
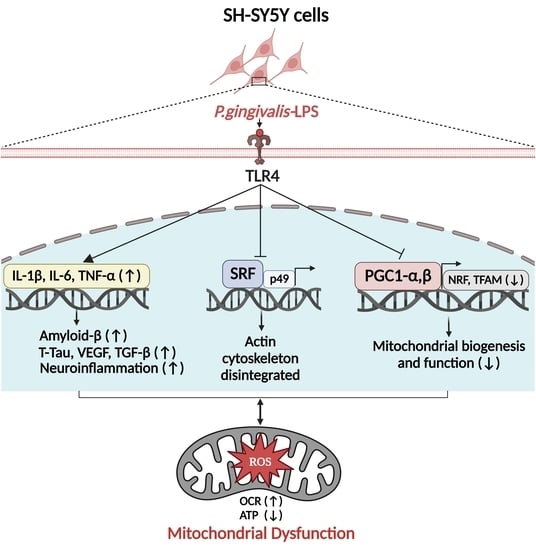
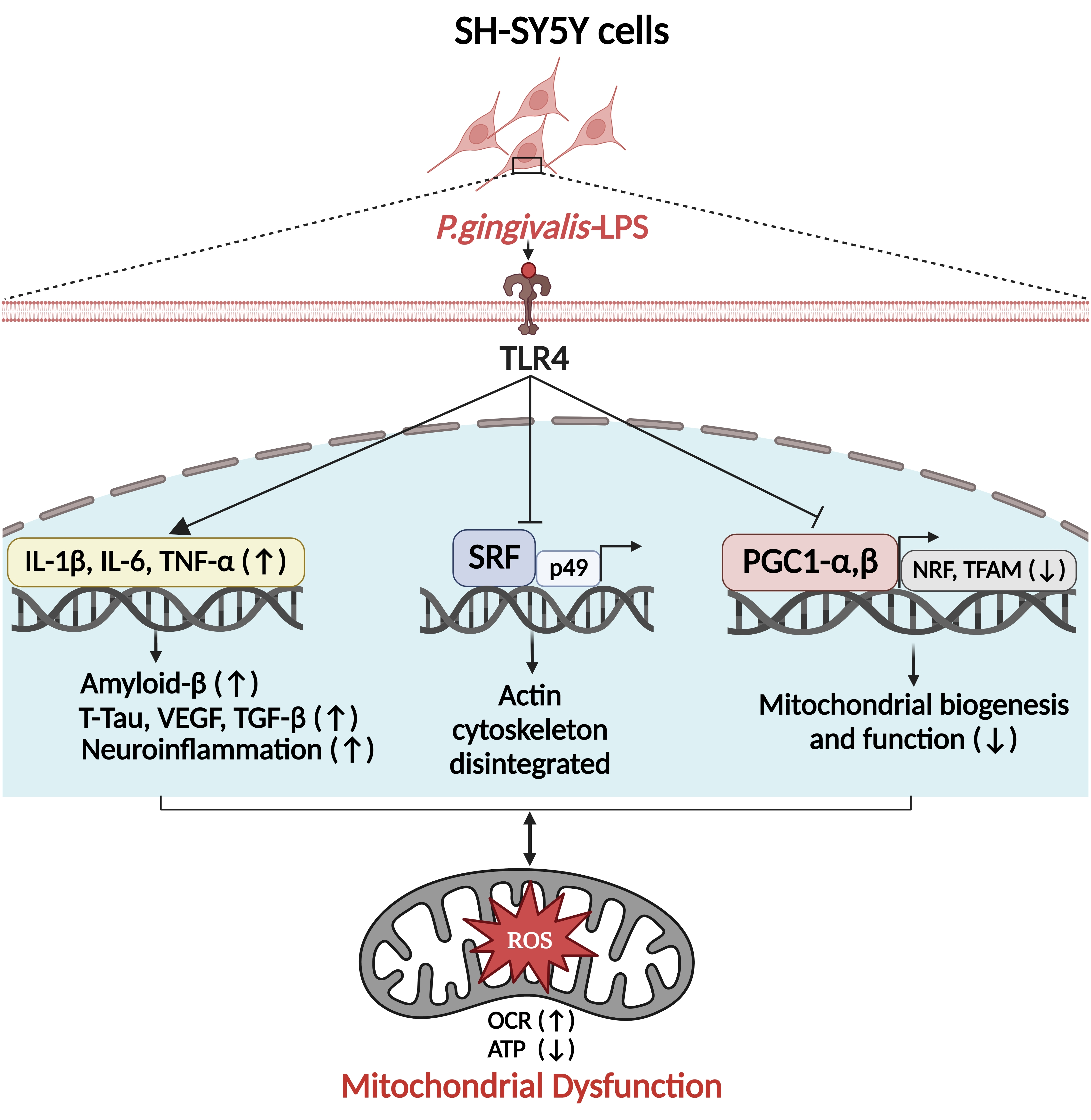
P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
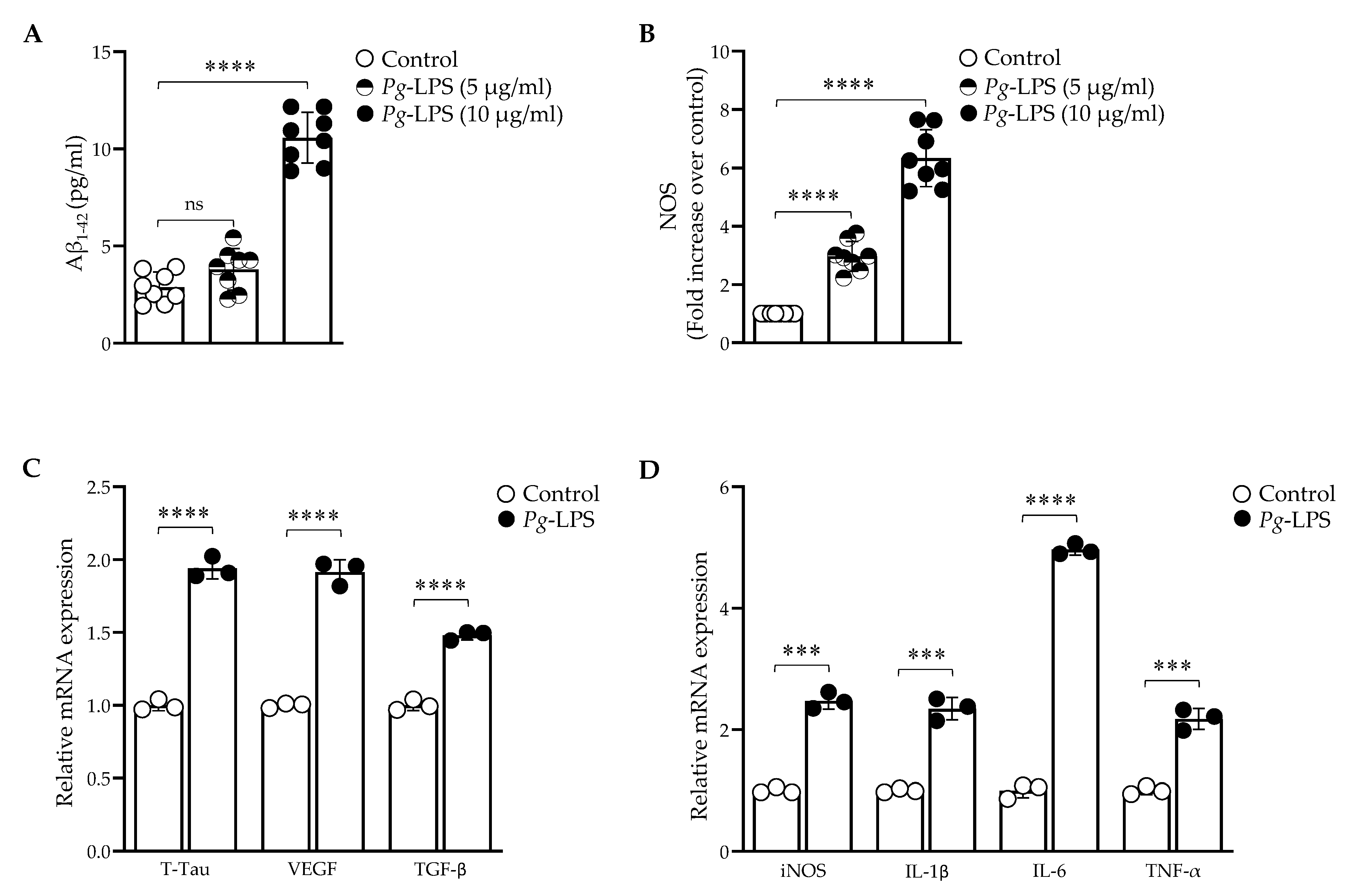
2.1. P. gingivalis-LPS Upregulates the Expression of Alzheimer Disease Related Dementia and Neuroinflammatory Markers
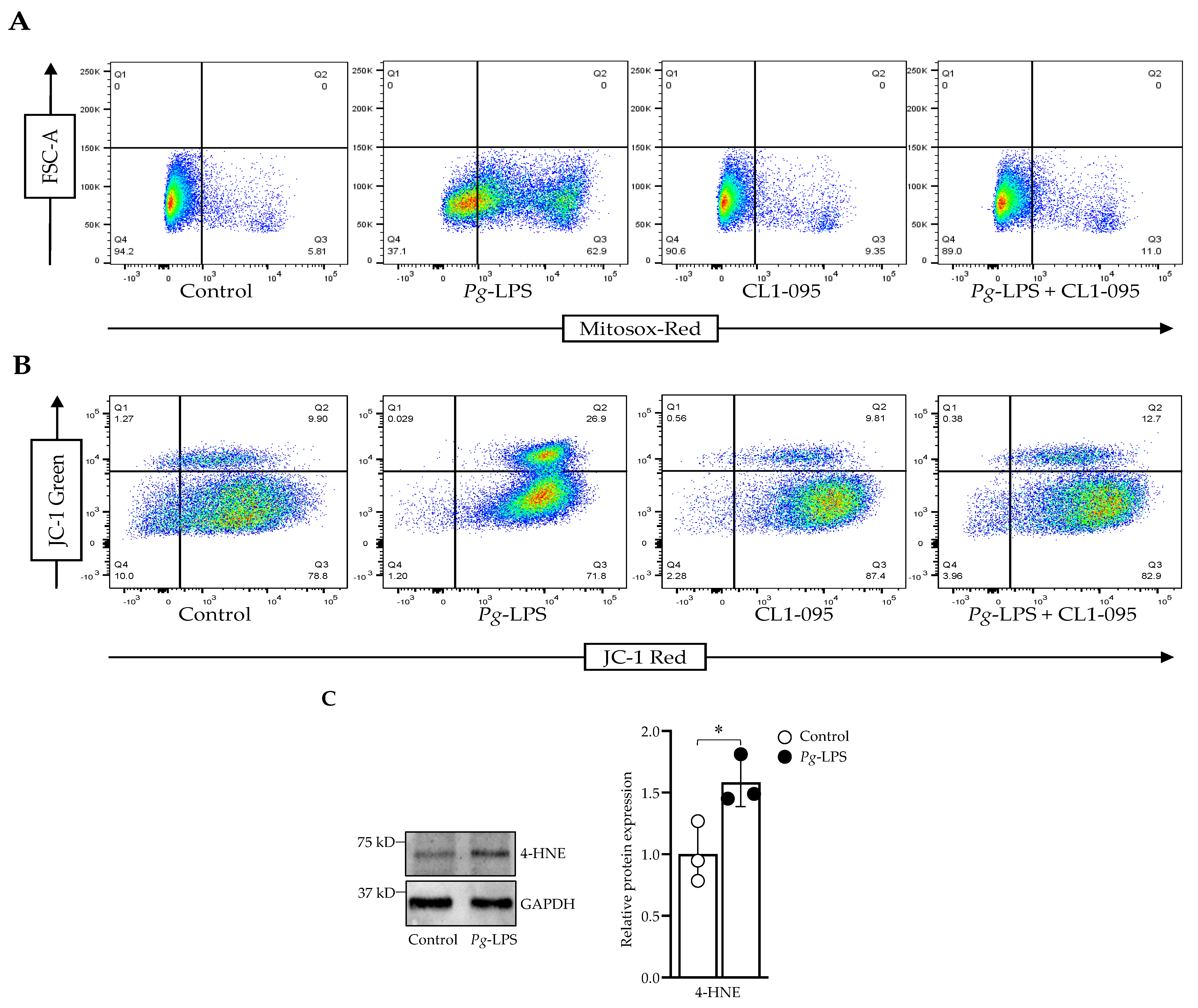
2.2. P. gingivalis-LPS Induces Mitochondrial ROS and Decreases Membrane Potential Mediated by TLR4
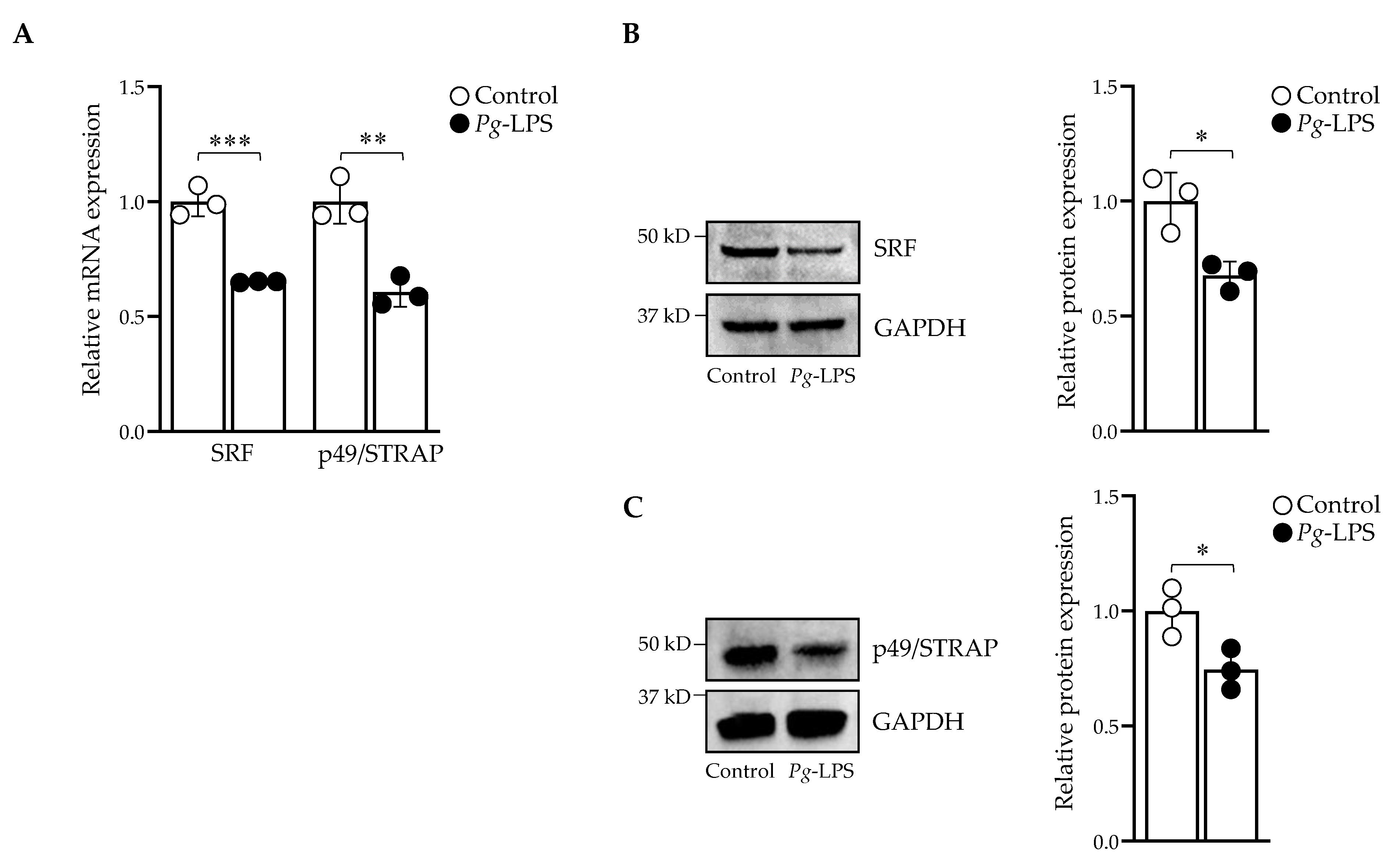
2.3. P. gingivalis-LPS Downregulates Serum Response Factor and p49/STRAP
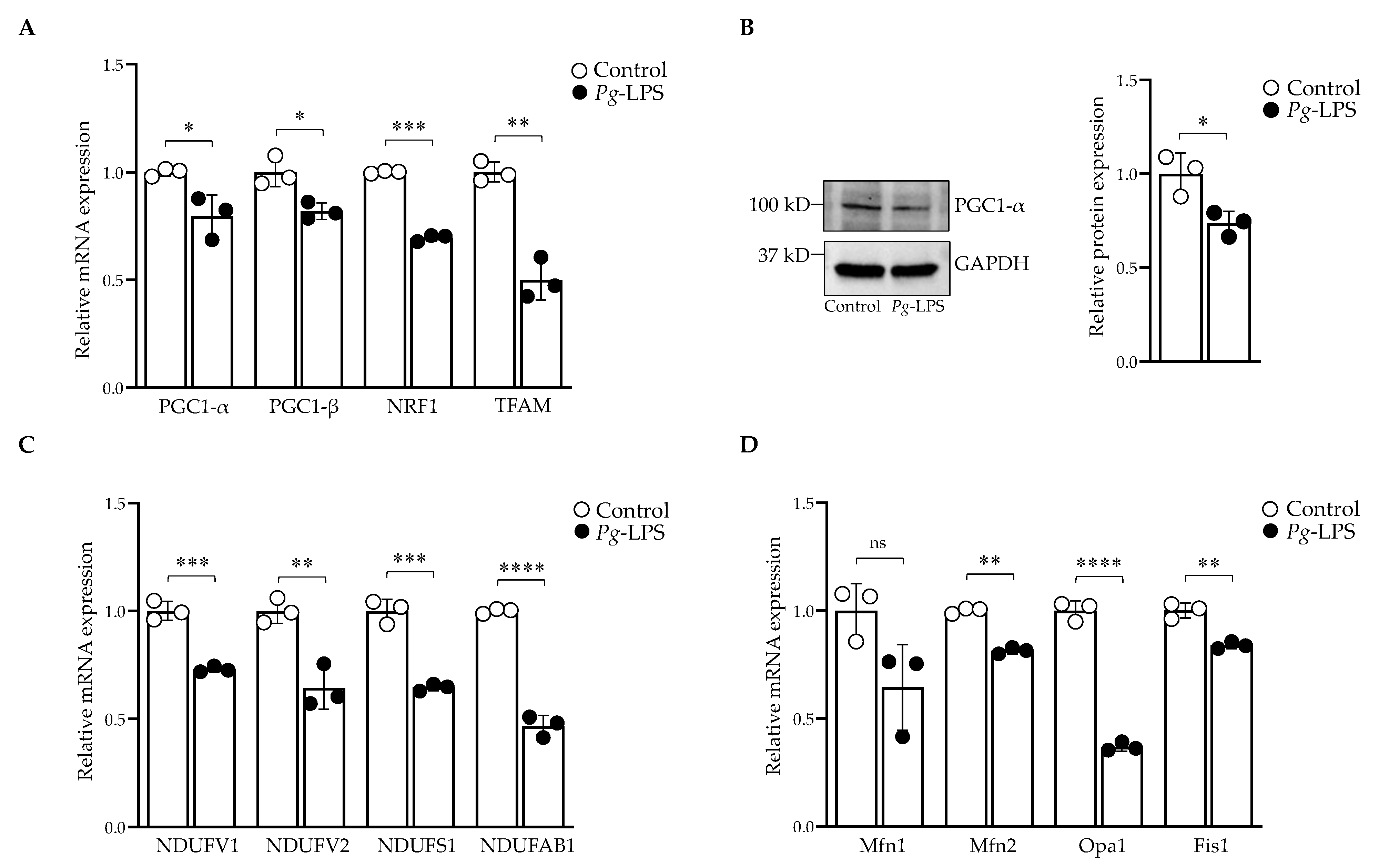
2.4. P. gingivalis-LPS Represses the Mitochondrial Biogenesis, Fission and Fusion Genes
2.5. P. gingivalis-LPS Alters Oxidative Phosphorylation, Glycolysis and Reduces the ATP
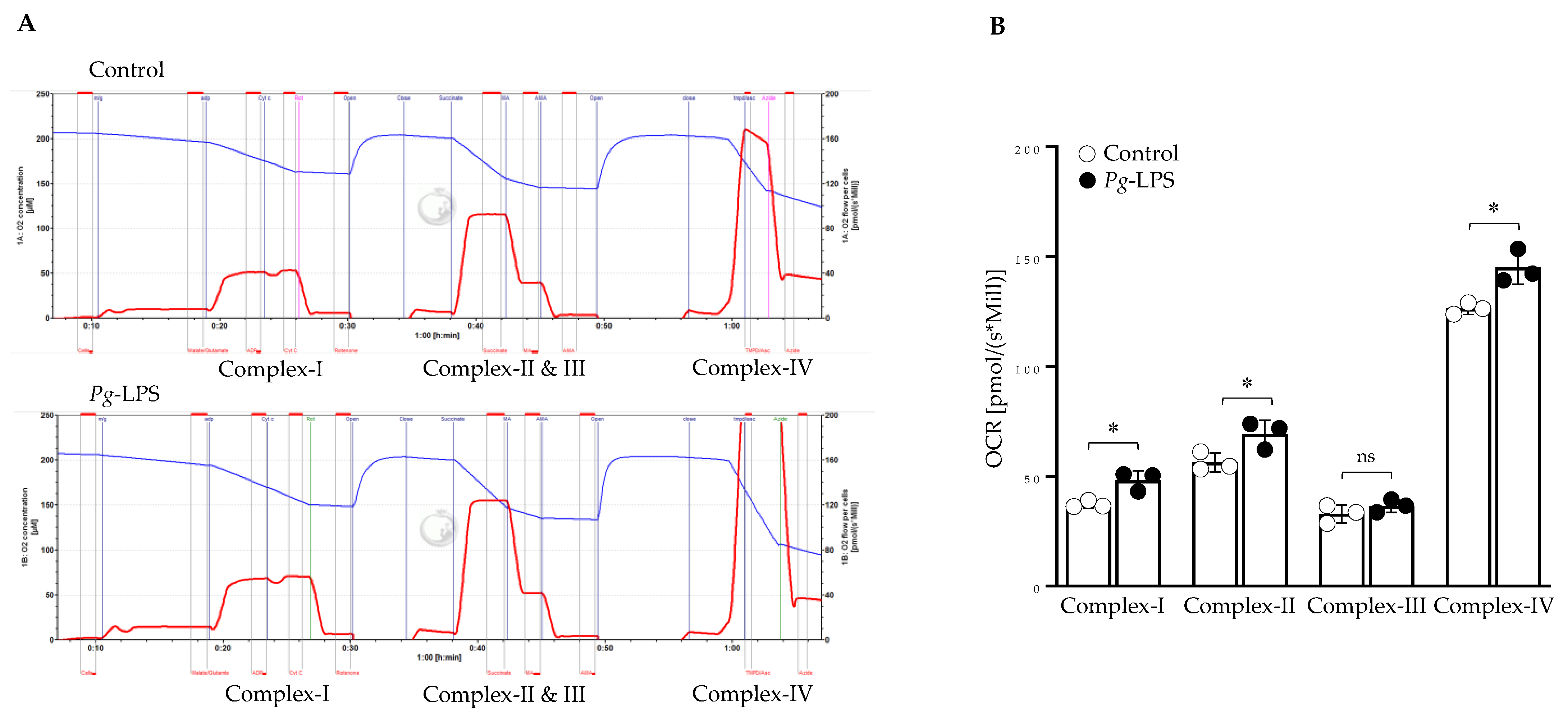
2.6. P. gingivalis-LPS Specifically Alters the Mitochondrial Function in Complex I, II, and IV
2.7. TLR4 Expression, Actin Assembly and Mitochondrial Morphology
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Cell Viability and LDH Assay
4.2. Quantification of Aβ1–42, NOS and Reverse-Transcriptase qPCR
4.3. Flow Cytometry
4.4. Western Blot
4.5. Mitochondrial Oxygen Consumption Rate, Glycolysis and ATP Production
4.6. High-Resolution Respirometry
4.7. Immunofluorescence
4.8. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, C.K.; Wu, Y.T.; Chang, Y.C. Association between Chronic Periodontitis and the Risk of Alzheimer’s Disease: A Retrospective, Population-Based, Matched-Cohort Study. Alzheimer’s Res. Ther. 2017, 9, 56. [Google Scholar] [CrossRef]
- Arrivé, E.; Letenneur, L.; Matharan, F.; Laporte, C.; Helmer, C.; Barberger-Gateau, P.; Miquel, J.L.; Dartigues, J.F. Oral Health Condition of French Elderly and Risk of Dementia: A Longitudinal Cohort Study: Elderly’s Oral Health and Risk of Dementia. Community Dent. Oral Epidemiol. 2012, 40, 230–238. [Google Scholar] [CrossRef]
