1. Introduction
Pleural mesothelioma (PM) is an aggressive tumor arising from the mesothelial cell layer lining within the thoracic cavity. Exposure to the carcinogenic mineral asbestos is considered the general cause of PM [
1]. Although asbestos usage is banned in a total of 68 countries around the world as of 13 July 2022 (according to
www.ibasecretariat.org, accessed on 13 September 2022), the incidence of PM is still rising due to the long disease latency of about 40 years [
2]. Moreover, asbestos is still used in many developing countries of the world [
3]. In addition, modern materials with a similar structure to asbestos, such as some forms of multiwall carbon nanotubes, may induce asbestos-like disease [
4,
5].
The treatment of PM is challenging, and the prognosis remains poor, especially because of poor treatment response and tumor recurrence within a median time of 10–18 months after initial treatment [
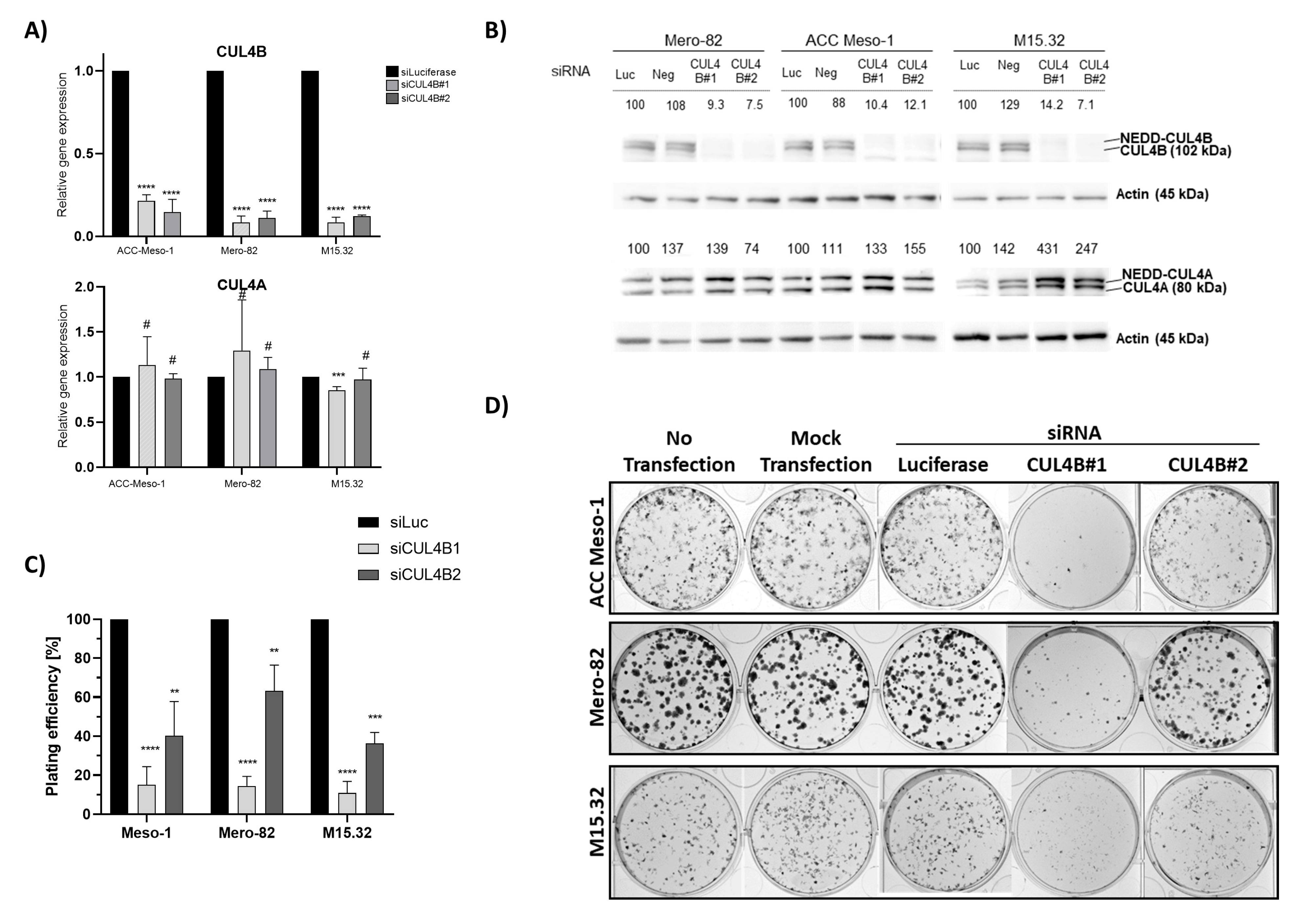
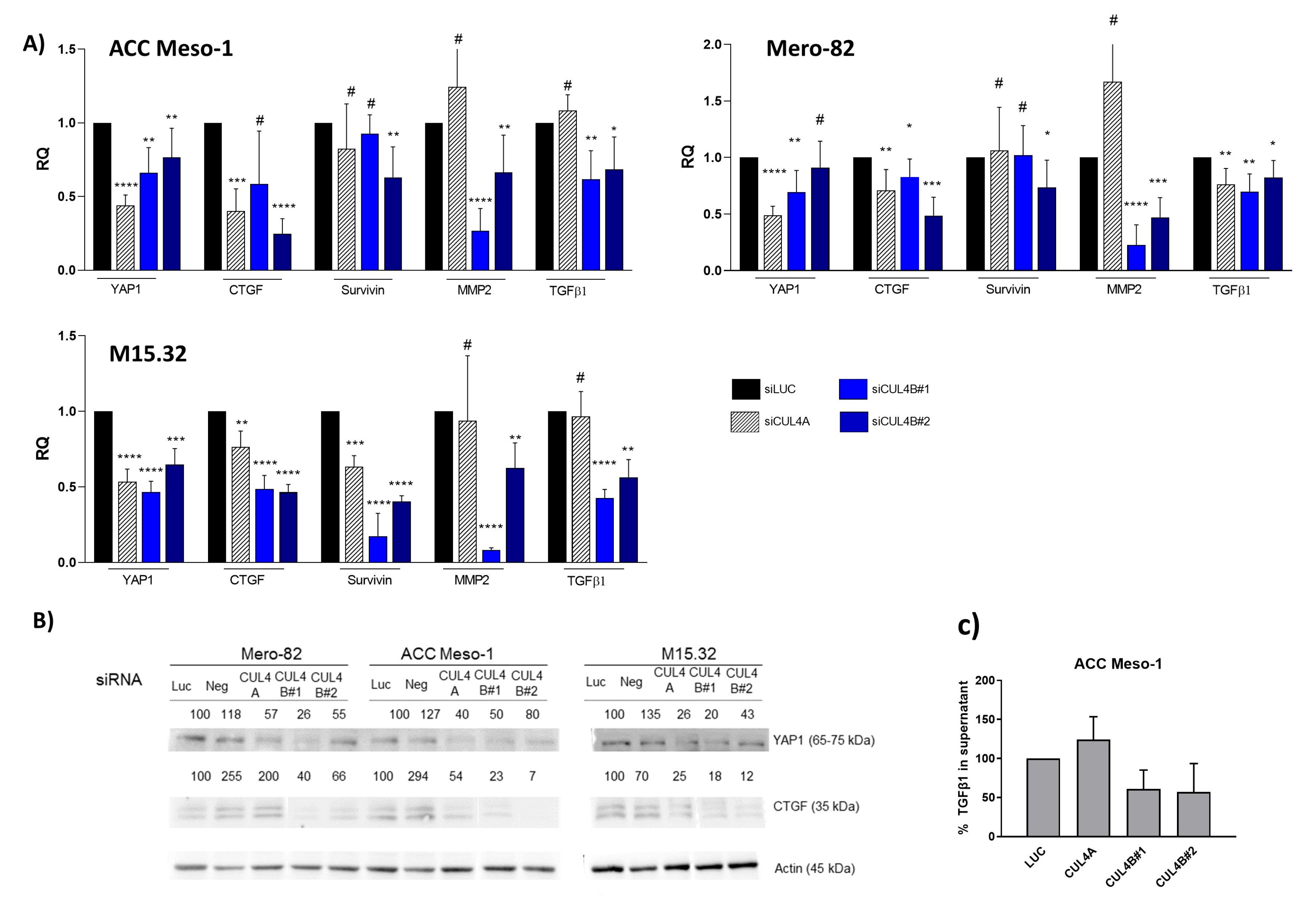
3]. Exploring factors driving aggressive PM phenotypes remains crucial for the identification of new treatments. We previously demonstrated that high expression of cullin 4B (CUL4B), a member of the cullins-RING ligase protein family, was associated with worse outcomes of PM patients [
6]. This prompted us to further investigate the mechanistic role of CUL4B in PM.
