigue syndrome. Metabolomics 2016, 13, 8. [Google Scholar] [CrossRef]
- Guo, C.; Che, X.; Briese, T.; Ranjan, A.; Allicock, O.; Yates, R.A.; Cheng, A.; March, D.; Hornig, M.; Komaroff, A.L.; et al. Deficient butyrate-producing capacity in the gut microbiome is associated with bacterial network disturbances and fatigue symptoms in ME/CFS. Cell Host Microbe 2023, 31, 288–304.e288. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Gunter, C.; Fleming, E.; Vernon, S.D.; Bateman, L.; Unutmaz, D.; Oh, J. Multi-’omics of gut microbiome-host interactions in short- and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients. Cell Host Microbe 2023, 31, 273–287.e275. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Mihaylova, I.; Leunis, J.C. Increased serum IgA and IgM against LPS of enterobacteria in chronic fatigue syndrome (CFS): Indication for the involvement of gram-negative enterobacteria in the etiology of CFS and for the presence of an increased gut-intestinal permeability. J. Affect. Disord. 2007, 99, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Cook, D.; Meyer, J.; Vernon, S.D.; Le, T.; Clevidence, D.; Robertson, C.E.; Schrodi, S.J.; Yale, S.; Frank, D.N. Changes in Gut and Plasma Microbiome following Exercise Challenge in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). PLoS ONE 2015, 10, e0145453. [Google Scholar] [CrossRef] [PubMed]
- Duchmann, R.; Neurath, M.F.; Meyer zum Büschenfelde, K.H. Responses to self and non-self intestinal microflora in health and inflammatory bowel disease. Res. Immunol. 1997, 148, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, H.J.; Pouwels, S.D.; Funke, A.; Bos, N.A.; Dijkstra, G. Crohn’s disease patients have more IgG-binding fecal bacteria than controls. Clin. Vaccine Immunol. 2012, 19, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Ruff, W.E.; Greiling, T.M.; Kriegel, M.A. Host-microbiota interactions in immune-mediated diseases. Nat. Rev. Microbiol. 2020, 18, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Ringel, K.; Kubera, M.; Anderson, G.; Morris, G.; Galecki, P.; Geffard, M. In myalgic encephalomyelitis/chronic fatigue syndrome, increased autoimmune activity against 5-HT is associated with immuno-inflammatory pathways and bacterial translocation. J. Affect. Disord. 2013, 150, 223–230. [Google Scholar] [CrossRef]
- Quigley, E.M. Gut bacteria in health and disease. Gastroenterol. Hepatol. 2013, 9, 560–569. [Google Scholar]
- Keyt, B.A.; Baliga, R.; Sinclair, A.M.; Carroll, S.F.; Peterson, M.S. Structure, Function, and Therapeutic Use of IgM Antibodies. Antibodies 2020, 9, 53. [Google Scholar] [CrossRef]
- de Sousa-Pereira, P.; Woof, J.M. IgA: Structure, Function, and Developability. Antibodies 2019, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Kalka, I.N.; Klompus, S.; Leviatan, S.; Weinberger, A.; Segal, E. Systemic antibody responses against human microbiota flagellins are overrepresented in chronic fatigue syndrome patients. Sci. Adv. 2022, 8, eabq2422. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Klompus, S.; Leviatan, S.; Kalka, I.N.; Weinberger, A.; Wijmenga, C.; Fu, J.; Zhernakova, A.; Weersma, R.K.; Segal, E. Population-wide diversity and stability of serum antibody epitope repertoires against human microbiota. Nat. Med. 2021, 27, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Conrey, P.E.; Denu, L.; O’Boyle, K.C.; Rozich, I.; Green, J.; Maslanka, J.; Lubin, J.B.; Duranova, T.; Haltzman, B.L.; Gianchetti, L.; et al. IgA deficiency destabilizes homeostasis toward intestinal microbes and increases systemic immune dysregulation. Sci. Immunol. 2023, 8, eade2335. [Google Scholar] [CrossRef] [PubMed]
