
EWS/FLI1 Characterization, Activation, Repression, Target Genes and Therapeutic Opportunities in Ewing Sarcoma



Abstract
1. Introduction
2. Characterization of EWS/FLI1
3. Mechanism of Gene Repression and Activation
3.1. EWS/FLI1 Mediated Gene Repression
3.2. EWS/FLI1 Mediated Genes Activation via GGAA Enhancer
4. Transcriptional Regulators and therapeutic opportunities
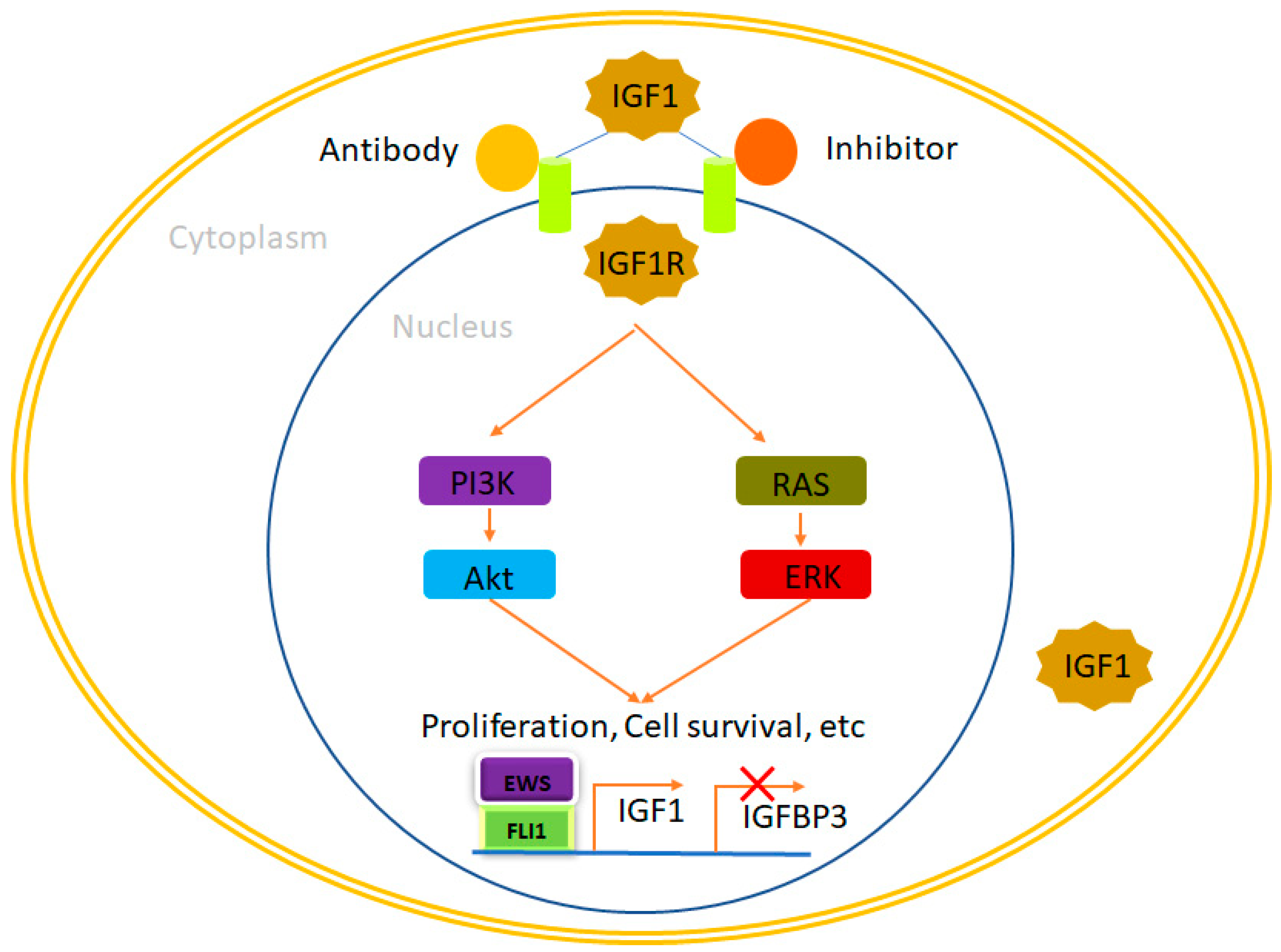
4.1. IGF1R
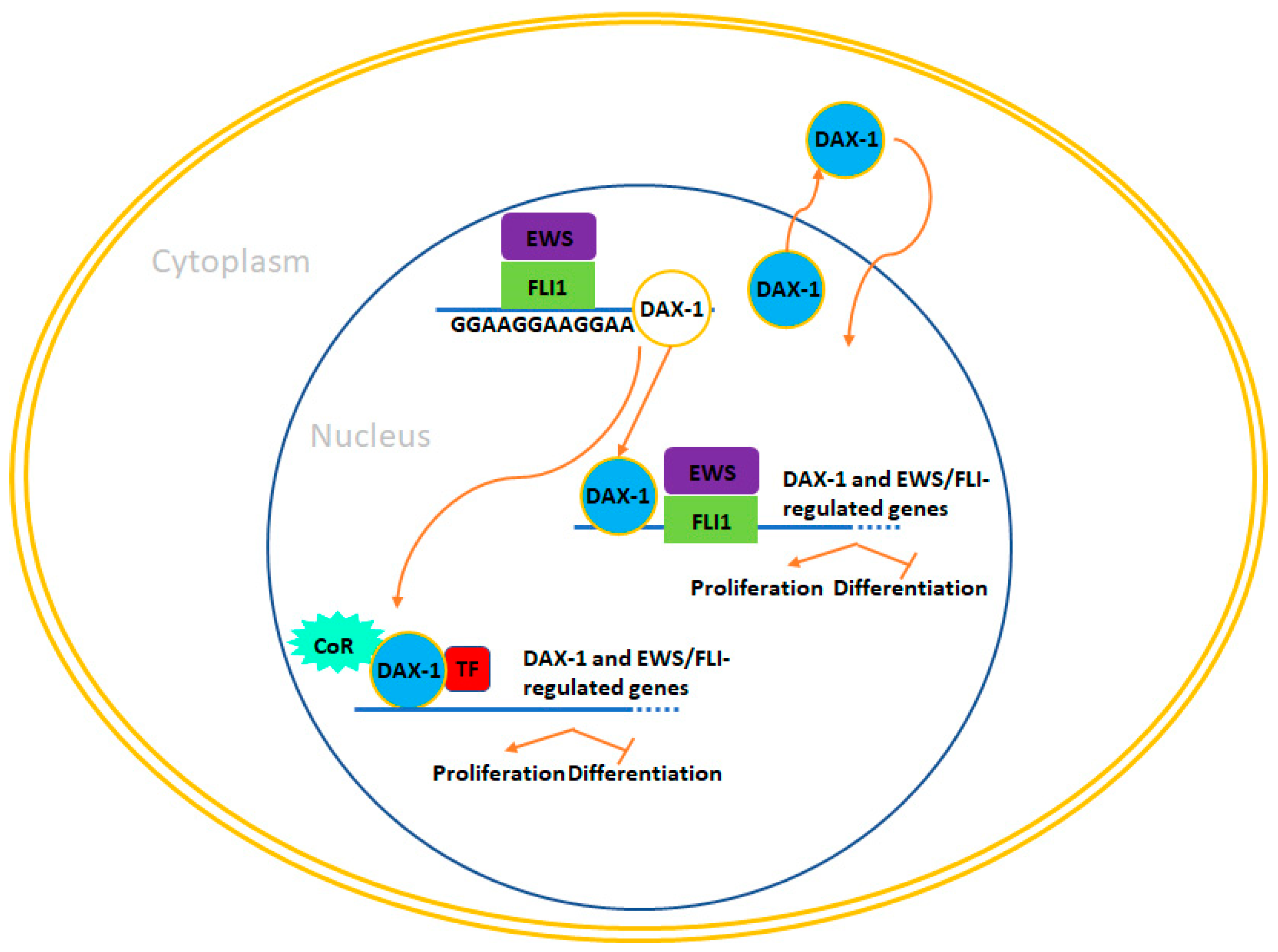
4.2. DAX-1 (NR0B1)
4.3. NKX2.2 (NK2 Homeobox 2)
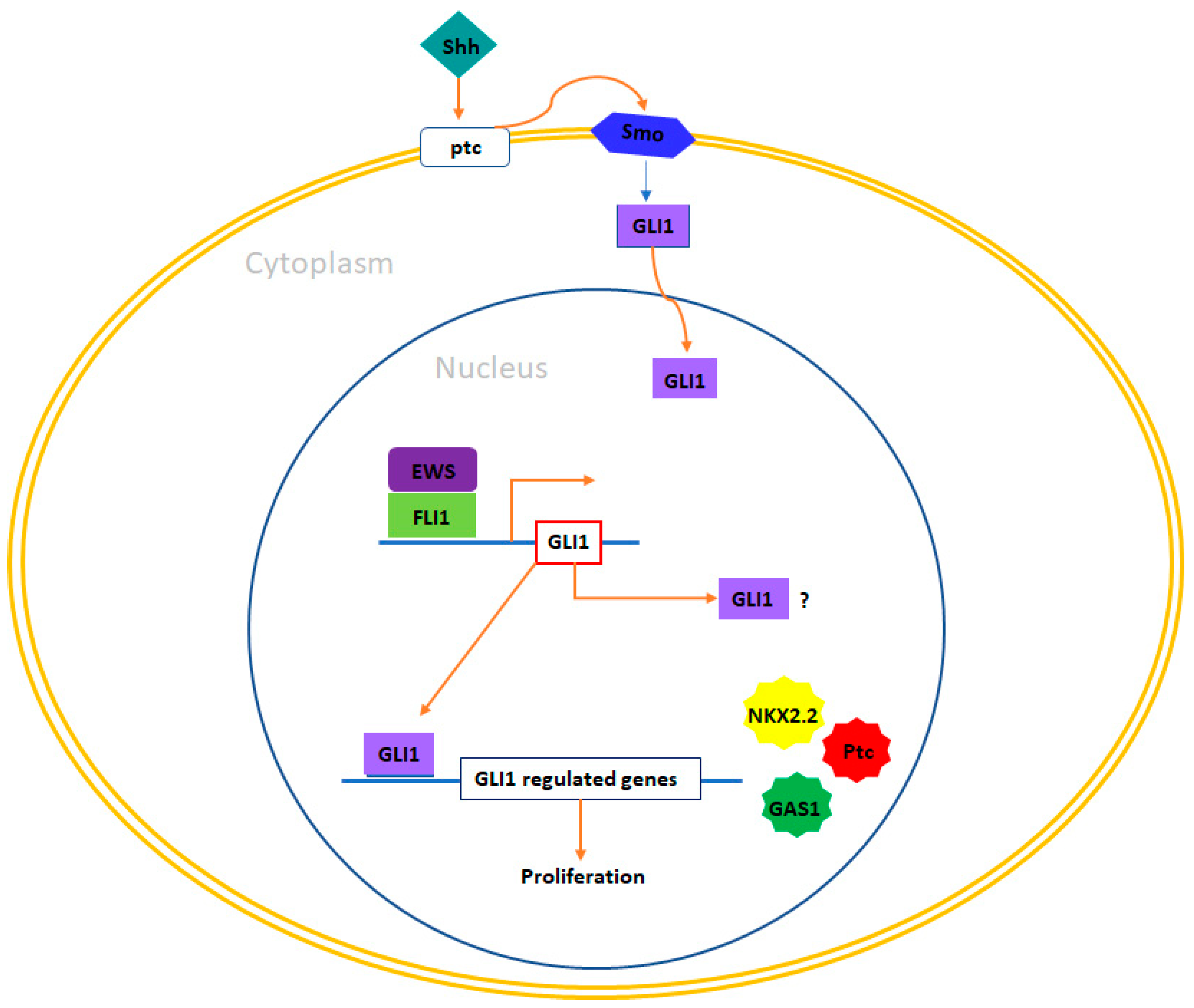
4.4. GLI1
4.5. EZH2 (Enhancer of Zeste)
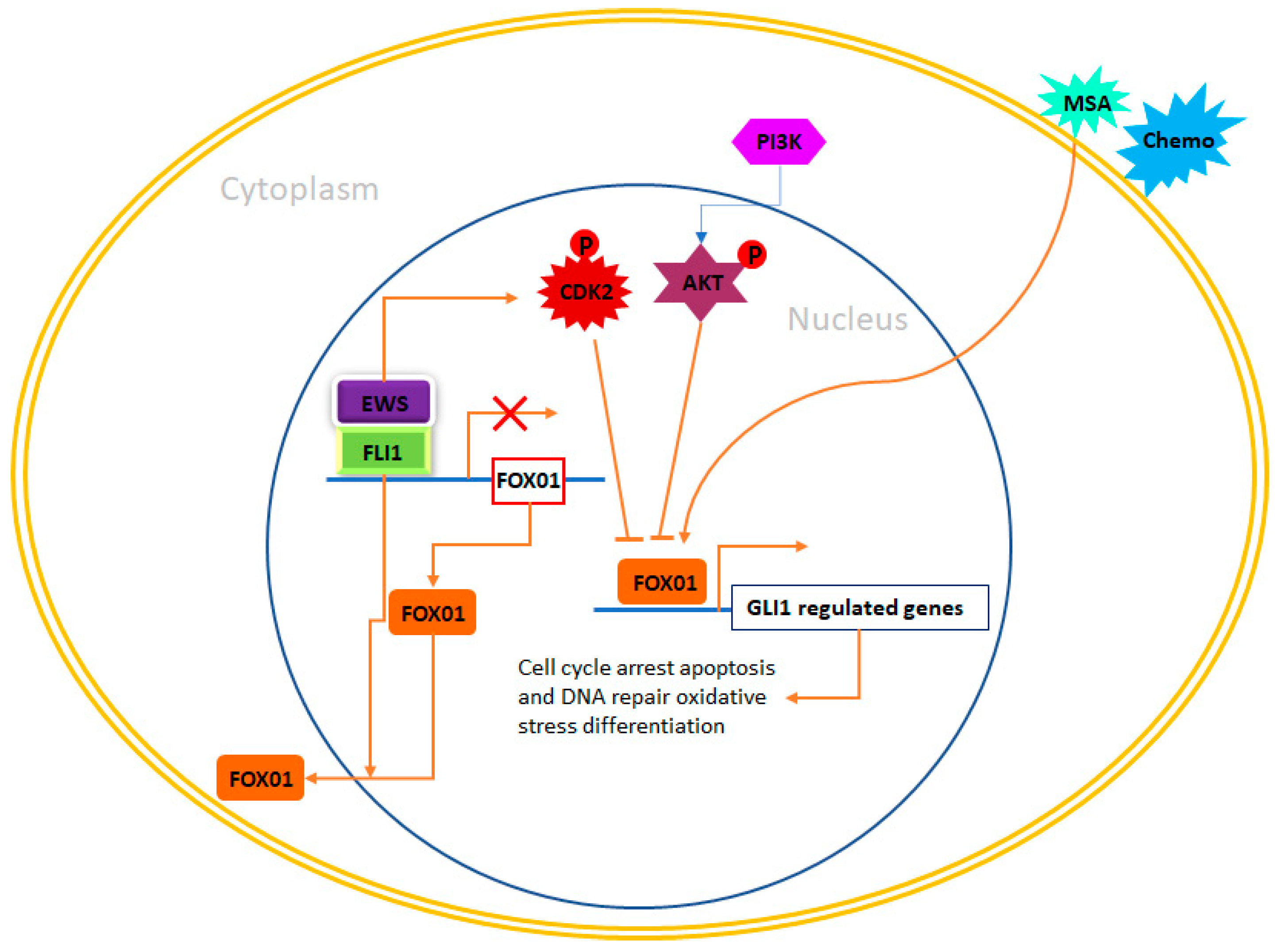
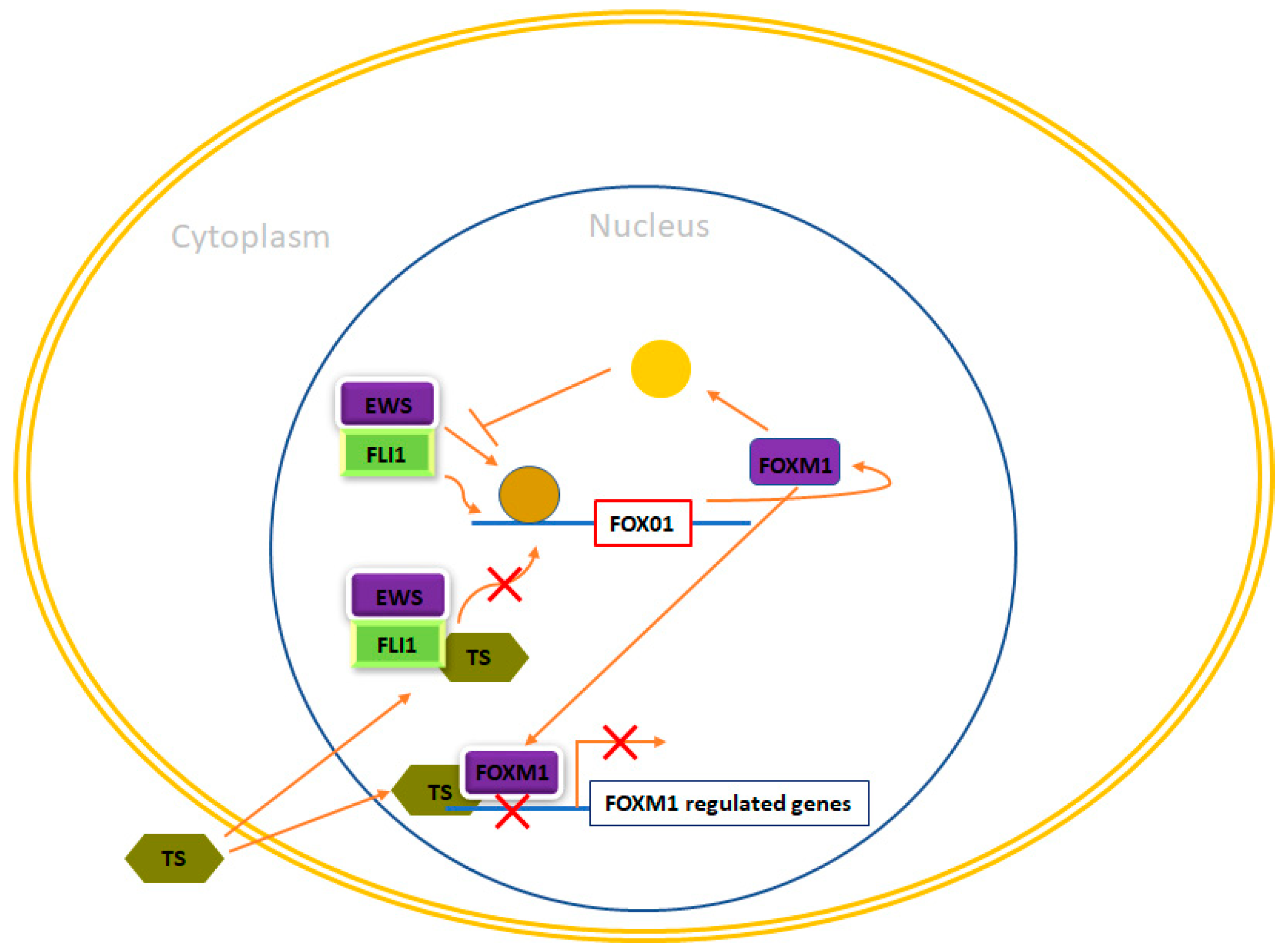
4.6. Forkhead Box (FOX) of Transcription Factors
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Becker, M.; Stefanelli, S.; Rougemont, A.-L.; Poletti, P.A.; Merlini, L. Non-odontogenic tumors of the facial bones in children and adolescents: Role of multiparametric imaging. Neuroradiology 2017, 59, 327–342. [Google Scholar] [CrossRef]
- Grünewald, T.G.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing Sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Hassan, M.; Kloczkowski, A.; Chun, W. Exploration of Flavonoids as Lead Compounds against Ewing Sarcoma through Molecular Docking, Pharmacogenomics Analysis, and Molecular Dynamics Simulations. Molecules 2023, 28, 414. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Yasir, M.; Shahzadi, S.; Kloczkowski, A. Exploration of Potential Ewing Sarcoma Drugs from FDA-Approved Pharmaceuticals through Computational Drug Repositioning, Pharmacogenomics, Molecular Docking, and MD Simulation Studies. ACS Omega 2022, 7, 19243–19260. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Hassan, M.; Kloczkowski, A.; Chun, W. Investigation of Flavonoid Scaffolds as DAX1 Inhibitors against Ewing Sarcoma through Pharmacoinformatic and Dynamic Simulation Studies. Molecules 2023, 24, 9332. [Google Scholar] [CrossRef] [PubMed]
- Je, E.M.; An, C.H.; Yoo, N.J.; Lee, S.H. Mutational analysis of PIK3CA, JAK2, BRAF, FOXL2, IDH1, AKT1 and EZH2 oncogenes in sarcomas. APMIS 2012, 120, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.; Ranft, A.; Esposito, I.; da Silva-Buttkus, P.; Aichler, M.; Baumhoer, D.; Schaefer, K.; Ottaviano, L.; Poremba, C.; Jundt, G. High STEAP1 expression is associated with improved outcome of Ewing’s sarcoma patients. Ann. Oncol. 2012, 23, 2185–2190. [Google Scholar] [CrossRef]
- Riggi, N.; Suvà, M.L.; Stamenkovic, I. Ewing’s sarcoma. N. Engl. Med. 2021, 384, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Seong, B.K.A.; Dharia, N.V.; Lin, S.; Donovan, K.A.; Chong, S.; Robichaud, A.; Conway, A.; Hamze, A.; Ross, L.; Alexe, G. TRIM8 modulates the EWS/FLI oncoprotein to promote survival in Ewing sarcoma. Cancer Cell 2021, 39, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Li, H.; Reidy, K.; Beers, L.F.; Christensen, B.L.; Lee, S. EWS/FLI1 oncogene activates caspase 3 transcription and triggers apoptosis in vivo. Cancer Res 2010, 70, 1154–1163. [Google Scholar] [CrossRef]
- Franzetti, G.; Laud-Duval, K.; Van Der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; De Pinieux, G.; Snaar-Jagalska, E. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Hoffman, L.M.; Welm, A.L.; Lessnick, S.L.; Beckerle, M. The EWS/FLI oncogene drives changes in cellular morphology, adhesion, and migration in Ewing sarcoma. Genes Cancer 2012, 3, 102–116. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Hoffman, L.M.; Jensen, C.C.; Lin, Y.-C.; Grossmann, A.H.; Randall, R.L.; Lessnick, S.L.; Welm, A.L.; Beckerle, M.C. Molecular dissection of the mechanism by which EWS/FLI expression compromises actin cytoskeletal integrity and cell adhesion in Ewing sarcoma. Mol. Biol. Cell 2014, 25, 2695–2709. [Google Scholar] [CrossRef]
- Fadul, J.; Bell, R.; Hoffman, L.M.; Beckerle, M.C.; Engel, M.E.; Lessnick, S.L. EWS/FLI utilizes NKX2-2 to repress mesenchymal features of Ewing sarcoma. Genes Cancer 2015, 6, 129. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Mazloom-Farsibaf, H.; Chang, B.-J.; Roudot, P.; Rajendran, D.; Daetwyler, S.; Fiolka, R.; Warren, M.; Amatruda, J.F.; Danuser, G.J. In vivo 3D profiling of site-specific human cancer cell morphotypes in zebrafish. J. Cell Biol. 2022, 221, e202109100. [Google Scholar] [CrossRef]
- Bierbaumer, L.; Katschnig, A.M.; Radic-Sarikas, B.; Kauer, M.O.; Petro, J.A.; Högler, S.; Gurnhofer, E.; Pedot, G.; Schäfer, B.W.; Schwentner, R.J.O. YAP/TAZ inhibition reduces metastatic potential of Ewing sarcoma cells. Oncogenesis 2021, 10, 2. [Google Scholar] [CrossRef]
- Katschnig, A.; Kauer, M.; Schwentner, R.; Tomazou, E.; Mutz, C.; Linder, M.; Sibilia, M.; Alonso, J.; Aryee, D.; Kovar, H. EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene 2017, 36, 5995–6005. [Google Scholar] [CrossRef]