Cullins form complexes with other proteins and catalyze the ubiquitination of target proteins for proteasomal degradation or activity changes [
7]. Two cullin 4 paralogs, CUL4A and CUL4B, share 82% similarity in protein sequence. Both paralogs have been shown to be associated with tumorigenesis and the progression of various tumors [
8,
9]. In PM, the CUL4A protein levels were elevated in 64% of tumors compared to normal tissues [
10], but there was no association between CUL4A and clinical outcomes [
6]. In our previous work, we showed that CUL4B was significantly upregulated in PM tumor tissues compared to non-cancerous inflammatory pleural tissues. High expression of CUL4B was associated with short progression-free survival (PFS), and this was further confirmed using the gene expression dataset from TCGA [
7].
The CUL4B protein contains an extra nuclear localization signal in the N-terminal sequence, compared to CUL4A [
11]. Thus, the majority of CUL4B is localized in the nucleus, whereas CUL4A is mainly localized in the cytoplasm of cells, including mesothelioma tumor cells [
6,
11]. Thus, it can be speculated that CUL4B has unique functions within the nucleus, which are distinct from CUL4A. Indeed, a nuclear function of CUL4B has been discovered, such as histone modification and epigenetic regulation of various target genes [
11]. In neuronal cells, CUL4B ubiquitinates WDR5, a component of the histone methyltransferase complex, for degradation [
12]. The depletion of CUL4B caused increased levels of H3K4me3 via WDR5 stabilization and, therefore, alteration of neuronal gene expression [
12]. CUL4B catalyzes histone ubiquitination (H2AK119) and facilitates the recruitment of polycomb repressive complex 2 (PRC2) to repress the expression of various tumor-suppressor genes, most importantly PTEN and p16 [
13].
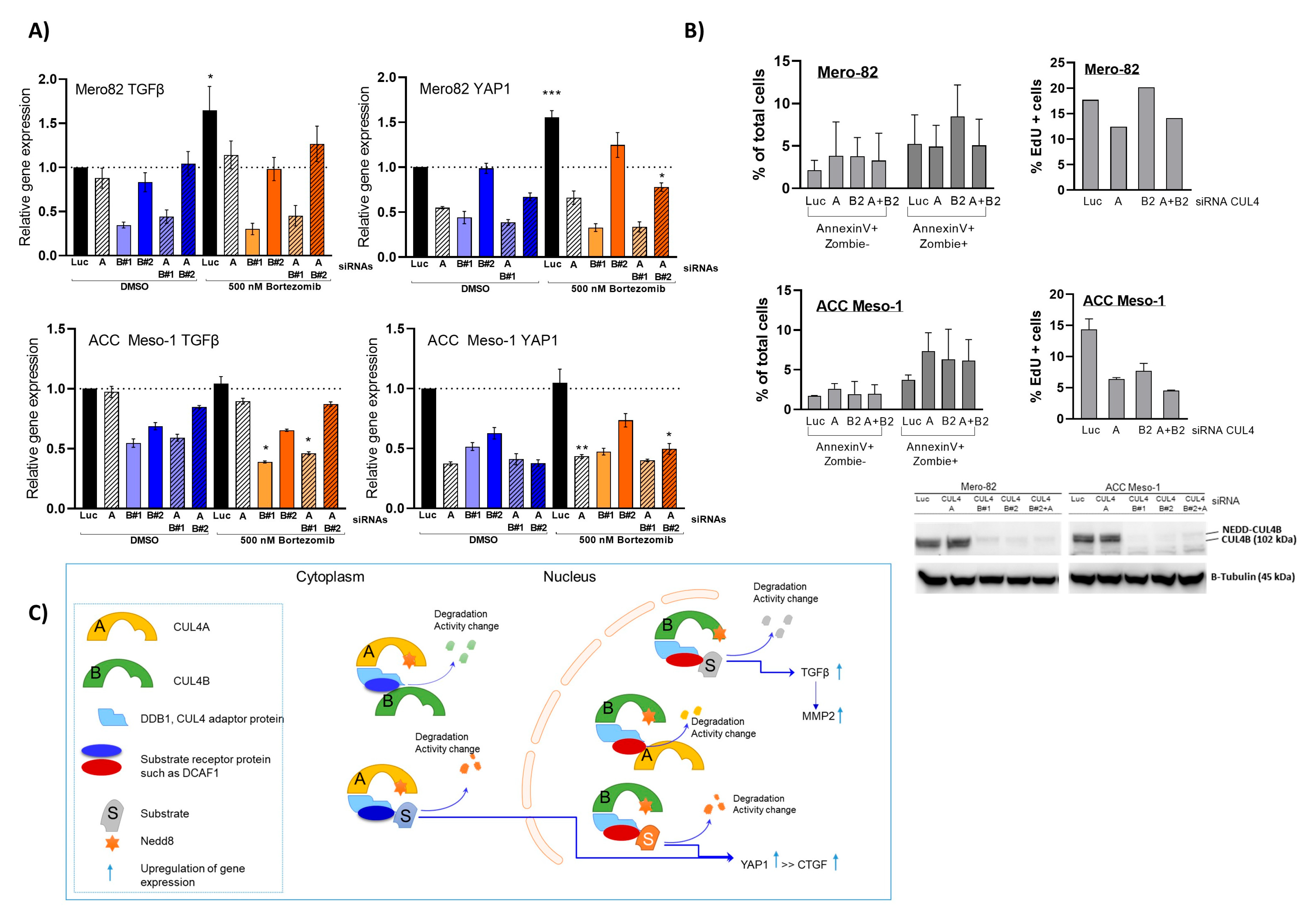
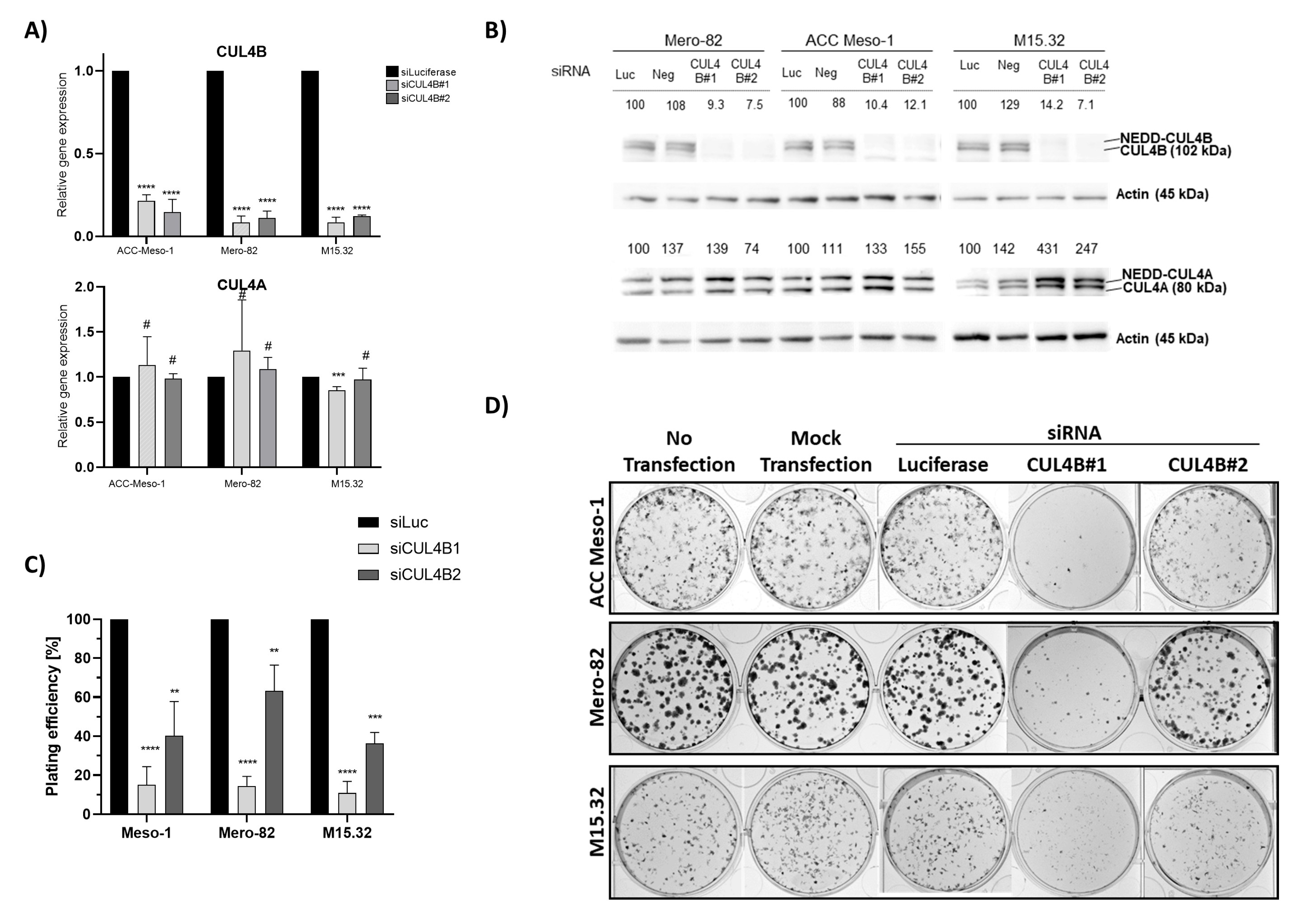
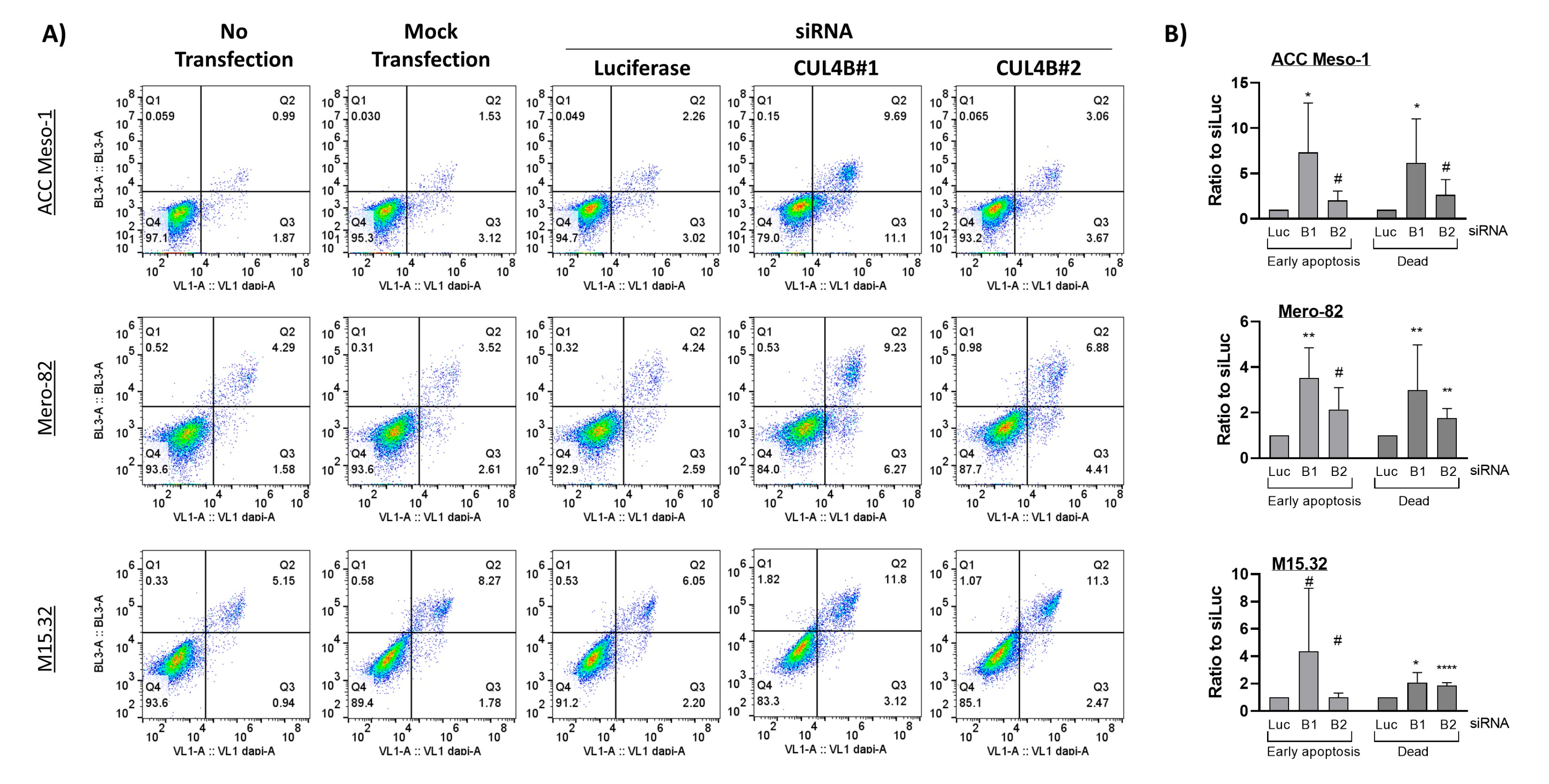
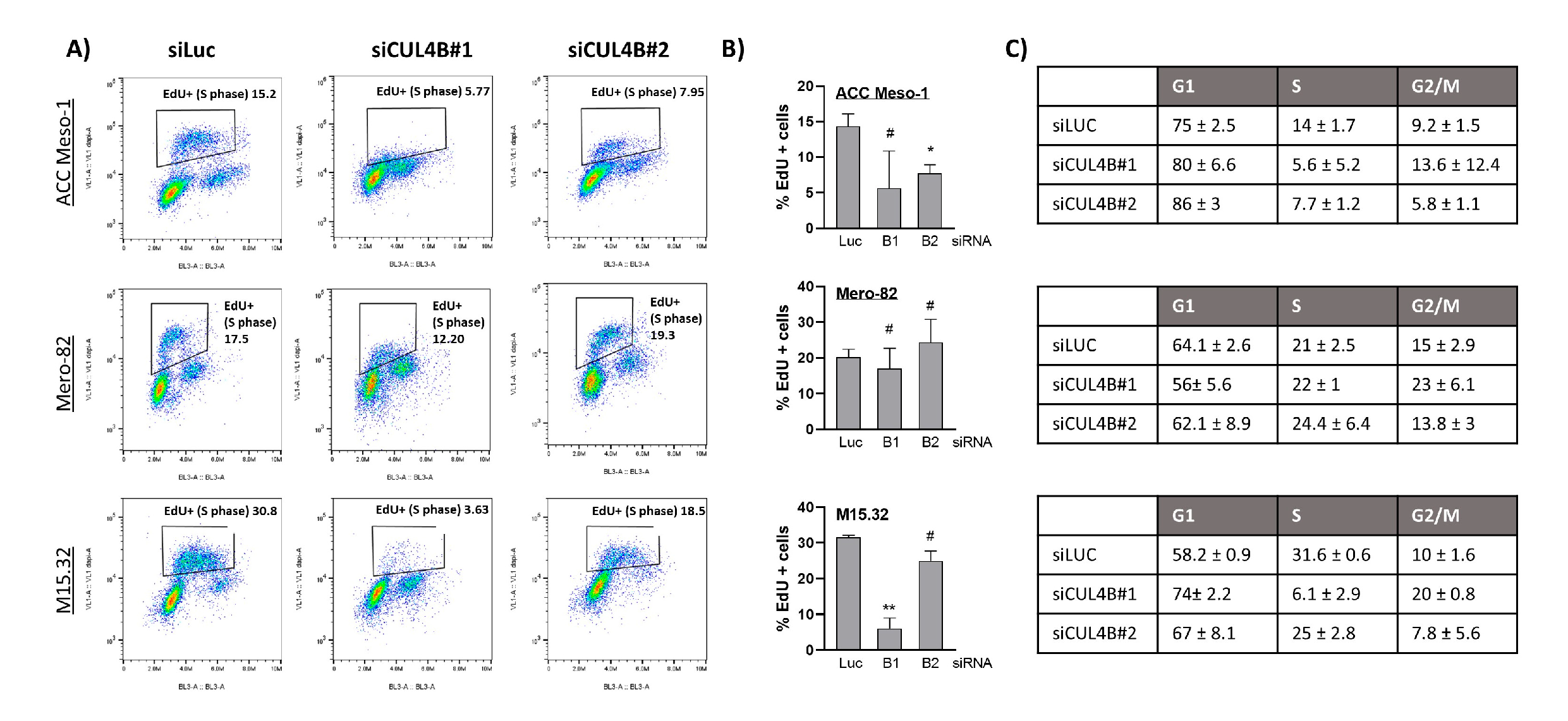
Here, we employed an siRNA approach to explore the role of CUL4B in cell lines and primary PM cells. We demonstrated that CUL4B knockdown resulted in cell death and reduced colony formation. Although various cellular effects are similar to CUL4A knockdown, we identified the unique effect of CUL4B knockdown in PM being the regulation of TGFβ1 and one of TGFβ-responsive genes, MMP2, expression. Overall, our data suggest that high expression of CUL4B may drive PM aggressiveness via regulation of TGFβ1 expression.