- Doron, I.; Leonardi, I.; Li, X.V.; Fiers, W.D.; Semon, A.; Bialt-DeCelie, M.; Migaud, M.; Gao, I.H.; Lin, W.Y.; Kusakabe, T.; et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell 2021, 184, 1017–1031.e1014. [Google Scholar] [CrossRef] [PubMed]
- Fadlallah, J.; Sterlin, D.; Fieschi, C.; Parizot, C.; Dorgham, K.; El Kafsi, H.; Autaa, G.; Ghillani-Dalbin, P.; Juste, C.; Lepage, P.; et al. Synergistic convergence of microbiota-specific systemic IgG and secretory IgA. J. Allergy Clin. Immunol. 2019, 143, 1575–1585.e1574. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Reiner, G.L.; Lugo, K.A.; Kreuk, L.S.; Stanbery, A.G.; Ansaldo, E.; Seher, T.D.; Ludington, W.B.; Barton, G.M. Maternal IgG and IgA Antibodies Dampen Mucosal T Helper Cell Responses in Early Life. Cell 2016, 165, 827–841. [Google Scholar] [CrossRef]
- Rengarajan, S.; Knoop, K.A.; Rengarajan, A.; Chai, J.N.; Grajales-Reyes, J.G.; Samineni, V.K.; Russler-Germain, E.V.; Ranganathan, P.; Fasano, A.; Sayuk, G.S.; et al. A Potential Role for Stress-Induced Microbial Alterations in IgA-Associated Irritable Bowel Syndrome with Diarrhea. Cell Rep. Med. 2020, 1, 100124. [Google Scholar] [CrossRef]
- Sterlin, D.; Larsen, M.; Fadlallah, J.; Parizot, C.; Vignes, M.; Autaa, G.; Dorgham, K.; Juste, C.; Lepage, P.; Aboab, J.; et al. Perturbed Microbiota/Immune Homeostasis in Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e997. [Google Scholar] [CrossRef]
- Vujkovic-Cvijin, I.; Welles, H.C.; Ha, C.W.Y.; Huq, L.; Mistry, S.; Brenchley, J.M.; Trinchieri, G.; Devkota, S.; Belkaid, Y. The systemic anti-microbiota IgG repertoire can identify gut bacteria that translocate across gut barrier surfaces. Sci. Transl. Med. 2022, 14, eabl3927. [Google Scholar] [CrossRef]
- Jackson, M.A.; Pearson, C.; Ilott, N.E.; Huus, K.E.; Hegazy, A.N.; Webber, J.; Finlay, B.B.; Macpherson, A.J.; Powrie, F.; Lam, L.H. Accurate identification and quantification of commensal microbiota bound by host immunoglobulins. Microbiome 2021, 9, 33. [Google Scholar] [CrossRef]
- Gacesa, R.; Kurilshikov, A.; Vich Vila, A.; Sinha, T.; Klaassen, M.A.Y.; Bolte, L.A.; Andreu-Sánchez, S.; Chen, L.; Collij, V.; Hu, S.; et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature 2022, 604, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Useros, N.; Nova, E.; González-Zancada, N.; Díaz, L.E.; Gómez-Martínez, S.; Marcos, A. Microbiota and Lifestyle: A Special Focus on Diet. Nutrients 2020, 12, 1776. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.F.D.; Rocchetti, G.; Lucini, L.; Lorusso, L.; Manara, E.; Bertelli, M.; Puglisi, E.; Capelli, E. Potential role of microbiome in Chronic Fatigue Syndrome/Myalgic Encephalomyelits (CFS/ME). Sci. Rep. 2021, 11, 7043. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Milan-Mattos, J.C.; Anibal, F.F.; Perseguini, N.M.; Minatel, V.; Rehder-Santos, P.; Castro, C.A.; Vasilceac, F.A.; Mattiello, S.M.; Faccioli, L.H.; Catai, A.M. Effects of natural aging and gender on pro-inflammatory markers. Braz. J. Med. Biol. Res. 2019, 52, e8392. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic-Cvijin, I.; Sklar, J.; Jiang, L.; Natarajan, L.; Knight, R.; Belkaid, Y. Host variables confound gut microbiota studies of human disease. Nature 2020, 587, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Lax, S.; Smith, D.P.; Hampton-Marcell, J.; Owens, S.M.; Handley, K.M.; Scott, N.M.; Gibbons, S.M.; Larsen, P.; Shogan, B.D.; Weiss, S.; et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 2014, 345, 1048–1052. [Google Scholar] [CrossRef]