- Apfelbaum, A.A.; Wu, F.; Hawkins, A.G.; Magnuson, B.; Jiménez, J.A.; Taylor, S.D.; Wrenn, E.D.; Waltner, O.; Pfaltzgraff, E.R.; Song, J.Y. EWS:: FLI1 and HOXD13 control tumor cell plasticity in Ewing sarcoma. Clin Cancer Res. 2022, 28, 4466–4478. [Google Scholar] [CrossRef] [PubMed]
- Aynaud, M.-M.; Mirabeau, O.; Gruel, N.; Grossetête, S.; Boeva, V.; Durand, S.; Surdez, D.; Saulnier, O.; Zaïdi, S.; Gribkova, S. Transcriptional programs define intratumoral heterogeneity of Ewing sarcoma at single-cell resolution. Cell Rep. 2020, 30, 1767–1779.e6. [Google Scholar] [CrossRef]
- Khoogar, R.; Li, F.; Chen, Y.; Ignatius, M.; Lawlor, E.R.; Kitagawa, K.; Huang, T.H.-M.; Phelps, D.A.; Houghton, P.J. Single-cell RNA profiling identifies diverse cellular responses to EWSR1/FLI1 downregulation in Ewing sarcoma cells. Cell Oncol 2022, 45, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Apfelbaum, A.A.; Wrenn, E.D.; Lawlor, E.R. The importance of fusion protein activity in Ewing sarcoma and the cell intrinsic and extrinsic factors that regulate it: A review. Front. Oncol. 2022, 12, 1044707. [Google Scholar] [CrossRef] [PubMed]
- Adane, B.; Alexe, G.; Seong, B.K.A.; Lu, D.; Hwang, E.E.; Hnisz, D.; Lareau, C.A.; Ross, L.; Lin, S.; Cruz, F. STAG2 loss rewires oncogenic and developmental programs to promote metastasis in Ewing sarcoma. Cencer Cell 2021, 39, 827–844.e10. [Google Scholar] [CrossRef]
- Sankar, S.; Tanner, J.M.; Bell, R.; Chaturvedi, A.; Randall, R.L.; Beckerle, M.C.; Lessnick, S.L. A novel role for keratin 17 in coordinating oncogenic transformation and cellular adhesion in Ewing sarcoma. Mol. Cell. Biol. 2013, 33, 4448–4460. [Google Scholar] [CrossRef]
- Owen, L.A.; Kowalewski, A.A.; Lessnick, S.L. EWS/FLI mediates transcriptional repression via NKX2. 2 during oncogenic transformation in Ewing’s sarcoma. PLoS ONE 2008, 3, e1965. [Google Scholar] [CrossRef] [PubMed]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schönegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef]
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007, 11, 421–429. [Google Scholar] [CrossRef]
- Sankar, S.; Bell, R.; Stephens, B.; Zhuo, R.; Sharma, S.; Bearss, D.J.; Lessnick, S. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene 2013, 32, 5089–5100. [Google Scholar] [CrossRef]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suvà, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G.; Volorio, A.; Iyer, S.; Broye, L.C.; Stamenkovic, I.; Riggi, N.; Rivera, M. Epigenome editing of microsatellite repeats defines tumor-specific enhancer functions and dependencies. Genes Dev. 2018, 32, 1008–1019. [Google Scholar] [CrossRef]
- Minas, T.Z.; Surdez, D.; Javaheri, T.; Tanaka, M.; Howarth, M.; Kang, H.-J.; Han, J.; Han, Z.-Y.; Sax, B.; Kream, B. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget 2017, 8, 34141. [Google Scholar] [CrossRef] [PubMed]
- Cidre-Aranaz, F.; Alonso, J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front. Oncol. 2015, 5, 163. [Google Scholar] [CrossRef]
- Kauer, M.; Ban, J.; Kofler, R.; Walker, B.; Davis, S.; Meltzer, P.; Kovar, H. A molecular function map of Ewing’s sarcoma. PloS ONE 2009, 4, e5415. [Google Scholar] [CrossRef]
- Monument, M.J.; Johnson, K.M.; Grossmann, A.H.; Schiffman, J.D.; Randall, R.L.; Lessnick, S.L. Microsatellites with macro-influence in ewing sarcoma. Genes 2012, 3, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, A.; Ohmura, S.; Orth, M.F.; Li, J.; Wehweck, F.S.; Knott, M.M.; Stein, S.; Saucier, D.; Arrigoni, C.; Gerke, J. Oncogenic hijacking of a developmental transcription factor evokes therapeutic vulnerability for ROS-induction in Ewing sarcoma. Nat. Commun. 2019, 578666. [Google Scholar]
- Theisen, E.R.; Selich-Anderson, J.; Miller, K.R.; Tanner, J.M.; Taslim, C.; Pishas, K.I.; Sharma, S.; Lessnick, S. Chromatin profiling reveals relocalization of lysine-specific demethylase 1 by an oncogenic fusion protein. Epigenetics 2021, 16, 405–424. [Google Scholar] [CrossRef]
- Guenther, L.M.; Dharia, N.V.; Ross, L.; Conway, A.; Robichaud, A.L.; Catlett, J.L., 2nd; Wechsler, C.S.; Frank, E.S.; Goodale, A.; Church, A.J.; et al. A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1343–1357. [Google Scholar] [CrossRef]
- Selles, M.C.; Fortuna, J.T.S.; Zappa-Villar, M.F.; de Faria, Y.P.R.; Souza, A.S.; Suemoto, C.K.; Leite, R.E.P.; Rodriguez, R.D.; Grinberg, L.T.; Reggiani, P.C.; et al. Adenovirus-Mediated Transduction of Insulin-Like Growth Factor 1 Protects Hippocampal Neurons from the Toxicity of Aβ Oligomers and Prevents Memory Loss in an Alzheimer Mouse Model. Mol. Neurobiol. 2020, 57, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aragoncillo, E.; Carrillo, J.; Lalli, E.; Agra, N.; Gomez-Lopez, G.; Pestana, A.; Alonso, J. DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell-cycle progression in Ewing’s tumor cells. Oncogene 2008, 27, 6034–6043. [Google Scholar] [CrossRef]