- Fiorillo, L.; Cervino, G.; Laino, L.; D’Amico, C.; Mauceri, R.; Tozum, T.F.; Gaeta, M.; Cicciù, M. Porphyromonas gingivalis, Periodontal and Systemic Implications: A Systematic Review. Dent. J. 2019, 7, 114. [Google Scholar] [CrossRef]
- Ilievski, V.; Zuchowska, P.K.; Green, S.J.; Toth, P.T.; Ragozzino, M.E.; Le, K.; Aljewari, H.W.; O’Brien-Simpson, N.M.; Reynolds, E.C.; Watanabe, K. Chronic Oral Application of a Periodontal Pathogen Results in Brain Inflammation, Neurodegeneration and Amyloid Beta Production in Wild Type Mice. PLoS ONE 2018, 13, e0204941. [Google Scholar] [CrossRef]
- Olsen, I.; Singhrao, S.K. Is There a Link between Genetic Defects in the Complement Cascade and Porphyromonas gingivalis in Alzheimer’s Disease? J. Oral Microbiol. 2020, 12, 1676486. [Google Scholar] [CrossRef]
- Holt, S.C.; Kesavalu, L.; Walker, S.; Genco, C.A. Virulence Factors of Porphyromonas gingivalis. Periodontology 2000 1999, 20, 168–238. [Google Scholar] [CrossRef]
- Kim, S.; Shin, S.J.; Park, Y.H.; Nam, Y.; Kim, S.-M.; Kim, S.-M.; Jung, I.D.; Yang, H.D.; Park, Y.-M.; Moon, M.; et al. Gram-Negative Bacteria and Their Lipopolysaccharides in Alzheimer’s Disease: Pathologic Roles and Therapeutic Implications. Transl. Neurodegener. 2021, 10, 49. [Google Scholar] [CrossRef]
- Carter, C.J.; France, J.; Crean, S.; Singhrao, S.K. The Porphyromonas gingivalis/Host Interactome Shows Enrichment in GWASdb Genes Related to Alzheimer’s Disease, Diabetes and Cardiovascular Diseases. Front. Aging Neurosci. 2017, 9, 408. [Google Scholar] [CrossRef]
- Paik, Y. Toll-Like Receptor 4 Mediates Inflammatory Signaling by Bacterial Lipopolysaccharide in Human Hepatic Stellate Cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef]
- Noble, J.M.; Scarmeas, N.; Celenti, R.S.; Elkind, M.S.V.; Wright, C.B.; Schupf, N.; Papapanou, P.N. Serum IgG Antibody Levels to Periodontal Microbiota Are Associated with Incident Alzheimer Disease. PLoS ONE 2014, 9, e114959. [Google Scholar] [CrossRef]