3. Discussion
Previously observed correlations between CUL4B levels and clinical outcomes suggested that CUL4B likely plays a role in PM progression [
6,
17], but the mechanism has been so far unexplored. Here, we employed PM cell culture and primary cells to demonstrate the mechanism of CUL4B in the regulation of PM progression. We showed that CUL4B is important for tumor cell growth, similar to its paralog, CUL4A. In addition, we demonstrated that CUL4B regulates TGF-β1 signaling in PM cells and this may affect the tumor microenvironment in part by signaling macrophage recruitment. Our data further confirmed the important role of CUL4B in cancer and support the oncogenic role of CUL4B in PM.
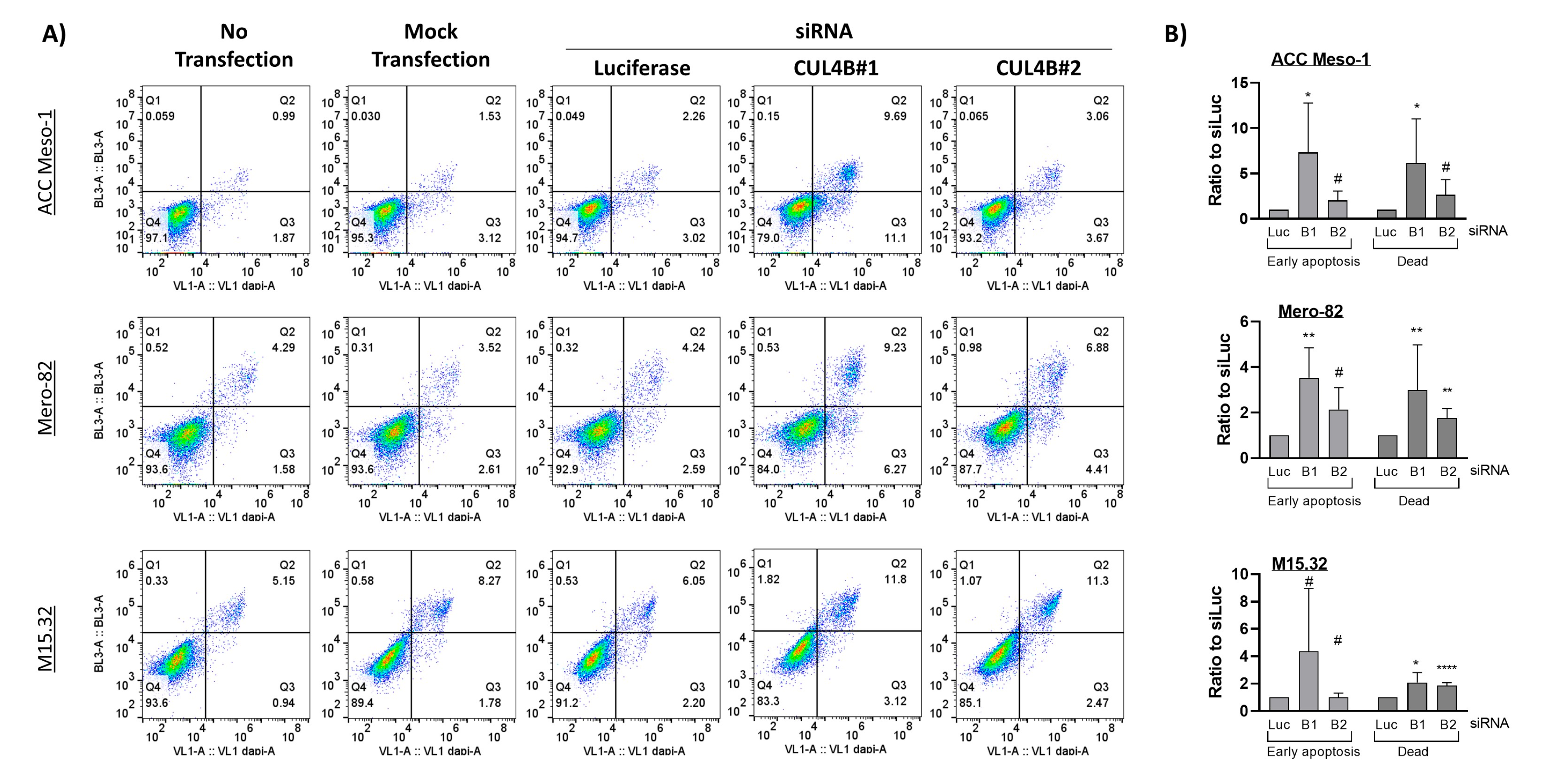
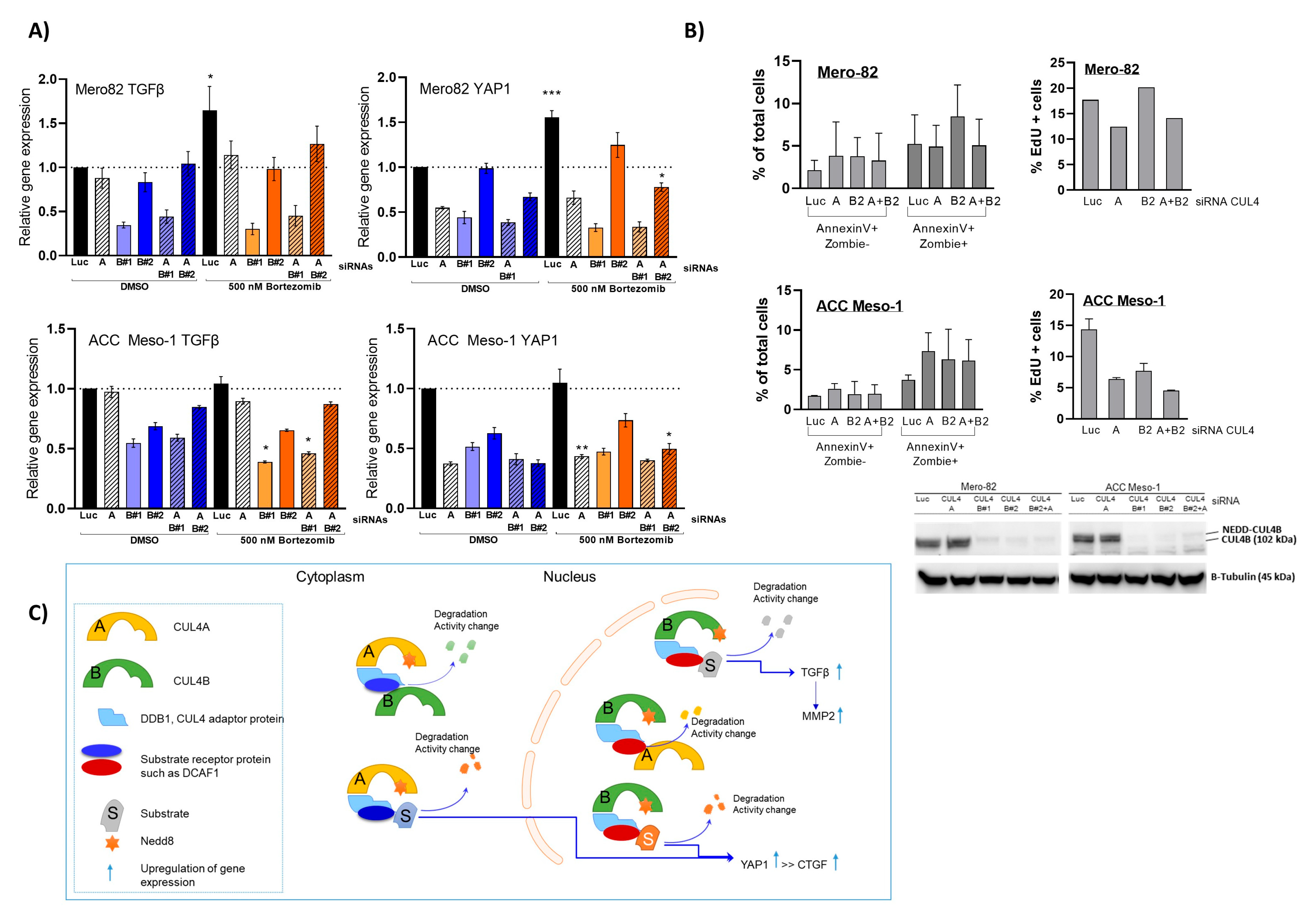
Similar to its paralog, CUL4B knockdown resulted in immediate alteration of the cell cycle and cell viability. This aspect of CUL4A and CUL4B in the regulation of cell survival is well known, mainly due to their shared functions in the degradation of crucial cell cycle regulators including p21, p27, and CDT1 [
6,
7,
11]. Nevertheless, in terms of cell cycle arrest, each cell line seemed to respond differently. This is most likely due to the different growth rate and the proportion of cells in each cell cycle phase during the treatment. In addition, this might depend on other cell cycle regulators that are individually altered in each cell line. Knockdown of both paralogs at the same time did not add up to the effects of the single-knockdown, suggesting that CUL4A and CUL4B employ different mechanisms to regulate PM cell growth and survival. This is most likely explained by the functional complexity, as CUL4A and CUL4B form a complex with DDB1 that links CUL4 to its substrate receptor protein [
18]. Thus far, there have been over 50 putative substrate receptors identified for CUL4 ubiquitin ligase that possess different biological functions [
19]. The increased protein expression of CUL4B following CUL4A knockdown and vice versa suggest that both paralogs might induce the degradation of each other. Nevertheless, this has to be further investigated in more detail.