- He, Y.; Zhou, J.; Ma, S.; Nie, Y.; Yue, D.; Jiang, Q.; Wali, A.R.; Tang, J.Z.; Gu, Z. Multi-Responsive “Turn-On” Nanocarriers for Efficient Site-Specific Gene Delivery In Vitro and In Vivo. Adv. Healthc. Mater. 2016, 5, 2799–2812. [Google Scholar] [CrossRef] [PubMed]
- Machiela, M.J.; Grünewald, T.G.; Surdez, D.; Reynaud, S.; Mirabeau, O.; Karlins, E.; Rubio, R.A.; Zaidi, S.; Grossetete-Lalami, S.; Ballet, S. Genome-wide association study identifies multiple new loci associated with Ewing sarcoma susceptibility. Nat. Commun. 2018, 9, 3184. [Google Scholar] [CrossRef]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: Imperfect specificity for Ewing sarcoma. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2016, 29, 370–380. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zuo, Q.; Hu, B.; Jin, H.; Wang, C.; Cheng, Z.; Deng, X.; Yang, C.; Ruan, H.; Yu, C.; et al. A novel, liver-specific long noncoding RNA LINC01093 suppresses HCC progression by interaction with IGF2BP1 to facilitate decay of GLI1 mRNA. Cancer Lett. 2019, 450, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Clymer, J.; Bell, J.B.; Beauchamp, E.M.; Blyth, G.T.; Goldman, S.; Platanias, L.C. Pharmacological mTOR targeting enhances the antineoplastic effects of selective PI3Kα inhibition in medulloblastoma. Sci. Rep. 2019, 9, 12822. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gu, J.; Ding, X.; Ge, G.; Zang, X.; Ji, R.; Shao, M.; Mao, Z.; Zhang, Y.; Zhang, J.; et al. LINC00978 promotes the progression of hepatocellular carcinoma by regulating EZH2-mediated silencing of p21 and E-cadherin expression. Cell Death Dis. 2019, 10, 752. [Google Scholar] [CrossRef]
- Chang, L.C.; Lin, H.Y.; Tsai, M.T.; Chou, R.H.; Lee, F.Y.; Teng, C.M.; Hsieh, M.T.; Hung, H.Y.; Huang, L.J.; Yu, Y.L.; et al. YC-1 inhibits proliferation of breast cancer cells by down-regulating EZH2 expression via activation of c-Cbl and ERK. Br. J. Pharmacol. 2014, 171, 4010–4025. [Google Scholar] [CrossRef]
- Svoboda, L.K.; Harris, A.; Bailey, N.J.; Schwentner, R.; Tomazou, E.; von Levetzow, C.; Magnuson, B.; Ljungman, M.; Kovar, H.; Lawlor, E.R. Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 2014, 9, 1613–1625. [Google Scholar] [CrossRef]
- Ren, B.; Rose, J.B.; Liu, Y.; Jaskular-Sztul, R.; Contreras, C.; Beck, A.; Chen, H. Heterogeneity of Vascular Endothelial Cells, De Novo Arteriogenesis and Therapeutic Implications in Pancreatic Neuroendocrine Tumors. J. Clin. Med. 2019, 8, 1980. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, Z.; Ge, C.; Chen, C.; Zhao, F.; Li, H.; Chen, T.; Yao, M.; Li, J. NK3 homeobox 1 (NKX3.1) up-regulates forkhead box O1 expression in hepatocellular carcinoma and thereby suppresses tumor proliferation and invasion. J. Biol. Chem. 2017, 292, 19146–19159. [Google Scholar] [CrossRef]
- Lewis, T.B.; Coffin, C.M.; Bernard, P.S. Differentiating Ewing’s sarcoma from other round blue cell tumors using a RT-PCR translocation panel on formalin-fixed paraffin-embedded tissues. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2007, 20, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhou, N.; Hu, T.; Zhao, W.; Wu, D.; Wang, S. LncRNA MEG3 negatively modified osteosarcoma development through regulation of miR-361-5p and FoxM1. J. Cell. Physiol. 2019, 234, 13464–13480. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.; Joo, J.; Lee, S.; Wai, D.; Triche, T.J.; May, W.A. FOXM1 is an oncogenic mediator in Ewing Sarcoma. PLoS ONE 2013, 8, e54556. [Google Scholar] [CrossRef]
- Galifi, C.A.; Wood, T.L. Insulin-like growth factor-1 receptor crosstalk with integrins, cadherins, and the tumor microenvironment: Sticking points in understanding IGF1R function in cancer. Endocr Relat Cancer 2023, 30. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, S.N.; Tang, N.; Holler, A.E.; Wang, N.; Wang, J.J. Targeting the IGF1R/PI3K/AKT pathway sensitizes Ewing sarcoma to BET bromodomain inhibitors. Mol Cancer Ther 2019, 18, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Biology, D. The role of tyrosine kinases as a critical prognostic parameter and its targeted therapies in ewing sarcoma. Front. Cell Dev Biol. 2020, 8, 613. [Google Scholar] [CrossRef]
- Poreba, E.; Durzynska, J. Nuclear localization and actions of the insulin-like growth factor 1 (IGF-1) system components: Transcriptional regulation and DNA damage response. Mutat. Res. Rev. Mutat. Res. 2020, 784, 108307. [Google Scholar] [CrossRef]
