
Effects of Mitochondrial Transplantation on Transcriptomics in a Polymicrobial Sepsis Model
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
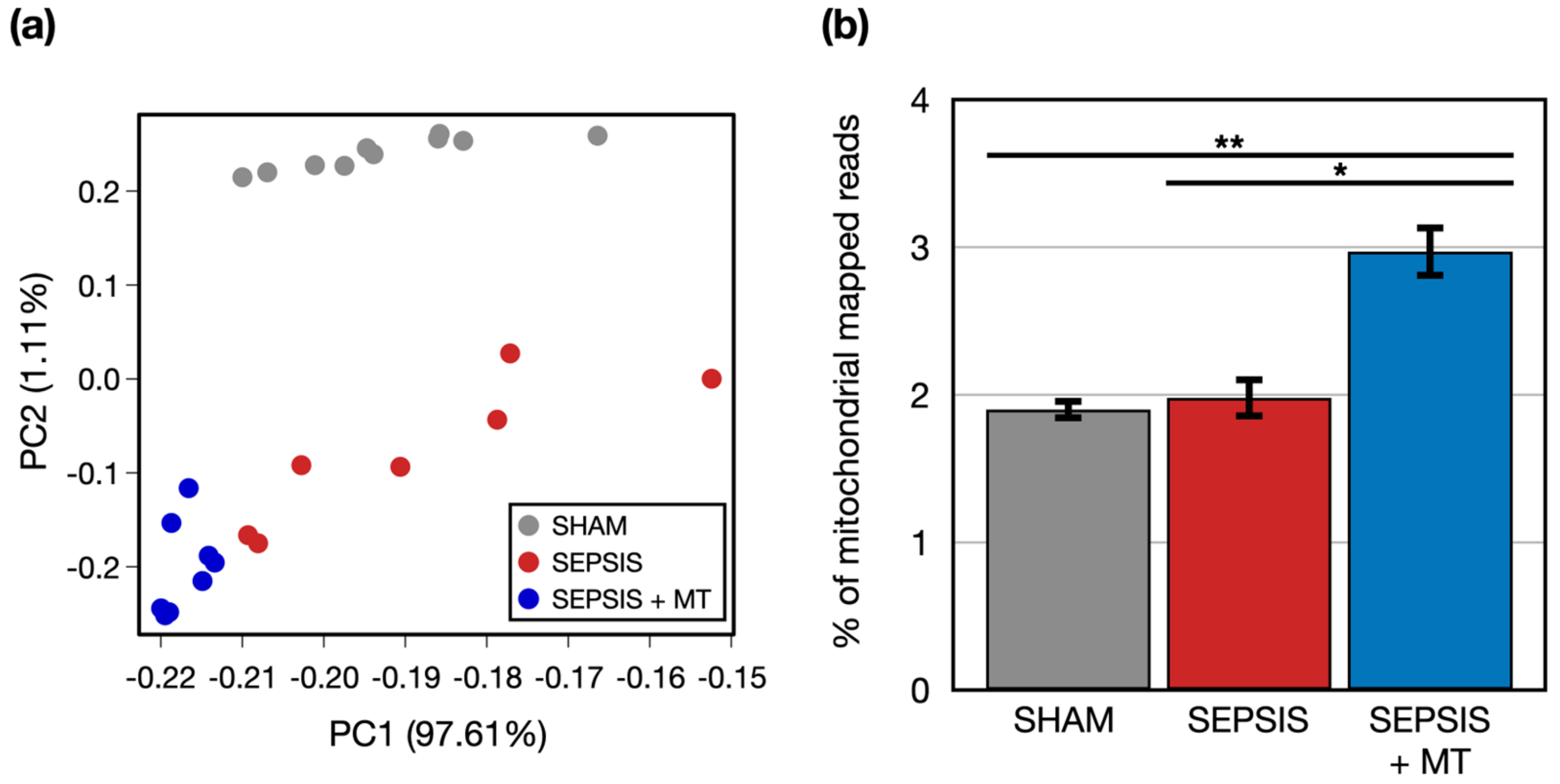
2.1. Transcriptome Analysis Based on RNA-Seq for Understanding the Effects of Mitochondrial Transplantation in Sepsis
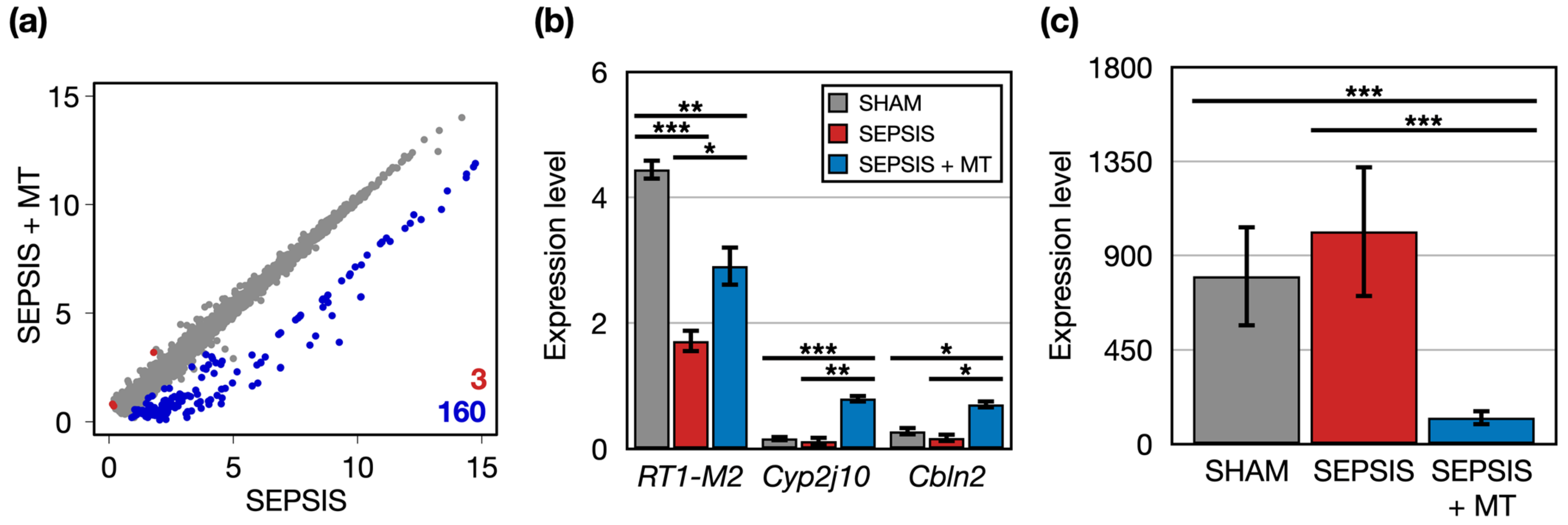
2.2. Altered Gene Expression Patterns in Response to Mitochondrial Transplantation
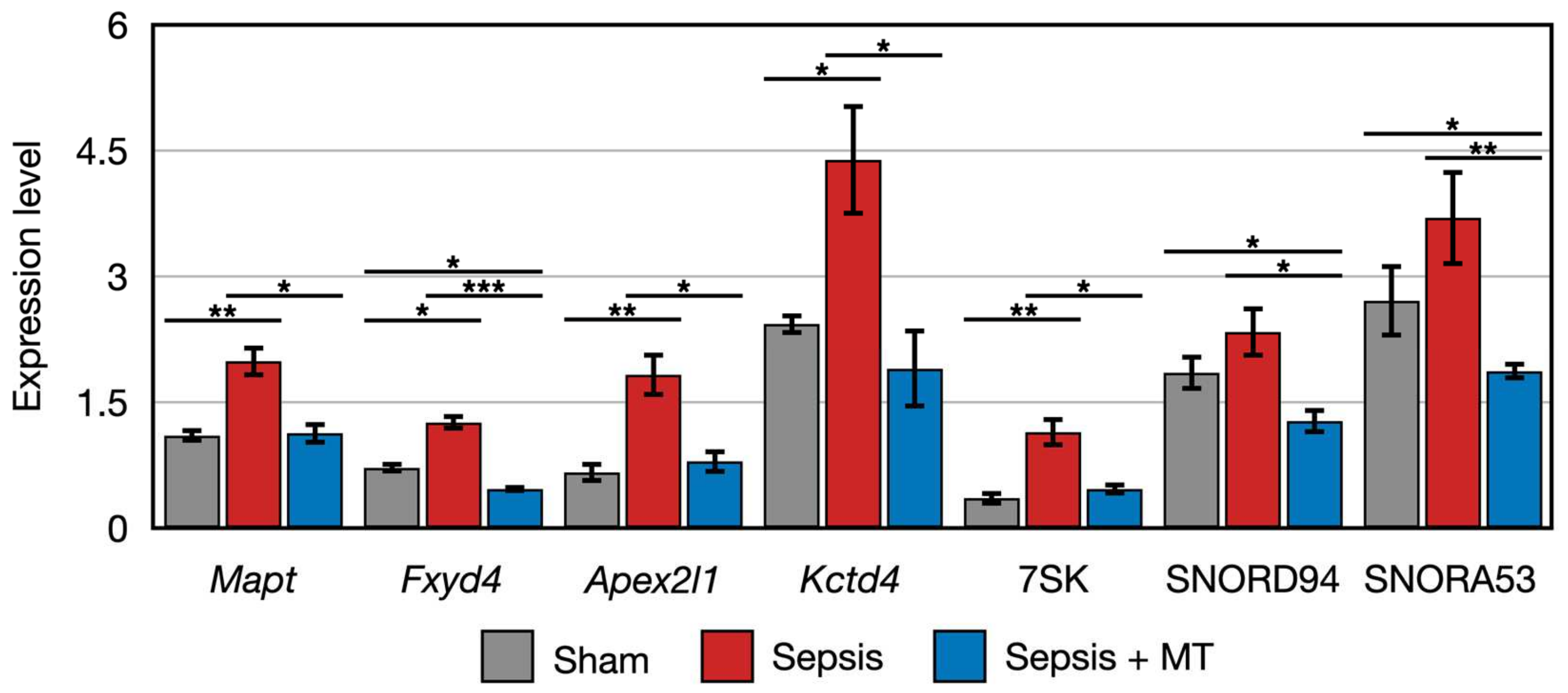
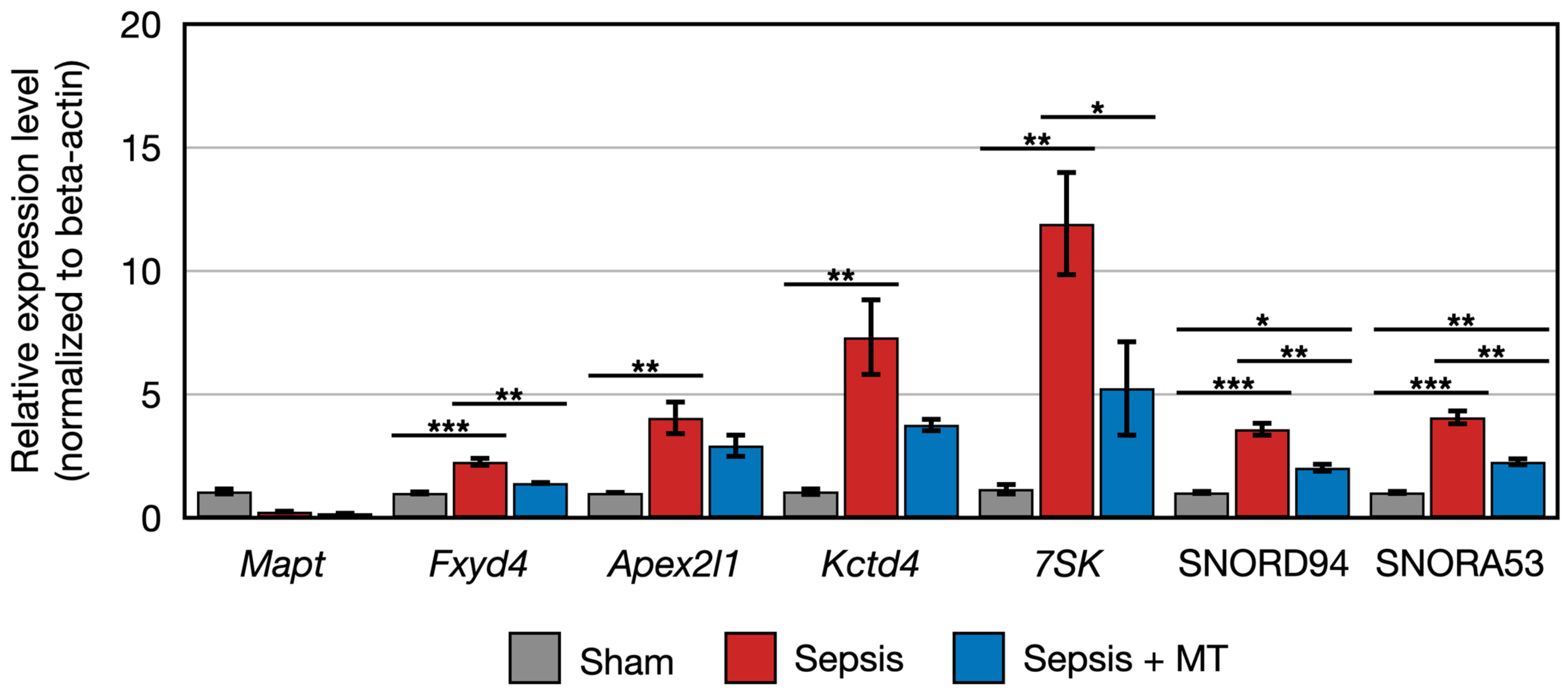
2.3. Quantitative PCR Validation of Transcriptome Sequencing Data
3. Discussion
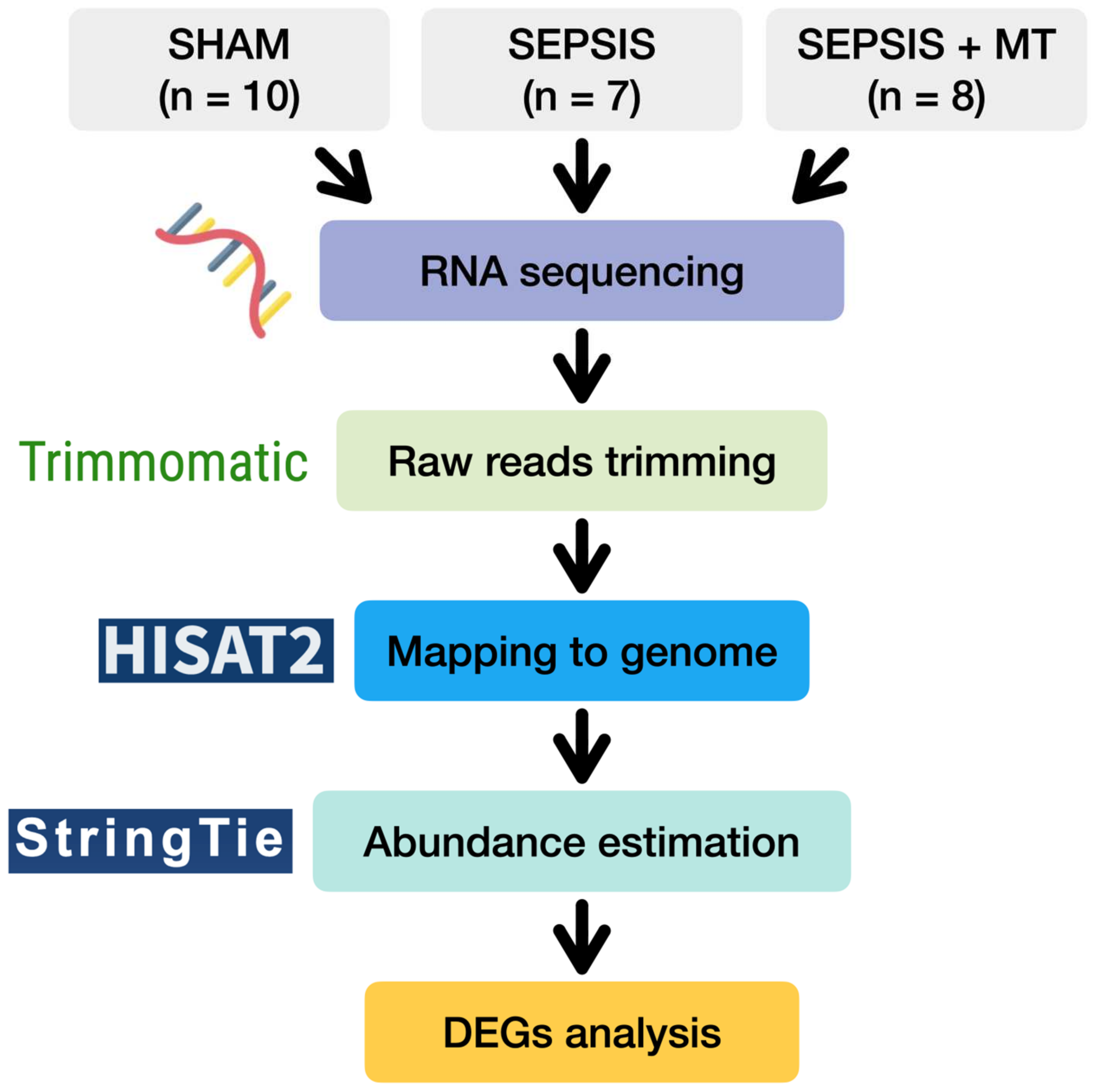
4. Materials and Methods
4.1. Mitochondrial Isolation from L6 Cell Lines
4.2. In Vivo Sepsis Model Induction
4.3. RNA Extraction and Sequencing
4.4. Genome-Wide Transcriptome Analysis
4.5. Quantitative PCR (qPCR)
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Ulger, O.; Kubat, G.B. Therapeutic applications of mitochondrial transplantation. Biochimie 2022, 195, 1–15. [Google Scholar] [CrossRef]
- Cloer, C.M.; Givens, C.S.; Buie, L.K.; Rochelle, L.K.; Lin, Y.T.; Popa, S.; Shelton, R.V.M.; Zhan, J.; Zimmerman, T.R.; Jones, B.G.; et al. Mitochondrial transplant after ischemia reperfusion promotes cellular salvage and improves lung function during ex-vivo lung perfusion. J. Heart Lung Transplant. 2023, 42, 575–584. [Google Scholar] [CrossRef]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef]