- Martin, M.; Katz, J.; Vogel, S.N.; Michalek, S.M. Differential Induction of Endotoxin Tolerance by Lipopolysaccharides Derived from Porphyromonas gingivalis and Escherichia coli. J. Immunol. 2001, 167, 5278–5285. [Google Scholar] [CrossRef]
- Jones, K.J.; Ekhlassi, S.; Montufar-Solis, D.; Klein, J.R.; Schaefer, J.S. Differential Cytokine Patterns in Mouse Macrophages and Gingival Fibroblasts After Stimulation with Porphyromonas gingivalis or Escherichia Coli Lipopolysaccharide. J. Periodontol. 2010, 81, 1850–1857. [Google Scholar] [CrossRef]
- Poole, S.; Singhrao, S.K.; Kesavalu, L.; Curtis, M.A.; Crean, S. Determining the Presence of Periodontopathic Virulence Factors in Short-Term Postmortem Alzheimer’s Disease Brain Tissue. J. Alzheimer’s Dis. 2013, 36, 665–677. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Harding, A.; Poole, S.; Kesavalu, L.; Crean, S. Porphyromonas gingivalis Periodontal Infection and Its Putative Links with Alzheimer’s Disease. Mediat. Inflamm. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment with Small-Molecule Inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Castegna, A.; Pocernich, C.B.; Drake, J.; Scapagnini, G.; Calabrese, V. Nutritional Approaches to Combat Oxidative Stress in Alzheimer’s Disease. J. Nutr. Biochem. 2002, 13, 444–461. [Google Scholar] [CrossRef]
- Cabezas-Opazo, F.A.; Vergara-Pulgar, K.; Pérez, M.J.; Jara, C.; Osorio-Fuentealba, C.; Quintanilla, R.A. Mitochondrial Dysfunction Contributes to the Pathogenesis of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2015, 2015, 1–12. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Carlson, E.A.; Rao, V.K.; Yan, S.S. From a Cell’s Viewpoint: Targeting Mitochondria in Alzheimer’s Disease. Drug Discov. Today Ther. Strateg. 2013, 10, e91–e98. [Google Scholar] [CrossRef][Green Version]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and Reactive Oxygen Species. Free. Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive Oxygen Species Production by Mitochondria. In Mitochondrial DNA; Stuart, J.A., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; Volume 554, pp. 165–181. ISBN 978-1-934115-60-2. [Google Scholar]