- Guenther, S.; Loebel, M.; Mooslechner, A.A.; Knops, M.; Hanitsch, L.G.; Grabowski, P.; Wittke, K.; Meisel, C.; Unterwalder, N.; Volk, H.D.; et al. Frequent IgG subclass and mannose binding lectin deficiency in patients with chronic fatigue syndrome. Hum. Immunol. 2015, 76, 729–735. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef]
- Eaton-Fitch, N.; du Preez, S.; Cabanas, H.; Staines, D.; Marshall-Gradisnik, S. A systematic review of natural killer cells profile and cytotoxic function in myalgic encephalomyelitis/chronic fatigue syndrome. Syst. Rev. 2019, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; Bright, A.T.; Alaynick, W.A.; Wang, L.; Baxter, A.; Nathan, N.; Anderson, W.; Gordon, E. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E5472–E5480. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M.; Berk, M.; Puri, B.K. Myalgic encephalomyelitis or chronic fatigue syndrome: How could the illness develop? Metab. Brain Dis. 2019, 34, 385–415. [Google Scholar] [CrossRef]
- Schlomann, B.H.; Parthasarathy, R. Timescales of gut microbiome dynamics. Curr. Opin. Microbiol. 2019, 50, 56–63. [Google Scholar] [CrossRef]
- Baker, R.; Shaw, E.J. Diagnosis and management of chronic fatigue syndrome or myalgic encephalomyelitis (or encephalopathy): Summary of NICE guidance. BMJ 2007, 335, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Straus, S.E.; Hickie, I.; Sharpe, M.C.; Dobbins, J.G.; Komaroff, A. The chronic fatigue syndrome: A comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann. Intern. Med. 1994, 121, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.J.; Heaton, K.W. Stool form scale as a useful guide to intestinal transit time. Scand. J. Gastroenterol. 1997, 32, 920–924. [Google Scholar] [CrossRef]
- Grüning, B.; Dale, R.; Sjödin, A.; Chapman, B.A.; Rowe, J.; Tomkins-Tinch, C.H.; Valieris, R.; Köster, J. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 2018, 15, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Telatin, A.; Fariselli, P.; Birolo, G. SeqFu: A Suite of Utilities for the Robust and Reproducible Manipulation of Sequence Files. Bioengineering 2021, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. vegan: Community Ecology Package, R package version v.2.6-4; 2022. Available online: https://cran.r-project.org/web/packages/vegan/ (accessed on 21 March 2023).
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef] [PubMed]
- McIver, L.J.; Abu-Ali, G.; Franzosa, E.A.; Schwager, R.; Morgan, X.C.; Waldron, L.; Segata, N.; Huttenhower, C. bioBakery: A meta’omic analysis environment. Bioinformatics 2018, 34, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaart, K.G.; Tolosana-Delgado, R.; van den Boogaart, M.B.K.G. compositions: Compositional Data Analysis, R package version 2.0-6; 2023. Available online: https://cran.r-project.org/web/packages/compositions/ (accessed on 21 March 2023).
- Rohart, F.; Gautier, B.; Singh, A.; KA, L.C. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wickham, H. Reshaping Data with the reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Dowle, M.; Srinivasan, A.; Short, T. data.table: Extension of ‘data.frame’, R package version 1.14.6; 2022. Available online: https://cran.r-project.org/web/packages/data.table/ (accessed on 21 March 2023).
- Wickham, H.; François, R.; Henry, L.; Müller, K.; Vaughan, D. dplyr: A Grammar of Data Manipulation, R package version 1.1.0; 2023. Available online: https://cran.r-project.org/web/packages/dplyr/ (accessed on 21 March 2023).
- Kassambara, A. ggpubr: ‘ggplot2′ Based Publication Ready Plots, R package version 0.6.0; 2023. Available online: https://cran.r-project.org/web/packages/ggpubr/index.html (accessed on 21 March 2023).