- Moreau, F.; Kirk, N.S.; Zhang, F.; Gelfanov, V.; List, E.O.; Chrudinová, M.; Venugopal, H.; Lawrence, M.C.; Jimenez, V.; Bosch, F. Interaction of a viral insulin-like peptide with the IGF-1 receptor produces a natural antagonist. Nat. Commun. 2022, 13, 6700. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marsiglia, W.M.; Chen, H.; Katigbak, J.; Erdjument-Bromage, H.; Kemble, D.J.; Fu, L.; Ma, J.; Sun, G.; Zhang, Y. Molecular basis for receptor tyrosine kinase A-loop tyrosine transphosphorylation. Nat. Chem. Biol. 2020, 16, 267–277. [Google Scholar] [CrossRef]
- Guan, J.; Borenäs, M.; Xiong, J.; Lai, W.-Y.; Palmer, R.H.; Hallberg, B. IGF1R Contributes to Cell Proliferation in ALK-Mutated Neuroblastoma with Preference for Activating the PI3K-AKT Signaling Pathway. Cancer (Basel) 2023, 15, 4252. [Google Scholar] [CrossRef]
- Nagao, H.; Cai, W.; Brandão, B.B.; Albrechtsen, N.J.W.; Steger, M.; Gattu, A.K.; Pan, H.; Dreyfuss, J.M.; Wunderlich, F.T.; Mann, M. Leucine-973 is a crucial residue differentiating insulin and IGF-1 receptor signaling. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Werner, H.; LeRoith, D. Hallmarks of cancer: The insulin-like growth factors perspective. Front. Oncol. 2022, 12, 1055589. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.; Lam, H.Y.; Yap, K.C.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Li, S.U.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiong, H. Correlation of LAGE3 with unfavorable prognosis and promoting tumor development in HCC via PI3K/AKT/mTOR and Ras/RAF/MAPK pathways. BMC Cancer 2022, 22, 1–13. [Google Scholar] [CrossRef]
- Garofalo, C.; Mancarella, C.; Grilli, A.; Manara, M.C.; Astolfi, A.; Marino, M.T.; Conte, A.; Sigismund, S.; Carè, A.; Belfiore, A.; et al. Identification of Common and Distinctive Mechanisms of Resistance to Different Anti-IGF-IR Agents in Ewing’s Sarcoma. Mol. Endocrinol. 2012, 26, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Manara, M.C.; Nicoletti, G.; Marino, M.T.; Lollini, P.L.; Astolfi, A.; Pandini, G.; López-Guerrero, J.A.; Schaefer, K.L.; Belfiore, A.; et al. Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene 2011, 30, 2730–2740. [Google Scholar] [CrossRef]
- Amaral, A.T.; Garofalo, C.; Frapolli, R.; Manara, M.C.; Mancarella, C.; Uboldi, S.; Giandomenico, S.D.; Ordóñez, J.L.; Sevillano, V.; Malaguarnera, R. Trabectedin efficacy in Ewing sarcoma is greatly increased by combination with anti-IGF signaling agents. Clin Cancer Res 2015, 21, 1373–1382. [Google Scholar] [CrossRef]
- Buonocore, F.; Achermann, J. Primary adrenal insufficiency: New genetic causes and their long-term consequences. Clin Endocrinol 2020, 92, 11–20. [Google Scholar] [CrossRef]
- Agrawal, S.; He, J.C.; Tharaux, P.-L. Nuclear receptors in podocyte biology and glomerular disease. Nat. Rev. Nephrol. 2021, 17, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Modi, D. The molecular genetics of testis determination. Genet. Male Infertil. 2020, 3–17. [Google Scholar] [CrossRef]
- Muscatelli, F.; Strom, T.M.; Walker, A.P.; Zanaria, E.; Récan, D.; Meindl, A.; Bardoni, B.; Guioli, S.; Zehetner, G.; Rabl, W.; et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 1994, 372, 672–676. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Li, Y.; Chow, S.T.; Xie, W.; Zhang, X.; Zhou, J.; Chan, F. Interplay between orphan nuclear receptors and androgen receptor-dependent or-independent growth signalings in prostate cancer. Mol. Aspects Med. 2021, 78, 100921. [Google Scholar] [CrossRef]
- Mizutani, T.; Orisaka, M.; Kawabe, S.; Morichika, R.; Uesaka, M.; Yoshida, Y. YAP/TAZ-TEAD is a novel transcriptional regulator of genes encoding steroidogenic enzymes in rat granulosa cells and KGN cells. Mol. Cell. Endocrinol. 2023, 559, 111808. [Google Scholar] [CrossRef]
- Indrieri, A.; Carrella, S.; Carotenuto, P.; Banfi, S.; Franco, B. The pervasive role of the miR-181 family in development, neurodegeneration, and cancer. Int. J. Mol. Sci. 2020, 21, 2092. [Google Scholar] [CrossRef]
- Martins, R.S.T.; Power, D.M.; Fuentes, J.; Deloffre, L.A.M.; Canário, A.V.M. DAX1 regulatory networks unveil conserved and potentially new functions. Gene 2013, 530, 66–74. [Google Scholar] [CrossRef]
- Erkizan, H.V.; Uversky, V.N.; Toretsky, J.A. Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing’s sarcoma. Clin. Cancer Res. 2010, 16, 4077–4083. [Google Scholar] [CrossRef]
- Liu, X.-F.; Li, X.-Y.; Zheng, P.-S.; Yang, W.-T. DAX1 promotes cervical cancer cell growth and tumorigenicity through activation of Wnt/β-catenin pathway via GSK3β. Cell Death Dis. 2018, 9, 339. [Google Scholar] [CrossRef]