- Hirst, J.; King, M.S.; Pryde, K.R. The Production of Reactive Oxygen Species by Complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Dröse, S. Superoxide Generation by Complex III: From Mechanistic Rationales to Functional Consequences. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M.-Y. Increased Lipid Peroxidation Precedes Amyloid Plaque Formation in an Animal Model of Alzheimer Amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef]
- Galbusera, C.; Facheris, M.; Magni, F.; Galimberti, G.; Sala, G.; Tremolada, L.; Isella, V.; Guerini, F.; Appollonio, I.; Galli-Kienle, M.; et al. Increased Susceptibility to Plasma Lipid Peroxidation in Alzheimer Disease Patients. Curr. Alzheimer Res. 2004, 1, 103–109. [Google Scholar] [CrossRef]
- Kinane, J.A.; Benakanakere, M.R.; Zhao, J.; Hosur, K.B.; Kinane, D.F. Porphyromonas gingivalis Influences Actin Degradation within Epithelial Cells during Invasion and Apoptosis: Actin Cytoskeleton Degradation by P. gingivalis. Cell. Microbiol. 2012, 14, 1085–1096. [Google Scholar] [CrossRef]
- Herath, T.D.K.; Darveau, R.P.; Seneviratne, C.J.; Wang, C.-Y.; Wang, Y.; Jin, L. Heterogeneous Porphyromonas gingivalis LPS Modulates Immuno-Inflammatory Response, Antioxidant Defense and Cytoskeletal Dynamics in Human Gingival Fibroblasts. Sci. Rep. 2016, 6, 29829. [Google Scholar] [CrossRef]
- Beck, H.; Flynn, K.; Lindenberg, K.S.; Schwarz, H.; Bradke, F.; Di Giovanni, S.; Knöll, B. Serum Response Factor (SRF)-Cofilin-Actin Signaling Axis Modulates Mitochondrial Dynamics. Proc. Natl. Acad. Sci. USA. 2012, 109, E2523–E2532. [Google Scholar] [CrossRef]
