

Small Molecule Inhibitors for Unc-51-like Autophagy-Activating Kinase Targeting Autophagy in Cancer


Abstract
1. Introduction
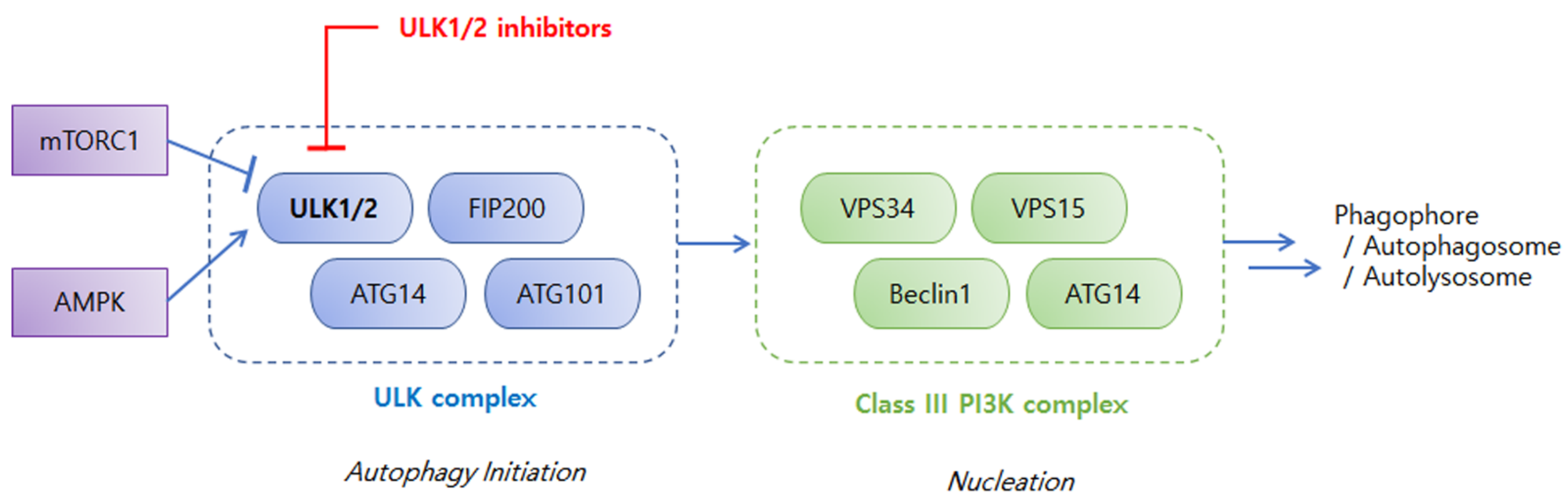
2. An Overview of ULKs in Autophagy and Cancers
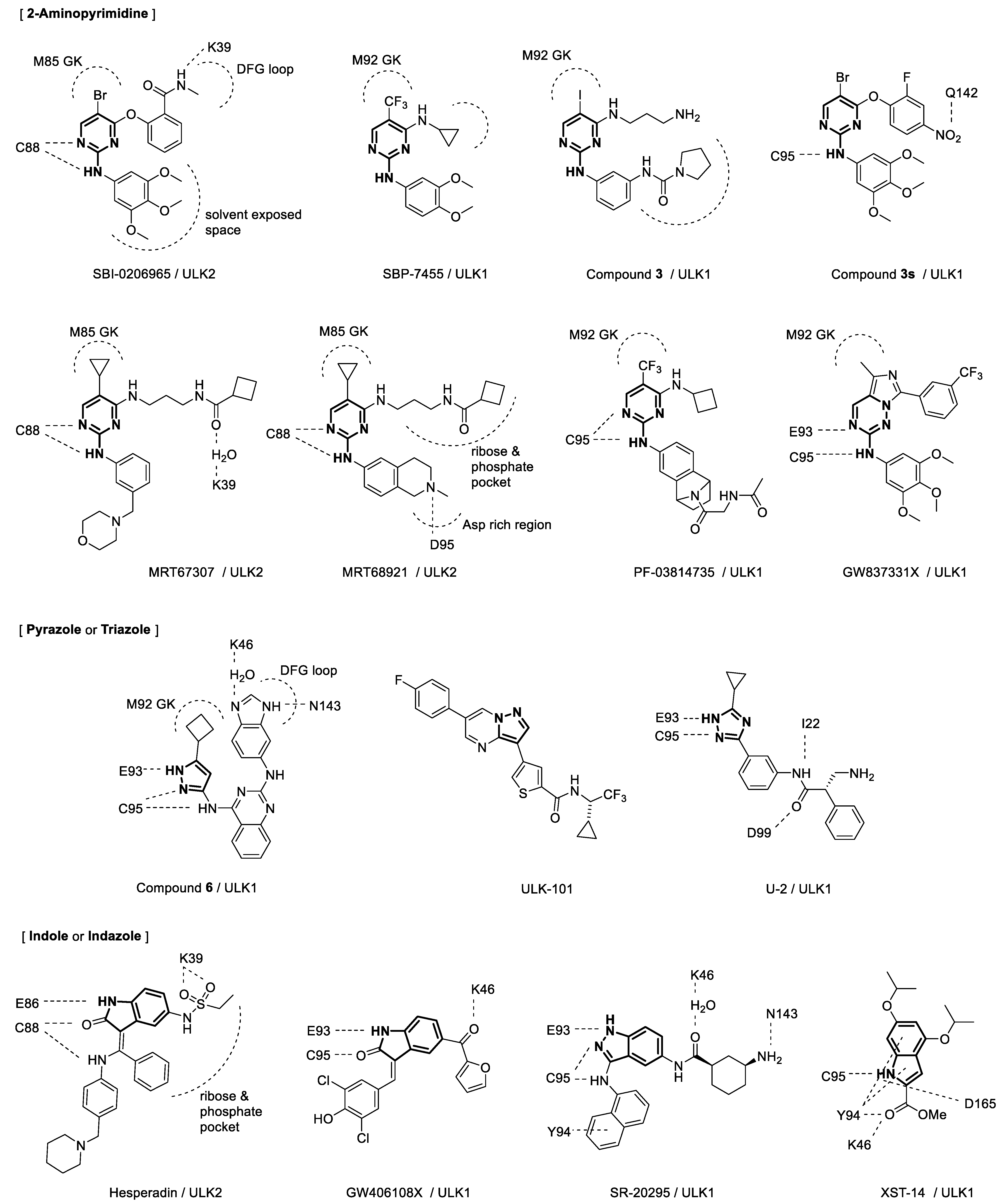
3. Discovery and Development of ULK Inhibitors
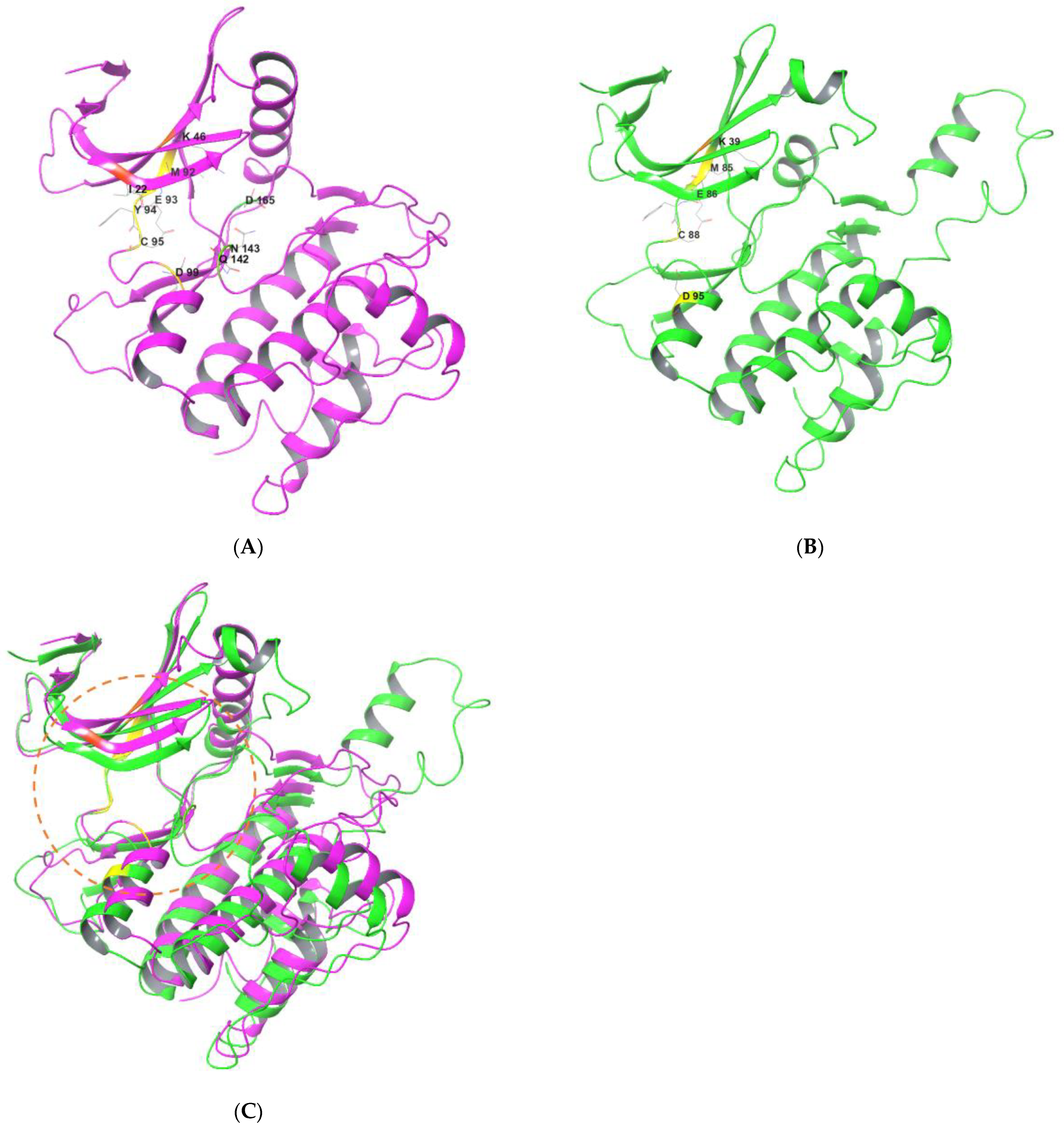
4. Binding Interactions between ULK1/2 and Their Inhibitors
5. Biological and Anticancer Effects of ULK Inhibitors

{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (ULK1) | IC50 (ULK2) | Cancer (Cell Line) | Synergistic Co-Treatment | |
|---|---|---|---|---|---|
| 1 | SBI-0206965 | 108 nM [38] 306 nM [22] 130 nM [39] | 711 nM [38] 3.88 μM [22] | Non-small cell lung cancer (A549, H460) [38,54] Neuroblastoma (SK-N-AS, SH-SY5Y, SK-N-DZ) [46] Renal cell carcinoma (A498, ACHN) [47] Leukemia (MV4-11, MOLM-13, HL-60, U937) [25,55] Breast cancer (MDA-MB-231, MCF-7) [56] | mTOR inhibitor AZD8055 [38], daunorubicin [55], doxorubicin [56], cisplatin [54], TRAIL a [46] |
| 2 | SBP-7455 | 13 nM [39] | Triple negative breast cancer (MDA-MB-468, BT549, MDA-MB-231) [39] | PARP inhibitors olaparib, niraparib [39] | |
| 3 | Compound 6 | 8 nM [21] | - | ||
| 4 | Compound 3 | 120 nM [40] | 360 nM [40] | - | |
| 5 | MRT67307 | 45 nM [24] 170 nM [40] 38.2 nM [22] | 38 nM [24] 230 nM [40] 92.3 nM [22] | Leukemia (THP-1, U939, Molt4, HEL92.1.7, K562, Raji, Jurkat, HL60) [57] | |
| 6 | MRT68921 | 2.9 nM [24] 17.0 nM [22] | 1.1 nM [24] 20.8 nM [22] | Mesothelioma (M28, REN) [58] b Leukemia (REH, MV4-11, MOLM-13, HL-60, U937, THP-1, Molt4, HEL92.1.7, K562, Raji, Jurkat, HL60) [25,49,57] Ovarian cancer (OVCAR3/4/8, COV318/362, CaOV3) [50] Various tumors [48] | Carboplatin and pemetrexed [58] |