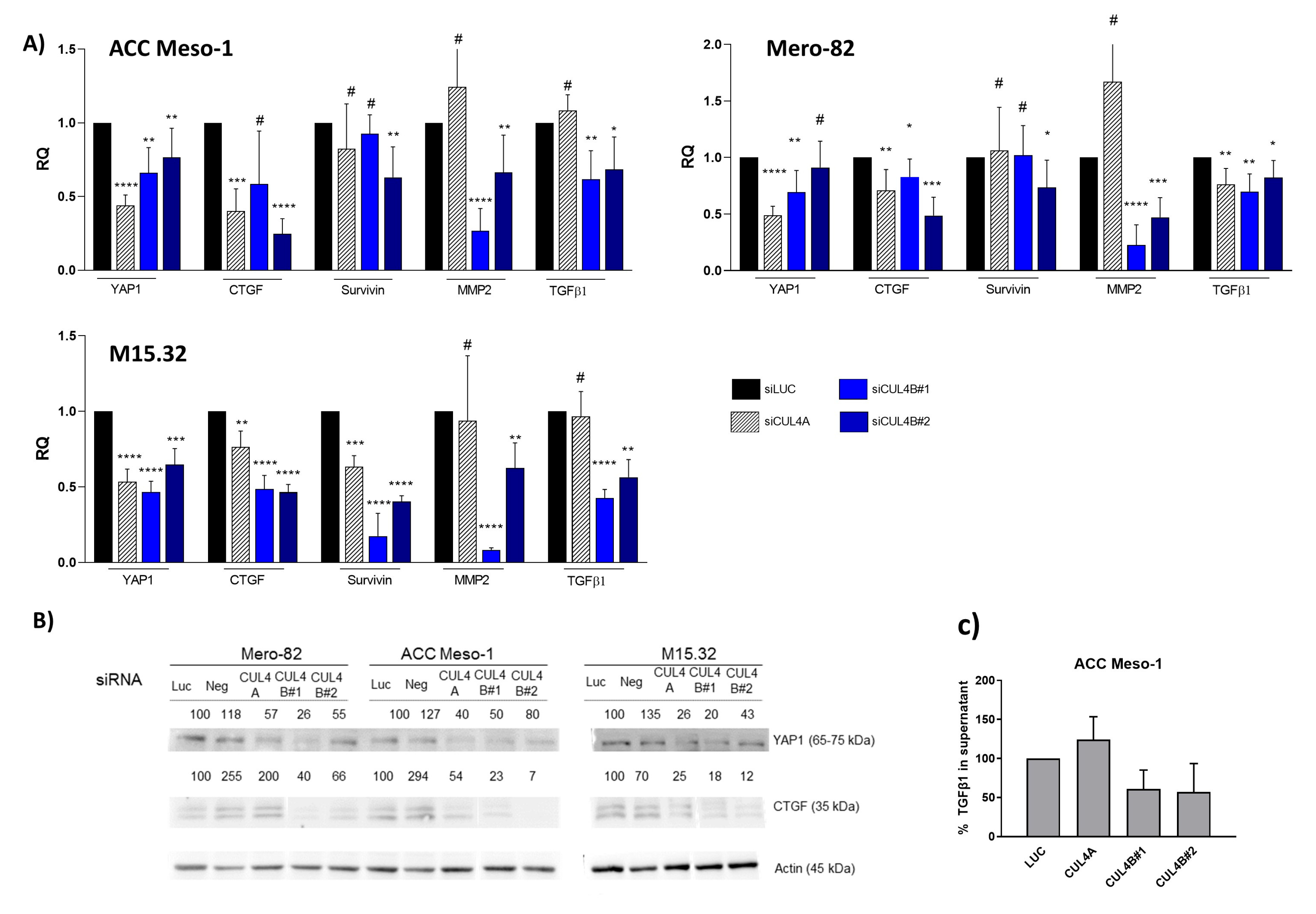
The novelty of our data is the unique effect of CUL4B in the regulation of TGFβ signaling in PM, which likely plays an important role in the regulation of the tumor microenvironment. Although the two paralogs share various similar functions in cells, we found that the expression of CUL4B correlated with PM disease outcomes, while CUL4A expression did not show any tendency [
6]. Thus, the unique action of CUL4B in the regulation of TGFβ signaling might represent an important aspect of CUL4B in the progression of PM. Our data are also supported by a recent study by Liu, L et al. that employed publicly available datasets to identify correlations between CUL4B and the PM tumor microenvironment. The study employed gene expression data from the TCGA and GEO databases to demonstrate significant correlations between CUL4B expression and clinical outcomes and immune cell infiltration [
20]. Confirming our previous data, the analyses showed that CUL4B expression was elevated in PM tumor tissue compared to normal pleura, and high CUL4B expression was associated with short the progression-free survival of PM patient cohorts. They further divided PM patients into two groups according to CUL4B expression and showed the enrichment of the TGF-β signaling pathway in the CUL4B-high compared to the CUL4B-low group. CUL4B expression was associated, both positively and negatively, with an abundance of some immune cells such as T-cell subpopulations and dendritic cells. Altogether, gene expression data from the publicly available databases support our hypothesis regarding the role of CUL4B in the regulation of TGF-β signaling, as well as the tumor microenvironment in PM tumors.