- Mendiola, M.; Carrillo, J.; García, E.; Lalli, E.; Hernández, T.; de Alava, E.; Tirode, F.; Delattre, O.; García-Miguel, P.; López-Barea, F. The orphan nuclear receptor DAX1 is up-regulated by the EWS/FLI1 oncoprotein and is highly expressed in Ewing tumors. Int. J. Cancer 2006, 118, 1381–1389. [Google Scholar] [CrossRef]
- Yu, L.; Davis, I.J.; Liu, P. Regulation of EWSR1-FLI1 Function by Post-Transcriptional and Post-Translational Modifications. Cancers (Basel) 2023, 15, 382. [Google Scholar] [CrossRef]
- Watanabe, M.; Kosaka, H.; Sugawara, M.; Maemoto, M.; Ono, Y.; Uemori, T.; Shizu, R.; Yoshinari, K. Screening for DAX1/EWS-FLI1 functional inhibitors identified dihydroorotate dehydrogenase as a therapeutic target for Ewing’s sarcoma. Cancer Med. 2023, 12, 9802–9814. [Google Scholar] [CrossRef]
- Ranhotra, H. The orphan nuclear receptors in cancer and diabetes. J. Recept. Signal Transduct. 2013, 33, 207–212. [Google Scholar] [CrossRef]
- Mutz, C.N.; Schwentner, R.; Kauer, M.O.; Ban, J.; Aryee, D.N.; Erhardt, S.; Fuchs, D.; Heitger, A.; Kovar, H. Investigating the NAD metabolome in Ewing Sarcoma. Int. J. Oncol. 2015, 75, 1162. [Google Scholar]
- Kinsey, M.; Smith, R.; Lessnick, S. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol. Cancer Res. 2006, 4, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Lalli, E. Role of orphan nuclear receptor DAX-1/NR0B1 in development, physiology, and disease. Adv. Biol. 2014. [Google Scholar] [CrossRef]
- Tirado, O.M.; Mateo-Lozano, S.; Villar, J.; Dettin, L.E.; Llort, A.; Gallego, S.; Ban, J.; Kovar, H.; Notario, V. Caveolin-1 (CAV1) is a target of EWS/FLI-1 and a key determinant of the oncogenic phenotype and tumorigenicity of Ewing’s sarcoma cells. Cancer Res. 2006, 66, 9937–9947. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Gangwal, K.; Sankar, S.; Boucher, K.; Thomas, D.; Lessnick, S. GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene 2009, 28, 4126–4132. [Google Scholar] [CrossRef]
- Guillon, N.; Tirode, F.; Boeva, V.; Zynovyev, A.; Barillot, E.; Delattre, O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS ONE 2009, 4, e4932. [Google Scholar] [CrossRef]
- Beck, R.; Monument, M.J.; Watkins, W.S.; Smith, R.; Boucher, K.M.; Schiffman, J.D.; Jorde, L.B.; Randall, R.L.; Lessnick, S.L. EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet. 2012, 205, 304–312. [Google Scholar] [CrossRef]
- Monument, M.J.; Johnson, K.M.; McIlvaine, E.; Abegglen, L.; Watkins, W.S.; Jorde, L.B.; Womer, R.B.; Beeler, N.; Monovich, L.; Lawlor, E. Clinical and biochemical function of polymorphic NR0B1 GGAA-microsatellites in Ewing sarcoma: A report from the Children’s Oncology Group. PLoS ONE 2014, 9, e104378. [Google Scholar] [CrossRef]
- Lalli, E.; Alonso, J. Targeting DAX-1 in embryonic stem cells and cancer. Expert Opin. Ther. Targets 2010, 14, 169–177. [Google Scholar] [CrossRef]
- Kinsey, M.; Smith, R.; Iyer, A.; McCabe, E.; Lessnick, S.; Kinsey, M.; Smith, R.; Iyer, A.K.; McCabe, E.R.; Lessnick, S.L. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res 69: 9047-9055. Cancer Res. 2009, 69, 9047–9055. [Google Scholar] [CrossRef]
- Davoodnejad, M.; Eshraghi, P.; Hamzehloie, T. Identification of mutations in Iranian patients’ DAX-1 gene with X-linked adrenal hypoplasia congenital. Egypt. J. Med. Hum. Genet. 2017, 18, 165–172. [Google Scholar] [CrossRef][Green Version]
- Martinez-Arguelles, D.B.; Papadopoulos, V. Prenatal phthalate exposure: Epigenetic changes leading to lifelong impact on steroid formation. Andrology 2016, 4, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingham, J.P.; Buonocore, F.; Duncan, A.J.; Achermann, J.C. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 607–619. [Google Scholar] [CrossRef]
- Albers, M.; Kranz, H.; Kober, I.; Kaiser, C.; Klink, M.; Suckow, J.; Kern, R.; Koegl, M.; Proteomics, C. Automated yeast two-hybrid screening for nuclear receptor-interacting proteins. Mol. Cell Proteomics 2005, 4, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Nagel, S.; MacLeod, R.A.; Pommerenke, C.; Meyer, C.; Kaufmann, M.; Drexler, H.G. NKL homeobox gene NKX2-2 is aberrantly expressed in Hodgkin lymphoma. Oncotarget 2018, 9, 37480. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006, 9, 405–416. [Google Scholar] [CrossRef]
- Qi, Y.; Cai, J.; Wu, Y.; Wu, R.; Lee, J.; Fu, H.; Rao, M.; Sussel, L.; Rubenstein, J.; Qiu, M. Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Dev. Camb. Engl. 2001, 128, 2723–2733. [Google Scholar] [CrossRef]
- Grunewald, T.G.; Diebold, I.; Esposito, I.; Plehm, S.; Hauer, K.; Thiel, U.; da Silva-Buttkus, P.; Neff, F.; Unland, R.; Müller-Tidow, C.; et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol. Cancer Res. MCR 2012, 10, 52–65. [Google Scholar] [CrossRef]