| 7 | Compound 3s | 99.15% inhibition at 10 μM [42] c | Lung cancer (A549) [42] Lymphoma (U937) [42] Breast cancer (HL60) [42] Acute myeloid leukemia (MDA-MB-469) [42] | ||
| 8 | PF-03814735 | KD 18.1 nM [22] d | KD 58.0 nM [22] d | Solid tumors [51,52] | |
| 9 | Hesperidin | KD 16.8 nM [22] d | KD 47.3 nM [22] d | Various tumors [53] | |
| 10 | SR-20295 | 45 nM [43] | - | ||
| 11 | ULK-101 | 8.3 nM [18] | 30 nM [18] | Osteosarcoma (U2OS) [18] Non-small cell lung cancer (H838, H727, H2030, A549) [18] | |
| 12 | XST-14 | 13.6 nM [44] | 70.9 nM [44] | Hepatocellular carcinoma (HepG2, Hep3B) [44] | sorafenib [44] |
| 13 | GW837331X | 646 nM [41] | Similar to ULK1 e [41] | - | |
| 14 | GW406108X | 427 nM [41] | Similar to ULK1 e [41] | - | |
| 15 | U-2 | 0.5 μM [45] | Hepatocellular carcinoma (SMMC-7721, HepG2, L02) [45] |
6. Selectivity Issues in ULK Inhibitors
7. Combination Therapy of ULK Inhibitors with Other Anticancer Agents
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kocak, M.; Erdi, S.E.; Jorba, G.; Maestro, I.; Farrés, J.; Kirkin, V.; Martinez, A.; Pless, O. Targeting autophagy in disease: Established and new strategies. Autophagy 2021, 18, 473–495. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. The incredible ULKs. Cell Commun. Signal. 2012, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autoph-agosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 1–15. [Google Scholar]
- Zhang, L.; Ouyang, L.; Guo, Y.; Zhang, J.; Liu, B. UNC-51-like Kinase 1: From an Autophagic Initiator to Multifunctional Drug Target. J. Med. Chem. 2018, 61, 6491–6500. [Google Scholar] [CrossRef]