TGF-β is an influential cytokine ubiquitously expressed and secreted by both inflammatory and cancer cells [
21]. There are three forms, of which TGF-β1 is the most-relevant [
22]. TGF-β can promote or suppress tumor growth depending on the cancer stage and cancer–microenvironment interactions [
23]. It has been long known that TGF-β plays an important role in PM biology. Gerwin et al. found that PM cells secrete both the active and inactive form of TGF-β [
24]. Furthermore, TGF-β is more prevalent in pleural effusions of patients with PM compared to patients with breast cancer or non-small cell lung cancer [
25]. TGF-β, furthermore, has a crucial role in epithelial-to-mesenchymal transition (EMT) [
26]. PM cells can undergo EMT when stimulated by TGF-β [
27]. Fassina et al. were able to show that tumors of the most-aggressive sarcomatoid subtype showed higher expression of EMT transcriptional regulators (e.g., SNAIL, SLUG, TWIST) and higher expression of mesenchymal markers (such as vimentin and MMP9) compared with tumors of the biphasic and epithelioid subtypes [
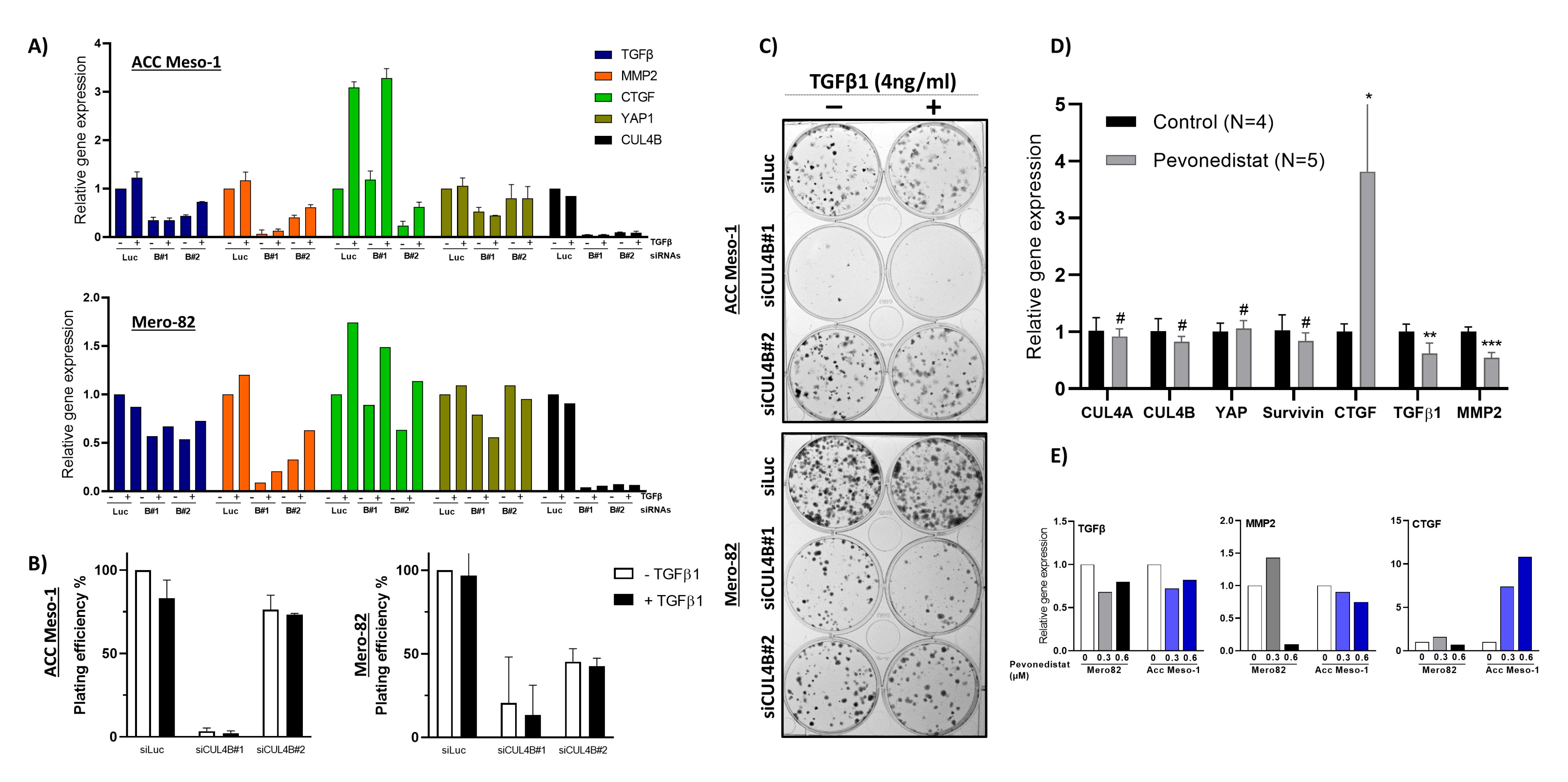
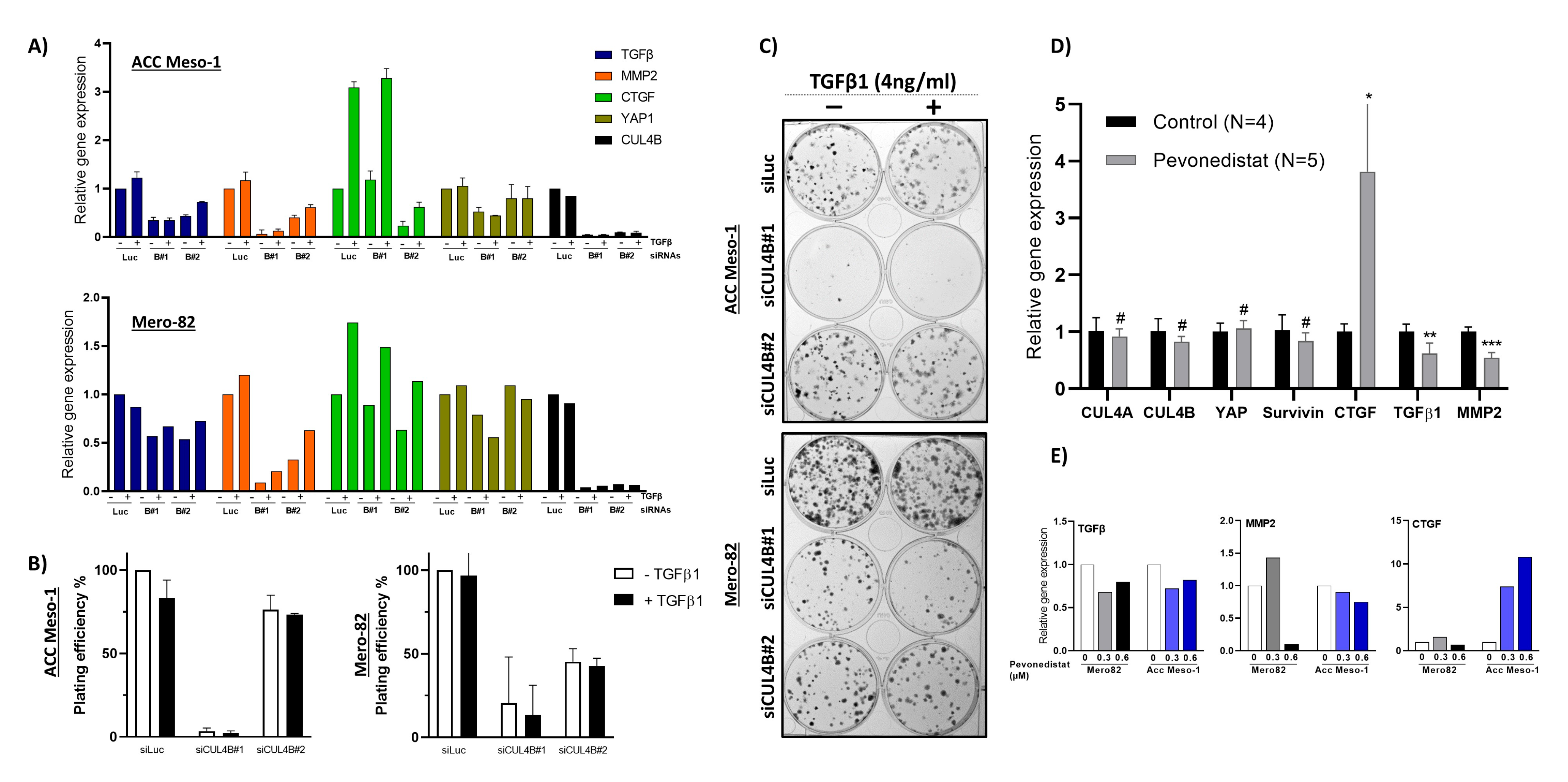
28], which further confirms the importance of TGF-β in PM tumor growth and invasion. Although TGF-β did not stimulate tumor cell growth (determined by 2D colony-formation assay) in our study, we noticed a change in the shape of colonies (
Figure 5C). TGF-β treated colonies were more diffuse compared to untreated ones. This might indicate increased motility related to EMT; nevertheless, this phenomenon cannot be concluded by our assays.
An immunosuppressive role (e.g., by inhibiting NK cells, weakening cytotoxic lymphocytes) of TGF-β has been demonstrated [
29] previously. TGF-β also has an important role with respect to tumor associated macrophages (TAMs), but the exact mechanism of regulation remains elusive [
30]. It has been shown that TGF-β secreted from tumor cells (e.g., in oral squamous cell carcinoma) induced the polarization of macrophages to the M2 tumor-promoting type [
31,
32]. The relationship between TGF-β1 signaling and tumor-associated macrophages has also been recently demonstrated in PM. Gene expression analysis in PM demonstrated that a high expression of TGF-β1 and MMP2 was associated with the presence of tumor-promoting M2 macrophages. Patients with high TGF-β1 expression and MMP2 showed worse survival [
33]. Another and the most-recent finding using a high-dimensional transcriptomic approach further identified a strong positive correlation between TGF-β1 and tumor-associated macrophage genes, as well as M2 macrophages in the PM biphasic subtype [
34]. Altogether, these data strengthen the relationship between CUL4B, TGF-β1 signaling, and the regulation of macrophages in PM progression.