- Ullah, A.; Sinkler, M.A.; Velasquez Zarate, L.; Clavijo, A.; White, J. Ewing Sarcoma and Ewing-Like Sarcoma and the Role of NKX2.2 Immunoreactivity. Cureus 2021, 13, e17391. [Google Scholar] [CrossRef] [PubMed]
- McCuiston, A.; Bishop, J. Usefulness of NKX2. 2 immunohistochemistry for distinguishing Ewing sarcoma from other sinonasal small round blue cell tumors. Head Neck Pathol. 2018, 12, 89–94. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yamazaki, K.; Ishida, Y. Upregulation of NKX2. 2, a target of EWSR1/FLI1 fusion transcript, in primary renal Ewing sarcoma. J. Cytol. 2015, 32, 30. [Google Scholar]
- Moustakas, A.; Heldin, C.-H. Mechanisms of TGFβ-induced epithelial–mesenchymal transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical hedgehog signaling pathway in cancer: Activation of GLI transcription factors beyond smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef]
- Doheny, D.; Manore, S.G.; Wong, G.L.; Lo, H.-W. Hedgehog signaling and truncated GLI1 in cancer. Cells 2020, 9, 2114. [Google Scholar] [CrossRef] [PubMed]
- Sigafoos, A.N.; Paradise, B.D.; Fernandez-Zapico, M.E. Hedgehog/GLI signaling pathway: Transduction, regulation, and implications for disease. Cancers 2021, 13, 3410. [Google Scholar] [CrossRef] [PubMed]
- Mannino, G.; Russo, C.; Maugeri, G.; Musumeci, G.; Vicario, N.; Tibullo, D.; Giuffrida, R.; Parenti, R.; Lo Furno, D. Adult stem cell niches for tissue homeostasis. J. Cell. Physiol. 2022, 237, 239–257. [Google Scholar] [CrossRef]
- Das, D.; Fletcher, R.B.; Ngai, J.J. Cellular mechanisms of epithelial stem cell self-renewal and differentiation during homeostasis and repair. Wiley Interdiscip Rev. Biol. 2020, 9, e361. [Google Scholar] [CrossRef]
- Zwerner, J.; Joo, J.; Warner, K.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.; May, W. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008, 27, 3282–3291. [Google Scholar] [CrossRef]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Üren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef]
- Joo, J.; Christensen, L.; Warner, K.; States, L.; Kang, H.-G.; Vo, K.; Lawlor, E.R.; May, W.A. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS ONE 2009, 4, e7608. [Google Scholar] [CrossRef] [PubMed]
- Mahindroo, N.; Punchihewa, C.; Fujii, N. Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J. Med. Chem. 2009, 52, 3829–3845. [Google Scholar] [CrossRef] [PubMed]
- Avery, J.T.; Zhang, R.; Boohaker, R. GLI1: A therapeutic target for cancer. Front. Oncol. 2021, 11, 673154. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Barkat, M.; Syed, S.; Shah, S.; Abbas, G.; Xu, C.; Mahdy, A.; Hussain, N.; Majeed, A.; Khan, K.-U.-R. HedgehogSignaling: Linking Embryonic Lung Development andAsthmatic Airway Remodeling. Cells 2022, 11, 1774. [Google Scholar] [CrossRef]
- Kongkham, P.N. Epigenetic Silencing of Novel Tumour Suppressor Genes in Medulloblastoma; University of Toronto: Toronto, ON, Canada, 2012. [Google Scholar]
- Maitah, M.I.Y.; Ali, S.; Ahmad, A.; Gadgeel, S.; Sarkar, F. Up-regulation of sonic hedgehog contributes to TGF-β1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS ONE 2011, 6, e16068. [Google Scholar] [CrossRef]
- Chaudhary, A.; Raza, S.S.; Haque, R. Transcriptional factors targeting in cancer stem cells for tumor modulation. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 1969. [Google Scholar]
- Ang, H.L.; Mohan, C.D.; Shanmugam, M.K.; Leong, H.C.; Makvandi, P.; Rangappa, K.S.; Bishayee, A.; Kumar, A.P.; Sethi, G. Mechanism of epithelial-mesenchymal transition in cancer and its regulation by natural compounds. Med. Res. Rev. 2023, 43, 1141–1200. [Google Scholar] [CrossRef]
- Nguyen, N.M.; Cho, J.J. Hedgehog pathway inhibitors as targeted cancer therapy and strategies to overcome drug resistance. Int. J. Mol. Sci. 2022, 23, 1733. [Google Scholar] [CrossRef]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.-W. Targeting the sonic hedgehog signaling pathway: Review of smoothened and GLI inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Peukert, S.; Miller-Moslin, K. Small-molecule inhibitors of the hedgehog signaling pathway as cancer therapeutics. ChemMedChem Chem. Enabling Drug Discov. 2010, 5, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.Z.; Peng, L.F. Small-molecule modulators of the Sonic Hedgehog signaling pathway. Mol. Biosyst. 2010, 6, 44–54. [Google Scholar] [CrossRef]
- Niyaz, M.; Khan, M.S.; Mudassar, S.J. Hedgehog signaling: An Achilles’ heel in cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Ray, H.J. Safety and tolerability of sonic hedgehog pathway inhibitors in cancer. Drug Saf. 2019, 42, 263–279. [Google Scholar] [CrossRef]