- Schratt, G.; Philippar, U.; Berger, J.; Schwarz, H.; Heidenreich, O.; Nordheim, A. Serum Response Factor Is Crucial for Actin Cytoskeletal Organization and Focal Adhesion Assembly in Embryonic Stem Cells. J. Cell Biol. 2002, 156, 737–750. [Google Scholar] [CrossRef]
- Zhang, X.; Azhar, G.; Zhong, Y.; Wei, J.Y. Identification of a Novel Serum Response Factor Cofactor in Cardiac Gene Regulation. J. Biol. Chem. 2004, 279, 55626–55632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Azhar, G.; Helms, S.; Zhong, Y.; Wei, J.Y. Identification of a Subunit of NADH-Dehydrogenase as a P49/STRAP-Binding Protein. BMC Cell Biol. 2008, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Williams, E.D.; Azhar, G.; Rogers, S.C.; Wei, J.Y. Does P49/STRAP, a SRF-Binding Protein (SRFBP1), Modulate Cardiac Mitochondrial Function in Aging? Exp. Gerontol. 2016, 82, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. In Neuronal Cell Culture; Amini, S., White, M.K., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1078, pp. 9–21. ISBN 978-1-62703-639-9. [Google Scholar] [CrossRef]
- Xun, Z.; Lee, D.-Y.; Lim, J.; Canaria, C.A.; Barnebey, A.; Yanonne, S.M.; McMurray, C.T. Retinoic Acid-Induced Differentiation Increases the Rate of Oxygen Consumption and Enhances the Spare Respiratory Capacity of Mitochondria in SH-SY5Y Cells. Mech. Ageing Dev. 2012, 133, 176–185. [Google Scholar] [CrossRef]
- Forster, J.I.; Köglsberger, S.; Trefois, C.; Boyd, O.; Baumuratov, A.S.; Buck, L.; Balling, R.; Antony, P.M.A. Characterization of Differentiated SH-SY5Y as Neuronal Screening Model Reveals Increased Oxidative Vulnerability. SLAS Discov. 2016, 21, 496–509. [Google Scholar] [CrossRef]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed Mitochondrial Dynamics, and Neurodegenerative Disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial Dynamics and Transport in Alzheimer’s Disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Loo, G. Inflammasomes in Neuroinflammatory and Neurodegenerative Diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Vande Walle, L.; Jordao, M.J.C.; et al. A20 Critically Controls Microglia Activation and Inhibits Inflammasome-Dependent Neuroinflammation. Nat. Commun. 