- Lee, J.M.; Hwang, J.W.; Kim, M.J.; Jung, S.Y.; Kim, K.S.; Ahn, E.H.; Min, K.; Choi, Y.S. Mitochondrial Transplantation Modulates Inflammation and Apoptosis, Alleviating Tendinopathy Both In Vivo and In Vitro. Antioxidants 2021, 10, 696. [Google Scholar] [CrossRef]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef]
- Mokhtari, B.; Yavari, R.; Badalzadeh, R.; Mahmoodpoor, A. An Overview on Mitochondrial-Based Therapies in Sepsis-Related Myocardial Dysfunction: Mitochondrial Transplantation as a Promising Approach. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 3277274. [Google Scholar] [CrossRef]
- Park, A.; Oh, M.; Lee, S.J.; Oh, K.J.; Lee, E.W.; Lee, S.C.; Bae, K.H.; Han, B.S.; Kim, W.K. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 4793. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Ma, Z.; Ma, H.; Li, Q.; Zhai, Q.; Jiang, T.; Zhang, Z.; Wang, Q. Mitochondrial Transplantation Attenuates Brain Dysfunction in Sepsis by Driving Microglial M2 Polarization. Mol. Neurobiol. 2020, 57, 3875–3890. [Google Scholar] [CrossRef]
- Hwang, J.W.; Lee, M.J.; Chung, T.N.; Lee, H.A.R.; Lee, J.H.; Choi, S.Y.; Park, Y.J.; Kim, C.H.; Jin, I.; Kim, S.H.; et al. The immune modulatory effects of mitochondrial transplantation on cecal slurry model in rat. Crit. Care 2021, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Liang, M.Z.; Chen, L. Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl. Neurodegener. 2019, 8, 17. [Google Scholar] [CrossRef]