A better understanding of the mechanisms through which CUL4B regulates TGF-β signaling in PM will better elucidate its oncogenic role in cancer. Given the known role of CUL4B in transcriptional regulation, the regulation of TGF-β signaling is likely through this mechanism. This hypothesis is supported by a recent study in breast cancer showing that CUL4B interacts with several HDAC-containing complexes and regulates EMT also in part via TGF-β regulation [
35]. An increased CUL4B protein level after CUL4A knockdown did not compensate for the downregulation of TGF-β and YAP expression. Both CUL4A and CUL4B regulate protein ubiquitination and degradation by forming a complex with DDB1 and substrate-specific adaptors. The substrate-specific adaptor is the key molecule that regulates substrate specificity [
19]. We hypothesized that, in the CUL4A knockdown condition, although CUL4B protein was increased, the gene expression could not be rescued due to the limited amount of substrate-specific adaptor.
Furthermore, it is important to identify whether CUL4B upregulation regulates other cells and immune cells in the tumor microenvironment. Immunotherapy was approved by the FDA for patients with unresectable PM and is being tested in several clinical trials; nevertheless, not all patients benefitted from the treatment [
36]. Thus, understanding additional players in the regulation of the tumor immune microenvironment may help to identify markers or additional targets that can be exploited to improve the treatment efficacy. Pevonedistat, a protein neddylation inhibitor also targeting cullins, is currently being tested in PM in a clinical trial, NCT03319537. Our work provides additional data for the better understanding of the mechanism of the clinical response to pevonedistat and its treatment efficacy in PM.
4. Materials and Methods
4.1. Cell Lines and Primary Cells
The malignant cell lines Mero-82 (The European Collection of Cell Cultures (ECACC), Salisbury, UK) and ACC-Meso-1 (RIKEN Bioresource, Saitama, Japan) were acquired from the indicated providers. M15.32 is a primary cell line cultured from pleural effusion from a patient with PM treated at the University Hospital Zurich. The patient signed an informed consent, and this study was approved by the cantonal ethical committee of Canton Zürich (BASEC-No. 2020-02566). Primary cells were cultured in an RPMI medium ATCC modification (Gibco
TM, A1049101, Life Technology Europe BV, Zug, Switzerland) containing 2 mM glutamine (Sigma-Aldrich, Buchs, Switzerland), 10% fetal bovine serum (FBS) (Biowest, Nuaillé, France), penicillin/streptomycin (Biowest, Nuaillé, France), heparin 2 µg/mL (SigmaAldrich, Buchs, Switzerland), hydrocortisone 2 µg/mL (SigmaAldrich, Buchs, Switzerland), and human epidermal growth factor (20 ng/mL, hEGF) (Peprotech, London, UK) and maintained at 37 °C and 5% CO
2. The identification of PM was achieved using immunohistochemical staining of epithelial and PM markers including pan-Cytokeratin, podoplanin, calretinin, and BAP1 staining. M15.32 showed nuclear BAP1 loss, identical to that of the original tumor (
Supplementary Figure S1A) and no BAP1 detectable by Western blot (
Supplementary Figure S1B). For the experiments, all cells were cultured in an RPMI medium ATCC modification (Gibco
TM, A1049101, Life Technology Europe BV, Zug, Switzerland) containing 2 mM glutamine (Sigma-Aldrich, Buchs, Switzerland) and 10% fetal bovine serum (FBS) (Biowest, Nuaillé, France) at 37 °C and 5% CO
2. To avoid superposed growth, the cells were regularly detached and passaged using a mild enzymatic solution (trypsin-EDTA, Biowest, Nuaillé, France). All cell lines were regularly tested for the absence of mycoplasmas.
4.2. siRNA Transfection
Forward transfection was performed with 20 nM siRNA using Lipofectamine
® RNAiMAX (Invitrogen 13778150, Life Technology Europe BV, Zug, Switzerland). The siRNA sequences are provided in
Supplementary Table S1. Cells were seeded in a 6-well plate (ACC Meso-1 30,000 cells/well, Mero82 and M15.32 40,000 cells/well) in the growth medium to reach 40% confluency of adherent cells after 24 h. On the next day, the siRNA/Lipofectamine complex mixture was prepared as follows: per well, 5 µL Lipofectamine
® was diluted in 250 µL Opti-MEM
® medium (Invitrogen, 31985-062 Life Technology Europe BV, Zug, Switzerland) and mixed with 200 nM siRNA duplex suspension in 250 µL Opti-MEM
®. The transfection complex was allowed to form for 15 min at room temperature. Next, a total of 500 µL of siRNA Lipofectamine
® complex mixture was pipetted dropwise onto the cells containing 2 mL growth medium (total volume 2.5 mL) in culture and cultured for 72 h for RNA extraction, Western blotting, and cell cycle analysis.
4.3. Double-siRNA Transfection and Treatment with Bortezomib
We performed siRNA forward transfection as described above. For double-transfection, 20 nM of each siRNA was used with 5 µL Lipofectamine® per well. Cells were cultured afterwards for 48 h when Bortezomib (Selleckchem PS-341, S1013, Houston, TX, USA) was added to each well. We added 6.25 µL Bortezomib (200 µM in 25% DMSO/water) to each well containing 2.5 mL culture medium (final concentration 500 nM) medium and incubated for another 6 h prior to RNA extraction. Control wells were treated with an equal amount of 25% DMSO/water.
4.4. Colony-Formation Assay
The colony-formation assay was used to determine the ability of a single cell to undergo division and form a colony. At 48 h after siRNA transfection as described above, cells were trypsinized and counted. We seeded different numbers of cells (250, 500, 1000 cells) per well (each in duplicate) of a 6-well plate and cultured in 2 mL of culture medium. On Day 4 after seeding, 500 µL of fresh culture medium was added. On Day 6, 1.5 mL of culture medium was aspirated and replenished with 1.5 mL of fresh culture medium. On day 8, cells were washed with ice-cold Dulbecco’s phosphate-buffered saline (DPBS, Biowest, Nuaillé, France) and fixed in ice-cold absolute methanol for 20 min at −20 °C. Afterwards, 1.5 mL of crystal violet was applied to each well and incubated for a minimum of 1 h. Crystal violet was washed with water, and the colonies were imaged using Fusion FX7 (Witec AG, Sursee, Switzerland). Colony counting was performed manually using the Image J [
37] manual cell counting function. From different seeding numbers, we only counted the wells with clearly visible isolated colonies. Finally, we used the average data of plating efficiency (number of colonies/number of cells seeded) of the duplication for further analysis. For the rescue experiment, we prepared a stock solution of 100 µg/mL of recombinant human TGF-β1 (Peprotech 100-21, London, UK) in 10 mM citric acid pH 3.0 and diluted to 1 µg/mL of working stock (250X) in DPBS containing 0.1% BSA. Then, 48 h after transfection, cells were counted and the cell suspension was seeded into 6-well plates in 2 mL of complete culture medium, for the control and TGF-β1-treated. Recombinant human TGF-β1 was added to the culture medium immediately after seeding at the final concentration of 4 ng/mL. The same amount of protein diluent was applied to the no-TGF-β1 control. Medium change was performed on Day 3 after seeding. After aspiration of 1 mL of medium, we replenished with 1 mL of fresh medium without or with 4 ng/mL of TGF-β1. Colonies were fixed and stained on Day 7 after seeding.
4.5. Analysis of Apoptosis and Proliferation by Flow Cytometry
Cells were seeded in 6-well plates and transfected with siRNAs as described above. At 72 h after transfection, they were collected for apoptosis and the cell cycle assay.