2018, 9, 2036. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 Inflammasomes Are Activated in Alzheimer’s Disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and Tau Toxicities in Alzheimer’s Are Linked via Oxidative Stress-Induced P38 Activation: Protective Role of Vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Pandur, E.; Varga, E.; Tamási, K.; Pap, R.; Nagy, J.; Sipos, K. Effect of Inflammatory Mediators Lipopolysaccharide and Lipoteichoic Acid on Iron Metabolism of Differentiated SH-SY5Y Cells Alters in the Presence of BV-2 Microglia. Int. J. Mol. Sci. 2018, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Nativel, B.; Couret, D.; Giraud, P.; Meilhac, O.; d’Hellencourt, C.L.; Viranaïcken, W.; Da Silva, C.R. Porphyromonas gingivalis Lipopolysaccharides Act Exclusively through TLR4 with a Resilience between Mouse and Human. Sci. Rep. 2017, 7, 15789. [Google Scholar] [CrossRef] [PubMed]
- Rolls, A.; Shechter, R.; London, A.; Ziv, Y.; Ronen, A.; Levy, R.; Schwartz, M. Toll-like Receptors Modulate Adult Hippocampal Neurogenesis. Nat. Cell Biol. 2007, 9, 1081–1088. [Google Scholar] [CrossRef]
- Acosta, C.; Davies, A. Bacterial Lipopolysaccharide Regulates Nociceptin Expression in Sensory Neurons. J. Neurosci. Res. 2008, 86, 1077–1086. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Gölz, L.; Memmert, S.; Rath-Deschner, B.; Jäger, A.; Appel, T.; Baumgarten, G.; Götz, W.; Frede, S. LPS from P. gingivalis and Hypoxia Increases Oxidative Stress in Periodontal Ligament Fibroblasts and Contributes to Periodontitis. Mediat. Inflamm. 2014, 2014, 986264. [Google Scholar] [CrossRef]