- Kim, K. Future of sepsis: Perspective on diagnosis. Clin. Exp. Emerg. Med. 2022, 9, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Nedel, W.; Deutschendorf, C.; Portela, L.V.C. Sepsis-induced mitochondrial dysfunction: A narrative review. World J. Crit. Care Med. 2023, 12, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Feng, Y.W.; Yao, Y.M. Potential therapy strategy: Targeting mitochondrial dysfunction in sepsis. Mil. Med. Res. 2018, 5, 41. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, H.A.R.; Lee, M.J.; Park, Y.J.; Mun, S.; Yune, C.J.; Chung, T.N.; Bae, J.; Kim, M.J.; Choi, Y.S.; et al. The Effects of Mitochondrial Transplantation on Sepsis Depend on the Type of Cell from Which They Are Isolated. Int. J. Mol. Sci. 2023, 24, 10113. [Google Scholar] [CrossRef]
- Davenport, E.E.; Burnham, K.L.; Radhakrishnan, J.; Humburg, P.; Hutton, P.; Mills, T.C.; Rautanen, A.; Gordon, A.C.; Garrard, C.; Hill, A.V.; et al. Genomic landscape of the individual host response and outcomes in sepsis: A prospective cohort study. Lancet Respir. Med. 2016, 4, 259–271. [Google Scholar] [CrossRef]
- Burnham, K.L.; Davenport, E.E.; Radhakrishnan, J.; Humburg, P.; Gordon, A.C.; Hutton, P.; Svoren-Jabalera, E.; Garrard, C.; Hill, A.V.S.; Hinds, C.J.; et al. Shared and Distinct Aspects of the Sepsis Transcriptomic Response to Fecal Peritonitis and Pneumonia. Am. J. Respir. Crit. Care Med. 2017, 196, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Scicluna, B.P.; van Vught, L.A.; Zwinderman, A.H.; Wiewel, M.A.; Davenport, E.E.; Burnham, K.L.; Nurnberg, P.; Schultz, M.J.; Horn, J.; Cremer, O.L.; et al. Classification of patients with sepsis according to blood genomic endotype: A prospective cohort study. Lancet Respir. Med. 