- Chmurska, A.; Matczak, K.; Marczak, A. Two Faces of Autophagy in the Struggle against Cancer. Int. J. Mol. Sci. 2021, 22, 2981. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy re-sponses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Kumar, P.; Jagtap, Y.A.; Patwa, S.M.; Kinger, S.; Dubey, A.R.; Prajapati, V.K.; Dhiman, R.; Poluri, K.M.; Mishra, A. Autophagy based cellular physiological strategies target oncogenic progression. J. Cell. Physiol. 2021, 237, 258–277. [Google Scholar] [CrossRef]
- Liu, L.; Yan, L.; Liao, N.; Wu, W.-Q.; Shi, J.-L. A Review of ULK1-Mediated Autophagy in Drug Resistance of Cancer. Cancers 2020, 12, 352. [Google Scholar] [CrossRef]
- Lin, C.; Tsao, Y.; Shu, C. Autophagy modulation as a potential targeted cancer therapy: From drug repurposing to new drug development. Kaohsiung J. Med Sci. 2021, 37, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Limpert, A.S.; Lambert, L.J.; Bakas, N.A.; Bata, N.; Brun, S.N.; Shaw, R.J.; Cosford, N.D. Autophagy in Cancer: Regulation by Small Molecules. Trends Pharmacol. Sci. 2018, 39, 1021–1032. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, Y.; Zhang, J.; Chen, L. Inhibiting Cytoprotective Autophagy in Cancer Therapy: An Update on Pharmacological Small-Molecule Compounds. Front. Pharmacol. 2022, 13, 966012. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed]
- Kondapuram, S.K.; Sarvagalla, S.; Coumar, M.S. Targeting autophagy with small molecules for cancer therapy. J. Cancer Metastasis Treat. 2019, 5, 32. [Google Scholar] [CrossRef]
- Gillson, J.; El-Aziz, Y.S.A.; Leck, L.Y.W.; Jansson, P.J.; Pavlakis, N.; Samra, J.S.; Mittal, A.; Sahni, S. Autophagy: A Key Player in Pancreatic Cancer Progression and a Potential Drug Target. Cancers 2022, 14, 3528. [Google Scholar] [CrossRef]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2020, 11, 1422. [Google Scholar] [CrossRef]