- Grinnon, S.T.; Miller, K.; Marler, J.R.; Lu, Y.; Stout, A.; Odenkirchen, J.; Kunitz, S. National Institute of Neurological Disorders and Stroke Common Data Element Project—Approach and methods. Clin. Trials 2012, 9, 322–329. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Participants Affected (%) | Mean (SD) | Range | |
|---|---|---|---|
| Age of ME/CFS onset (years) | - | 25.0 (9.34) | 12–38 |
| Length of ME/CFS (years) | - | 8.4 (6.83) | 2–21 |
| Symptoms | |||
| Post exertional malaise | 100 | - | - |
| Non-restorative sleep | 100 | - | - |
| Headaches of a new onset, pattern and severity | 80 | - | - |
| Recurrent sore throat with enlarged glands in neck | 40 | - | - |
| Impaired concentration | 100 | - | - |
| Impaired memory | 80 | - | - |
| Joint pain | 60 | - | - |
| Muscle pain | 80 | - | - |
| Visual and/or auditory hypersensitivity | 100 | - | - |
| Irritable bowel syndrome | 100 | - | - |
| Questionnaire (maximum score) | |||
| Shortened SF-36 (30) * | - | 11.0 (1.73) | 10–14 |
| Chalder fatigue–physical (28) * | - | 24.0 (3.67) | 18–27 |
| Chalder fatigue–mental (16) * | - | 13.5 (1.12) | 12–15 |
| HADS–anxiety (21) * | - | 7.8 (4.55) | 3–15 |
| HADS–depression (21) * | - | 6.5 (5.50) | 3–16 |
| Self-efficacy (60) ** | - | 12.0 (7.35) | 3–21 |
| Visual analogue (100) * | - | 62.5 (36.31) | 0–90 |
| Epworth sleepiness (24) *** | - | 7.5 (14) | 1–14 |
| Pair | Participant | Number of Species Detected per Fraction | Proportion of Species IgG+ | ||
|---|---|---|---|---|---|
| Only IgG− | Only IgG+ | Both IgG− and IgG+ | |||
| 1 | Patient | 7 | 7 | 201 | 0.967 |
| Control | 23 | 7 | 173 | 0.887 | |
| 2 | Patient | 18 | 7 | 190 | 0.916 |
| Control | 21 | 6 | 202 | 0.908 | |
| 3 | Patient | 107 | 4 | 154 | 0.596 |
| Control | 14 | 12 | 145 | 0.918 | |
| 4 | Patient | 4 | 19 | 138 | 0.975 |
| Control | 29 | 6 | 168 | 0.857 | |
| 5 | Patient | 9 | 18 | 162 | 0.952 |
| Control | 15 | 31 | 206 | 0.940 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seton, K.A.; Defernez, M.; Telatin, A.; Tiwari, S.K.; Savva, G.M.; Hayhoe, A.; Noble, A.; de Carvalho-KoK, A.L.S.; James, S.A.; Bansal, A.; et al. Investigating Antibody Reactivity to the Intestinal Microbiome in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Feasibility Study. Int. J. Mol. Sci. 2023, 24, 15316. https://doi.org/10.3390/ijms242015316
Seton KA, Defernez M, Telatin A, Tiwari SK, Savva GM, Hayhoe A, Noble A, de Carvalho-KoK ALS, James SA, Bansal A, et al. Investigating Antibody Reactivity to the Intestinal Microbiome in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Feasibility Study. International Journal of Molecular Sciences. 2023; 24(20):15316. https://doi.org/10.3390/ijms242015316
Chicago/Turabian StyleSeton, Katharine A., Marianne Defernez, Andrea Telatin, Sumeet K. Tiwari, George M. Savva, Antonietta Hayhoe, Alistair Noble, Ana L. S. de Carvalho-KoK, Steve A. James, Amolak Bansal, and et al. 2023. "Investigating Antibody Reactivity to the Intestinal Microbiome in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Feasibility Study" International Journal of Molecular Sciences 24, no. 20: 15316. https://doi.org/10.3390/ijms242015316
APA StyleSeton, K. A., Defernez, M., Telatin, A., Tiwari, S. K., Savva, G. M., Hayhoe, A., Noble, A., de Carvalho-KoK, A. L. S., James, S. A., Bansal, A., Wileman, T., & Carding, S. R. (2023). Investigating Antibody Reactivity to the Intestinal Microbiome in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Feasibility Study. International Journal of Molecular Sciences, 24(20), 15316. https://doi.org/10.3390/ijms242015316