2017, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.E.; Perumal, T.M.; Henao, R.; Nichols, M.; Howrylak, J.A.; Choi, A.M.; Bermejo-Martin, J.F.; Almansa, R.; Tamayo, E.; Davenport, E.E.; et al. A community approach to mortality prediction in sepsis via gene expression analysis. Nat. Commun. 2018, 9, 694. [Google Scholar] [CrossRef]
- Baghela, A.; Pena, O.M.; Lee, A.H.; Baquir, B.; Falsafi, R.; An, A.; Farmer, S.W.; Hurlburt, A.; Mondragon-Cardona, A.; Rivera, J.D.; et al. Predicting sepsis severity at first clinical presentation: The role of endotypes and mechanistic signatures. EBioMedicine 2022, 75, 103776. [Google Scholar] [CrossRef] [PubMed]
- Cano-Gamez, E.; Burnham, K.L.; Goh, C.; Allcock, A.; Malick, Z.H.; Overend, L.; Kwok, A.; Smith, D.A.; Peters-Sengers, H.; Antcliffe, D.; et al. An immune dysfunction score for stratification of patients with acute infection based on whole-blood gene expression. Sci. Transl. Med. 2022, 14, eabq4433. [Google Scholar] [CrossRef]
- Gabelein, C.G.; Feng, Q.; Sarajlic, E.; Zambelli, T.; Guillaume-Gentil, O.; Kornmann, B.; Vorholt, J.A. Mitochondria transplantation between living cells. PLoS Biol. 2022, 20, e3001576. [Google Scholar] [CrossRef]
- Wu, T.H.; Sagullo, E.; Case, D.; Zheng, X.; Li, Y.; Hong, J.S.; TeSlaa, T.; Patananan, A.N.; McCaffery, J.M.; Niazi, K.; et al. Mitochondrial Transfer by Photothermal Nanoblade Restores Metabolite Profile in Mammalian Cells. Cell Metab. 2016, 23, 921–929. [Google Scholar] [CrossRef]