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience 2018, 8, 74–84. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Ro, S.-H.; Jung, C.H.; Hahn, W.S.; Xu, X.; Kim, Y.-M.; Yun, Y.S.; Park, J.-M.; Kim, K.H.; Seo, M.; Ha, T.-Y. Distinct functions of Ulk1 and Ulk2 in the regulation of lipid metabolism in adipocytes. Autophagy 2013, 9, 2103–2114. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the Human Autophagy Initiating Kinase ULK1 in Complex with Potent Inhibitors. ACS Chem. Biol. 2015, 10, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Koschade, S.E.; Stolz, A.; Zivkovic, K.; Pohl, C.; Shaid, S.; Ren, H.; Lambert, L.J.; Cosford, N.D.P.; Brandts, C.H.; et al. Conservation of structure, function and inhibitor binding in UNC-51-like kinase 1 and 2 (ULK1/2). Biochem. J. 2019, 476, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.G.; Hurley, J.H. Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 2016, 39, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef]
- Hwang, D.Y.; Eom, J.-I.; Jang, J.E.; Jeung, H.-K.; Chung, H.; Kim, J.S.; Cheong, J.-W.; Min, Y.H. ULK1 inhibition as a targeted therapeutic strategy for FLT3-ITD-mutated acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2020, 39, 85. [Google Scholar] [CrossRef]
- Demeter, A.; Romero-Mulero, M.C.; Csabai, L.; Ölbei, M.; Sudhakar, P.; Haerty, W.; Korcsmáros, T. ULK1 and ULK2 are less redundant than previously thought: Computational analysis uncovers distinct regulation and functions of these autophagy induction proteins. Sci. Rep. 2020, 10, 10940. [Google Scholar] [CrossRef]