- Riggi, N.; Suvà, M.L.; Suvà, D.; Cironi, L.; Provero, P.; Tercier, S.; Joseph, J.M.; Stehle, J.C.; Baumer, K.; Kindler, V.; et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008, 68, 2176–2185. [Google Scholar] [CrossRef]
- von Levetzow, C.; Jiang, X.; Gwye, Y.; von Levetzow, G.; Hung, L.; Cooper, A.; Hsu, J.H.; Lawlor, E.R. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS ONE 2011, 6, e19305. [Google Scholar] [CrossRef]
- Richter, G.H.; Plehm, S.; Fasan, A.; Rössler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Löwel, D.; von Luettichau, I.; Mossbrugger, I. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef]
- Burdach, S.; Plehm, S.; Unland, R.; Borkhardt, A.; Staege, M.S.; Müller-Tidow, C.; Richter, G. Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle 2009, 8, 1991–1996. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Noh, B.J.; Jung, W.W.; Kim, H.S.; Park, Y.K. Pathogenetic implications of early growth response 1 in Ewing sarcoma. Pathology 2019, 51, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Lalmansingh, A.S.; Karmakar, S.; Jin, Y.; Nagaich, A.K. Multiple modes of chromatin remodeling by Forkhead box proteins. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1819, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.-F.; Brosens, J.J.; Gomes, A.R.; Koo, C.-Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.-F.; Gomes, A.R.J. Forkhead box transcription factors in cancer initiation, progression and chemotherapeutic drug response. Front. Media SA 2014, 4, 305. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Corno, C.; Zaffaroni, N.; Perego, P. Role of FoxO proteins in cellular response to antitumor agents. Cancers 2019, 11, 90. [Google Scholar] [CrossRef]
- Jiramongkol, Y.; Lam, E.W.-F. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. 2020, 39, 681–709. [Google Scholar] [CrossRef]
- Kim, C.G.; Lee, H.; Gupta, N.; Ramachandran, S.; Kaushik, I.; Srivastava, S.; Kim, S.-H.; Srivastava, S.K. Role of Forkhead Box Class O proteins in cancer progression and metastasis. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 1969; pp. 142–151. [Google Scholar]
- Li, F.; Dong, X.; Lin, P.; Jiang, J. Regulation of Akt/FoxO3a/Skp2 axis is critically involved in berberine-induced cell cycle arrest in hepatocellular carcinoma cells. Int. J. Mol. Sci. 2018, 19, 327. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Nepal, S.; Kim, M.J.; Chang, J.H.; Kim, S.H.; Jeong, G.S.; Jeong, C.H.; Park, G.H.; Jung, S.; Lim, J. Critical role of AMPK/FoxO3A axis in globular adiponectin-induced cell cycle arrest and apoptosis in cancer cells. J. Cell Physiol. 2016, 231, 357–369. [Google Scholar] [CrossRef]
- Shi, Y.Y.; Meng, X.T.; Xu, Y.N.; Tian, X.J. Role of FOXO protein’s abnormal activation through PI3K/AKT pathway in platinum resistance of ovarian cancer. J. Obstet. Gynaecol. Res. 2021, 47, 1946–1957. [Google Scholar] [CrossRef]
- Gheghiani, L.; Shang, S.; Fu, Z. Targeting the PLK1-FOXO1 pathway as a novel therapeutic approach for treating advanced prostate cancer. Sci. Rep. 2020, 10, 12327. [Google Scholar] [CrossRef]
- Tang, Y.; Jiang, L.; Zhao, X.; Hu, D.; Zhao, G.; Luo, S.; Du, X.; Tang, W. FOXO1 inhibits prostate cancer cell proliferation via suppressing E2F1 activated NPRL2 expression. Cell Biol. Int. 2021, 45, 2510–2520. [Google Scholar] [CrossRef]
- Dang, F.; Wei, W. Targeting the acetylation signaling pathway in cancer therapy. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 1969; pp. 209–218. [Google Scholar]
- Sissaoui, S.; Egginton, S.; Ting, L.; Ahmed, A.; Hewett, P. Hyperglycaemia up-regulates placental growth factor (PlGF) expression and secretion in endothelial cells via suppression of PI3 kinase-Akt signalling and activation of FOXO1. Sci. Rep. 2021, 11, 16344. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Wang, H.; Yang, L.; Liu, M.; Luo, J. Protein post-translational modifications in the regulation of cancer hallmarks. Cancer Gene Ther. 2023, 30, 529–547. [Google Scholar] [CrossRef]
- Tian, Y.-N.; Chen, H.-D.; Tian, C.-Q.; Wang, Y.-Q.; Miao, Z.-H. Polymerase independent repression of FoxO1 transcription by sequence-specific PARP1 binding to FoxO1 promoter. Disease 2020, 11, 71. [Google Scholar] [CrossRef]
- Jayabal, P.; Zhou, F.; Lei, X.; Ma, X.; Blackman, B.; Weintraub, S.T.; Houghton, P.J.; Shiio, Y.J. NELL2-cdc42 signaling regulates BAF complexes and Ewing sarcoma cell growth. Cell. Rep. 2021, 36. [Google Scholar] [CrossRef]
- McCalla, A.C. Targeting and Characterization of Dysregulations of the Aurora Kinases A and B Family Members in Ewing Sarcoma Models; North Carolina State University: Raleigh, NC, USA, 2019. [Google Scholar]