- Sercel, A.J.; Patananan, A.N.; Man, T.; Wu, T.H.; Yu, A.K.; Guyot, G.W.; Rabizadeh, S.; Niazi, K.R.; Chiou, P.Y.; Teitell, M.A. Stable transplantation of human mitochondrial DNA by high-throughput, pressurized isolated mitochondrial delivery. eLife 2021, 10, e63102. [Google Scholar] [CrossRef]
- Yoon, Y.G.; Koob, M.D. Intramitochondrial transfer and engineering of mammalian mitochondrial genomes in yeast. Mitochondrion 2019, 46, 15–21. [Google Scholar] [CrossRef]
- Guo, L.; Shen, S.; Rowley, J.W.; Tolley, N.D.; Jia, W.; Manne, B.K.; McComas, K.N.; Bolingbroke, B.; Kosaka, Y.; Krauel, K.; et al. Platelet MHC class I mediates CD8+ T-cell suppression during sepsis. Blood 2021, 138, 401–416. [Google Scholar] [CrossRef]
- Muntjewerff, E.M.; Meesters, L.D.; van den Bogaart, G.; Revelo, N.H. Reverse Signaling by MHC-I Molecules in Immune and Non-Immune Cell Types. Front. Immunol. 2020, 11, 605958. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Alvaro-Benito, M.; Stolzenberg, S.; Noe, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef]
- Yoo, J.S.; Sasaki, M.; Cho, S.X.; Kasuga, Y.; Zhu, B.; Ouda, R.; Orba, Y.; de Figueiredo, P.; Sawa, H.; Kobayashi, K.S. SARS-CoV-2 inhibits induction of the MHC class I pathway by targeting the STAT1-IRF1-NLRC5 axis. Nat. Commun. 2021, 12, 6602. [Google Scholar] [CrossRef]
- Klocke, J.; Kim, S.J.; Skopnik, C.M.; Hinze, C.; Boltengagen, A.; Metzke, D.; Grothgar, E.; Prskalo, L.; Wagner, L.; Freund, P.; et al. Urinary single-cell sequencing captures kidney injury and repair processes in human acute kidney injury. Kidney Int. 2022, 102, 1359–1370. [Google Scholar] [CrossRef]