- Charoensaensuk, V.; Chen, Y.-C.; Lin, Y.-H.; Ou, K.-L.; Yang, L.-Y.; Lu, D.-Y. Porphyromonas gingivalis Induces Proinflammatory Cytokine Expression Leading to Apoptotic Death through the Oxidative Stress/NF-ΚB Pathway in Brain Endothelial Cells. Cells 2021, 10, 3033. [Google Scholar] [CrossRef]
- Connelly, J.T.; Gautrot, J.E.; Trappmann, B.; Tan, D.W.-M.; Donati, G.; Huck, W.T.S.; Watt, F.M. Actin and Serum Response Factor Transduce Physical Cues from the Microenvironment to Regulate Epidermal Stem Cell Fate Decisions. Nat. Cell Biol. 2010, 12, 711–718. [Google Scholar] [CrossRef]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A Role for Peroxisome Proliferator-Activated Receptor γ Coactivator-1 in the Control of Mitochondrial Dynamics During Postnatal Cardiac Growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef]
- Chen, S.-D.; Yang, D.-I.; Lin, T.-K.; Shaw, F.-Z.; Liou, C.-W.; Chuang, Y.-C. Roles of Oxidative Stress, Apoptosis, PGC-1α and Mitochondrial Biogenesis in Cerebral Ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [PubMed]
- Patyal, P.; Nguyen, B.; Zhang, X.; Azhar, G.; Ameer, F.S.; Verma, A.; Crane, J.; Kc, G.; Che, Y.; Wei, J.Y. Rho/SRF Inhibitor Modulates Mitochondrial Functions. Int. J. Mol. Sci. 2022, 23, 11536. [Google Scholar] [CrossRef] [PubMed]
- Napa, K.; Baeder, A.C.; Witt, J.E.; Rayburn, S.T.; Miller, M.G.; Dallon, B.W.; Gibbs, J.L.; Wilcox, S.H.; Winden, D.R.; Smith, J.H.; et al. LPS from P. gingivalis Negatively Alters Gingival Cell Mitochondrial Bioenergetics. Int. J. Dent. 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Xu, T.; Dong, Q.; Luo, Y.; Liu, Y.; Gao, L.; Pan, Y.; Zhang, D. Porphyromonas gingivalis Infection Promotes Mitochondrial Dysfunction through Drp1-Dependent Mitochondrial Fission in Endothelial Cells. Int. J. Oral Sci. 2021, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Liao, Y.; Qiu, W.; Li, L.; Li, D.; Cao, X.; Ai, B. Targeting Toll-like Receptor 4 with CLI-095 (TAK-242) Enhances the Antimetastatic Effect of the Estrogen Receptor Antagonist Fulvestrant on Non-Small Cell Lung Cancer. Clin. Transl. Oncol. 