For apoptosis, we collected the cell culture supernatant (containing floating dead cells), and the adherent cells were detached with trypsin for 5 min. After stopping the trypsin reaction with fresh culture medium, the cell suspension was pooled with supernatant. Cells were washed with 3 mL of DPBS followed by 3 mL of Annexin V binding buffer containing 10 mM HEPES/NaOH pH7.4, 140 mM NaCl, and 2.5 mM CaCl2 and pelleted by centrifugation at 400× g for 5 min. Afterwards, we resuspended cells in 100 µL of Annexin V binding buffer and added 5 µL of Annexin V (Annexin V Pacific Blue conjugate, Molecular Probe; A35122, Life Technology Europe BV, Zug, Switzerland) and 1 µL of Zombie (Zombie NIR, Biolegend 423105, Amsterdam, The Netherlands). The complex was incubated at room temperature in the dark for 15 min. We then added 400 µL of Annexin V binding buffer to the suspension, kept on ice, filtrated through 0.22 µm filters and immediately analyzed with an Attune cytometer (Applied Biosystems, Life Technology Europe BV, Zug, Switzerland).
For the BrdU incorporation assay using the Click-iT™ EdU Pacific Blue™ Flow Cytometry Assay Kit (Invitrogen C10418), we treated the adherent cells with 10 µM EdU for 2 h in the complete medium. Afterwards, cells were washed and detached using trypsin for 5 min. The cell suspension was pelleted by centrifugation and washed with 3 mL DPBS. We resuspended the cells in 100 µL of 1%BSA in PBS and fixed with 100 µL of Click-iT® fixative by adding dropwise and incubated for 15 min. After fixation, we washed the cells with 3 mL of 1%BSA in PBS and permeabilized the cells with 100 µL of 1X Click-iT® saponin-based permeabilization buffer for 15 min. Afterwards 500 µL of Click-iT® reaction cocktail (prepared according to the manufacturer’s instructions) was added and incubated for another 30 min at room temperature in dark. We pelleted the cells, washed them once with 3 mL of permeabilization buffer, and discarded the supernatant. The cell pellet was resuspended in 500 μL of FxCycle PI/RNase (Invitrogen: F10797), filtrated, and immediately analyzed by the Attune cytometer.
We used an Attune acoustic focusing cytometer for data acquisition (Attune, Applied Biosystems, Life Technology Europe BV, Zug, Switzerland). Data analysis was performed with the FlowJo software v.10 (BD Biosciences, San Jose, CA, USA). We excluded debris and gated single cells for the analysis.
4.6. Viability MTT Assay for Drug Sensitivity
We seeded ACC Meso-1 at 2000 cells/100 µL/well in a 96-well plate. Cells were transfected with 20 nM siRNA at 24 h after seeding. We used 0.2 µL of Lipofectamine
® diluted in 10 µL of Opti-MEM
® medium per well and mixed with 240 nM siRNA duplexes diluted in 10 µL of Opti-MEM
®. The transfection complex (20 µL of complex containing 120 nM siRNA) was allowed to form for 15 min and was added dropwise to cells containing 100 µL of medium (final 20 nM). At 24 h after transfection, we aspirated the cell culture medium and replaced with complete medium containing an increasing concentration of cisplatin ranging from 2.5–40 µM (stock 1 mg/mL in 0.9% NaCl, Actavis, Regensdorf, Switzerland) using the diluent (0.9% NaCl) as the untreated control. At 48 h after cisplatin treatment, cell survival was measured by a colorimetric assay to rate the metabolic activity of a cell using tetrazole 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) using the protocol previously described [
38].
4.7. Protein Extraction and Western Blot
After removing the medium and floating cell debris, the adherent cells were enzymatically detached by trypsin, centrifuged, and washed with sterile DPBS. We resuspended the cell pellet in RIPA buffer (50 mM Tris HCl pH 8, 150 mM NaCl, 1% TritonX-100, 0.5% sodium deoxycholate, 0.1% SDS) containing a protease/phosphatase inhibitor cocktail (Cell Signaling, #5872, Danvers, MA, USA) for cell lysis for 20 min followed by pulse sonication 3 times of 10 s (Sonifier® S-450A, Branson Ultrasonics, Urdorf, Switzerland). We then centrifuged the cell extract at 15,000× g 4 °C for 20 min and collected the cell lysate. The protein concentration in the lysate was assessed by a Micro BCA Protein Assay Kit (Thermo Fisher, Reinach, Switzerland). Protein denaturation was performed with Laemmli buffer and heated at 95 °C for 10 min. We ran Western blots using homemade Tris-glycine gradient gels (4–20%) in Tris-glycine buffer. The gels were prepared with Rotiphorese® Gel 40 (29:1) (Carl Roth, Karlsruhe, Germany), Tris-HCl/SDS (pH 8.8), water, ammonium peroxodisulfate (APS) (Carl Roth, Karlsruhe, Germany), and tetramethylethylendiamine (TEMED) (Sigma Aldrich, Buchs, Switzerland). The Western blot analysis was conducted using equivalent amounts and volume of protein (range 10–40 µg), which were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred onto a polyvinylidene fluorid membrane (Immuno-Blot PVDF membrane, Bio-Rad, Cressier, Switzerland). Following transfer, the membranes were blocked with 5% blotting grade milk powder (Carl Roth, Karlsruhe, Germany) in Tris-buffered saline containing 0.1% TWEEN-20 (TBST, Sigma Aldrich, Buchs, Switzerland). Primary antibody incubations were carried out over night at 4 °C or for one hour at room temperature. The following primary antibodies were used for Western blot diluted in TBST containing 5% BSA and 0.01% sodium azide: CUL4B (HPA011880, Sigma-Aldrich Buchs, Switzerland, 1:250), CUL4A (2699S Cell Signaling, Danvers, MA, USA, 1:1000), and YAP/TAZ (D24E4 Cell Signaling, Danvers, MA, USA, 1:1000). CTGF (clone L-20 1:500), Actin (clone I-19 1-200), and secondary antibodies including mouse anti-rabbit IgG HRP (sc-2357 1:10,000), mouse anti-goat IgG HRP (sc-2354 1:10,000), and m-IgGκ BP-HRP (sc-516102 1:10,000) were from Santa Cruz, Heidelberg, Germany. The secondary horseradish peroxidase (HRP) antibodies were applied at room temperature at a 1:10,000 dilution in 1% milk in TBST for one hour. Subsequent and in between washes were carried out in TBST. Following the washes, the antigen/antibody complexes were detected by chemiluminescent detection (Clarity Western ECL Western blot substrate, Biorad, Cressier, Switzerland) using a chemiluminescent appliance (Fusion-FX7.826, program FusionCapt). The signal intensity of the bands was also quantified with the FusionCapt program.
4.8. RNA Extraction and Reverse Transcriptase Quantitative Real-Time PCR
For RNA isolation, cells were washed with 2 mL DPBS after removal of the culture medium. Immediately after DPBS removal, 1 mL of TRIzol reagent (Invitrogen 15596018, Life Technology Europe BV, Zug, Switzerland) was added for cell lysis. The cell lysate in TRIzol reagent was processed for RNA isolation according to the protocol suggested by the manufacturer. Then, 200–500 ng of RNA was used for cDNA synthesis using PrimeScript RT with gDNA Eraser (TAKARA Bio, S-86901-06-01, Saint-Germain-en-Laye, France). For quantitative real-time PCR, the KAPA SYBR FAST qPCR master mix with ROX (Kapa Biosystems, KK4621 Merck, Buchs, Switzerland) was used and run on a 7500 fast real-time PCR system (Life technology, Life Technology Europe BV, Zug, Switzerland). Gene expression was quantified using 2
-delta delta Ct using beta-Actin as the reference gene and compared to the non-silencing control (siLuciferase). The sequences of the used PCR primers are provided in
Supplementary Table S2. The primers were used in the PCR reaction at a final concentration of 0.2 µM.
4.9. ELISA
ELISA was performed using the cell culture supernatant collected at 48 h after siRNA transfection to minimize variation due to the increased number of dead cells in the siRNA-treated samples. We collected the cell culture supernatant and centrifuged at 400× g for 5 min at 4 °C to remove the floating cells. We harvested the supernatant and centrifuged again at 10,000× g for 10 min at 4 °C to remove the debris. The supernatant was frozen at −80 °C until the analysis. TGF-β1 ELISA with the activation of latent TGF-β1 was performed using Human TGF-β1 DuoSet ELISA (R&D Systems DY240-05) with DuoSet ELISA Ancillary Reagent Kit 1 (R&D Systems DY007) according to the protocol recommended by the manufacturer. We subtracted the TGF-β1 level with the level of medium from no-cell control wells, treated with mock transfection. Then, the level of TGF-β1 was corrected with the number of living cells and normalized to siLuc.
4.10. Statistical Analyses
We used GraphPad Prism v.8.0.0 (San Diego, CA, USA) for testing the differences of the means with parametric t-tests.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}