- Seigneur, E.; Wang, J.; Dai, J.; Polepalli, J.; Sudhof, T.C. Cerebellin-2 regulates a serotonergic dorsal raphe circuit that controls compulsive behaviors. Mol. Psychiatry 2021, 26, 7509–7521. [Google Scholar] [CrossRef]
- Seigneur, E.; Sudhof, T.C. Genetic Ablation of All Cerebellins Reveals Synapse Organizer Functions in Multiple Regions Throughout the Brain. J. Neurosci. 2018, 38, 4774–4790. [Google Scholar] [CrossRef]
- Wang, C.; Liang, G.; Shen, J.; Kong, H.; Wu, D.; Huang, J.; Li, X. Long Non-Coding RNAs as Biomarkers and Therapeutic Targets in Sepsis. Front. Immunol. 2021, 12, 722004. [Google Scholar] [CrossRef]
- Murphy, S.; Di Liegro, C.; Melli, M. The in vitro transcription of the 7SK RNA gene by RNA polymerase III is dependent only on the presence of an upstream promoter. Cell 1987, 51, 81–87. [Google Scholar] [CrossRef]
- Barboric, M.; Kohoutek, J.; Price, J.P.; Blazek, D.; Price, D.H.; Peterlin, B.M. Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J. 2005, 24, 4291–4303. [Google Scholar] [CrossRef]
- He, W.J.; Chen, R.; Yang, Z.; Zhou, Q. Regulation of two key nuclear enzymatic activities by the 7SK small nuclear RNA. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Reichow, S.L.; Hamma, T.; Ferre-D’Amare, A.R.; Varani, G. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007, 35, 1452–1464. [Google Scholar] [CrossRef] [PubMed]