2020, 22, 2074–2086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ameer, F.S.; Azhar, G.; Wei, J.Y. Alternative Splicing Increases Sirtuin Gene Family Diversity and Modulates Their Subcellular Localization and Function. Int. J. Mol. Sci. 2021, 22, 473. [Google Scholar] [CrossRef]
- Zhang, X.; Azhar, G.; Wei, J.Y. The Expression of MicroRNA and MicroRNA Clusters in the Aging Heart. PLoS ONE 2012, 7, e34688. [Google Scholar] [CrossRef]
- Wang, X.; Yao, W.; Wang, M.; Zhu, J.; Xia, L. TLR4-SIRT3 Mechanism Modulates Mitochondrial and Redox Homeostasis and Promotes EPCs Recruitment and Survival. Oxidative Med. Cell. Longev. 2022, 2022, 1282362. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control: An Introduction to OXPHOS Analysis, 5th ed.; Bioenergetics Communications: Axams, Austria, 2020. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, A.; Azhar, G.; Zhang, X.; Patyal, P.; Kc, G.; Sharma, S.; Che, Y.; Wei, J.Y. P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress. Int. J. Mol. Sci. 2023, 24, 950. https://doi.org/10.3390/ijms24020950
Verma A, Azhar G, Zhang X, Patyal P, Kc G, Sharma S, Che Y, Wei JY. P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress. International Journal of Molecular Sciences. 2023; 24(2):950. https://doi.org/10.3390/ijms24020950
Chicago/Turabian StyleVerma, Ambika, Gohar Azhar, Xiaomin Zhang, Pankaj Patyal, Grishma Kc, Shakshi Sharma, Yingni Che, and Jeanne Y. Wei. 2023. "P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress" International Journal of Molecular Sciences 24, no. 2: 950. https://doi.org/10.3390/ijms24020950
APA StyleVerma, A., Azhar, G., Zhang, X., Patyal, P., Kc, G., Sharma, S., Che, Y., & Wei, J. Y. (2023). P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress. International Journal of Molecular Sciences, 24(2), 950. https://doi.org/10.3390/ijms24020950