- Chen, Y.-H.; Li, C.-L.; Chen, W.-J.; Liu, J.; Wu, H.-T. Diverse roles of FOXO family members in gastric cancer. World J. Gastrointest. Oncol. 2021, 13, 1367. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Jiang, Q.; Li, J.; Guo, J.; Therapy, T. The potent roles of salt-inducible kinases (SIKs) in metabolic homeostasis and tumorigenesis. Signal Transduct Target Ther. 2020, 5, 150. [Google Scholar] [CrossRef]
- Caltavituro, A.; Buonaiuto, R.; Pietroluongo, E.; Morra, R.; Salomone, F.; De Placido, P.; Pagliuca, M.; Vaia, A.; Ottaviano, M.; Tortora, M. Shifting from a Biological-Agnostic Approach to a Molecular-Driven Strategy in Rare Cancers: Ewing Sarcoma Archetype. Biomedicines 2023, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, K.; Zhang, Z.; Xue, D.; Li, W.; Pan, Z. The role of forkhead box family in bone metabolism and diseases. Front. Pharmacol. 2022, 12, 772237. [Google Scholar] [CrossRef] [PubMed]
- Gargallo, P.; Juan, A.; Yáñez, Y.; Dolz, S.; Segura, V.; Castel, V.; Cañete, A.J. Precision medicine in Ewing sarcoma: A translational point of view. Clin. Transl. Oncol. 2020, 22, 1440–1454. [Google Scholar] [CrossRef]
- Xu, H.; Liu, L.; Li, W.; Zou, D.; Yu, J.; Wang, L.; Wong, C. Transcription factors in colorectal cancer: Molecular mechanism and therapeutic implications. Oncogene 2021, 40, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Borhani, S.; Gartel, A. FOXM1: A potential therapeutic target in human solid cancers. Expert Opin. Ther. Targets 2020, 24, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Khan, P.; Ahmad, A.; Fatima, M.; Nasser, M.W. FOXM1: A small fox that makes more tracks for cancer progression and metastasis. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 1969. [Google Scholar]
- Liu, S.-X.; Zhou, Y.; Zhao, L.; Zhou, L.-S.; Sun, J.; Liu, G.-J.; Du, Y.-S.; Zhou, Y.-N. Thiostrepton confers protection against reactive oxygen species-related apoptosis by restraining FOXM1-triggerred development of gastric cancer. Medicine 2022, 193, 385–404. [Google Scholar] [CrossRef] [PubMed]
- Kalathil, D.; John, S.; Nair, A. FOXM1 and cancer: Faulty cellular signaling derails homeostasis. Front. Oncol. 2021, 10, 3472. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. Thiostrepton, proteasome inhibitors and FOXM1. Cell Cycle 2011, 10, 4341–4342. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N.S.; Sanders, D.A.; Rodriguez, R.; Balasubramanian, S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat. Chem. 2011, 3, 725–731. [Google Scholar] [CrossRef]
- Sengupta, A.; Rahman, M.; Mateo-Lozano, S.; Tirado, O.M.; Notario, V.J. The dual inhibitory effect of thiostrepton on FoxM1 and EWS/FLI1 provides a novel therapeutic option for Ewing’s sarcoma. Int. J. Oncol. 2013, 43, 803–812. [Google Scholar] [CrossRef]
- Alimardan, Z.; Abbasi, M.; Khodarahmi, G.; Kashfi, K.; Hasanzadeh, F.; Mahmud, A. Identification of new small molecules as dual FoxM1 and Hsp70 inhibitors using computational methods. Res. Pharm. Sci. 2022, 17, 635. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr No | Genes | Association Type | Disease | References |
|---|---|---|---|---|
| 1 | IGF1R | Biomarker, Altered Expression | Ewing sarcoma, Alzheimer’s disease | [36,37] |
| 2 | DAX-1 | Biomarker | Ewing sarcoma, Neoplasm | [38,39] |
| 3 | NKX2.2 | Altered expression, Biomarker | Ewing sarcoma, soft tissue neoplasm | [40,41] |
| 4 | GLI1 | Biomarker | Liver carcinoma, Ewing sarcoma, childhood medulloblastoma | [42,43] |
| 5 | EZH2 | Altered expression, Biomarker | Liver carcinoma, breast carcinoma, Ewing sarcoma | [44,45,46] |
| 6 | FOXO1 | Biomarker, Altered expression | Breast carcinoma, Neoplasm, Ewing sarcoma | [47,48,49] |
| 7 | FOXM1 | Altered expression, Biomarker | Childhood osteosarcoma, Ewing sarcoma | [50,51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasir, M.; Park, J.; Chun, W. EWS/FLI1 Characterization, Activation, Repression, Target Genes and Therapeutic Opportunities in Ewing Sarcoma. Int. J. Mol. Sci. 2023, 24, 15173. https://doi.org/10.3390/ijms242015173
Yasir M, Park J, Chun W. EWS/FLI1 Characterization, Activation, Repression, Target Genes and Therapeutic Opportunities in Ewing Sarcoma. International Journal of Molecular Sciences. 2023; 24(20):15173. https://doi.org/10.3390/ijms242015173
Chicago/Turabian StyleYasir, Muhammad, Jinyoung Park, and Wanjoo Chun. 2023. "EWS/FLI1 Characterization, Activation, Repression, Target Genes and Therapeutic Opportunities in Ewing Sarcoma" International Journal of Molecular Sciences 24, no. 20: 15173. https://doi.org/10.3390/ijms242015173
APA StyleYasir, M., Park, J., & Chun, W. (2023). EWS/FLI1 Characterization, Activation, Repression, Target Genes and Therapeutic Opportunities in Ewing Sarcoma. International Journal of Molecular Sciences, 24(20), 15173. https://doi.org/10.3390/ijms242015173