- King, T.H.; Liu, B.; McCully, R.R.; Fournier, M.J. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell 2003, 11, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Piekna-Przybylska, D.; Przybylski, P.; Baudin-Baillieu, A.; Rousset, J.P.; Fournier, M.J. Ribosome performance is enhanced by a rich cluster of pseudouridines in the A-site finger region of the large subunit. J. Biol. Chem. 2008, 283, 26026–26036. [Google Scholar] [CrossRef]
- Noh, J.H.; Kim, K.M.; Abdelmohsen, K.; Yoon, J.H.; Panda, A.C.; Munk, R.; Kim, J.; Curtis, J.; Moad, C.A.; Wohler, C.M.; et al. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes. Dev. 2016, 30, 1224–1239. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, C.; Miao, J.; Pu, K.; Ma, H.; Wang, Q. Muscle-Derived Mitochondrial Transplantation Reduces Inflammation, Enhances Bacterial Clearance, and Improves Survival in Sepsis. Shock 2021, 56, 108–118. [Google Scholar] [CrossRef]
- Zhang, A.; Liu, Y.; Pan, J.; Pontanari, F.; Chia-Hao Chang, A.; Wang, H.; Gao, S.; Wang, C.; Chang, A.C. Delivery of mitochondria confers cardioprotection through mitochondria replenishment and metabolic compliance. Mol. Ther. 2023, 31, 1468–1479. [Google Scholar] [CrossRef]
- Xie, Q.; Zeng, J.; Zheng, Y.; Li, T.; Ren, J.; Chen, K.; Zhang, Q.; Xie, R.; Xu, F.; Zhu, J. Mitochondrial Transplantation Attenuates Cerebral Ischemia-Reperfusion Injury: Possible Involvement of Mitochondrial Component Separation. Oxid. Med. Cell Longev. 2021, 2021, 1006636. [Google Scholar] [CrossRef]
- Murando, F.; Peloso, A.; Cobianchi, L. Experimental Abdominal Sepsis: Sticking to an Awkward but Still Useful Translational Model. Mediators Inflamm. 2019, 2019, 8971036. [Google Scholar] [CrossRef]
- Lee, M.J.; Kim, K.; Jo, Y.H.; Lee, J.H.; Hwang, J.E. Dose-dependent mortality and organ injury in a cecal slurry peritonitis model. J. Surg. Res. 2016, 206, 427–434. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Shumate, A.; Wong, B.; Pertea, G.; Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 2022, 18, e1009730. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Noh, J.H.; Lee, M.J.; Park, Y.J.; Kim, B.M.; Kim, Y.-S.; Hwang, S.; Park, C.; Kim, K. Effects of Mitochondrial Transplantation on Transcriptomics in a Polymicrobial Sepsis Model. Int. J. Mol. Sci. 2023, 24, 15326. https://doi.org/10.3390/ijms242015326
Kim S, Noh JH, Lee MJ, Park YJ, Kim BM, Kim Y-S, Hwang S, Park C, Kim K. Effects of Mitochondrial Transplantation on Transcriptomics in a Polymicrobial Sepsis Model. International Journal of Molecular Sciences. 2023; 24(20):15326. https://doi.org/10.3390/ijms242015326
Chicago/Turabian StyleKim, Seongmin, Ji Heon Noh, Min Ji Lee, Ye Jin Park, Bo Mi Kim, Yun-Seok Kim, Sangik Hwang, Chungoo Park, and Kyuseok Kim. 2023. "Effects of Mitochondrial Transplantation on Transcriptomics in a Polymicrobial Sepsis Model" International Journal of Molecular Sciences 24, no. 20: 15326. https://doi.org/10.3390/ijms242015326
APA StyleKim, S., Noh, J. H., Lee, M. J., Park, Y. J., Kim, B. M., Kim, Y.-S., Hwang, S., Park, C., & Kim, K. (2023). Effects of Mitochondrial Transplantation on Transcriptomics in a Polymicrobial Sepsis Model. International Journal of Molecular Sciences, 24(20), 15326. https://doi.org/10.3390/ijms242015326