- Preuss, F.; Chatterjee, D.; Mathea, S.; Shrestha, S.; St-Germain, J.; Saha, M.; Kannan, N.; Raught, B.; Rottapel, R.; Knapp, S. Nucleotide Binding, Evolutionary Insights, and Interaction Partners of the Pseudokinase Unc-51-like Kinase 4. Structure 2020, 28, 1184–1196.e6. [Google Scholar] [CrossRef]
- Mathea, S.; Salah, E.; Tallant, C.; Chatterjee, D.; Berger, B.-T.; Konietzny, R.; Müller, S.; Kessler, B.M.; Knapp, S. Conformational plasticity of the ULK3 kinase domain. Biochem. J. 2021, 478, 2811–2823. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Cheray, M.; Keane, L.; Engskog-Vlachos, P.; Joseph, B. ULK3-dependent activation of GLI1 promotes DNMT3A expression upon autophagy induction. Autophagy 2022, 18, 2769–2780. [Google Scholar] [CrossRef]
- Maloverjan, A.; Piirsoo, M.; Kasak, L.; Peil, L.; Østerlund, T.; Kogerman, P. Dual Function of UNC-51-like Kinase 3 (Ulk3) in the Sonic Hedgehog Signaling Pathway. J. Biol. Chem. 2010, 285, 30079–30090. [Google Scholar] [CrossRef]
- Kasak, L.; Näks, M.; Eek, P.; Piirsoo, A.; Bhadoria, R.; Starkov, P.; Saarma, M.; Kasvandik, S.; Piirsoo, M. Characterization of Protein Kinase ULK3 Regulation by Phosphorylation and Inhibition by Small Molecule SU6668. Biochemistry 2018, 57, 5456–5465. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S.; et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171. [Google Scholar] [CrossRef] [PubMed]
- Goruppi, S.; Procopio, M.-G.; Jo, S.; Clocchiatti, A.; Neel, V.; Dotto, G.P. The ULK3 Kinase Is Critical for Convergent Control of Cancer-Associated Fibroblast Activation by CSL and GLI. Cell Rep. 2017, 20, 2468–2479. [Google Scholar] [CrossRef] [PubMed]
- Khamrui, S.; Ung, P.M.; Secor, C.; Schlessinger, A.; Lazarus, M.B. High-resolution structure and inhibition of the schizophrenia-linked pseudokinase ULKJ. Am. Chem. Soc. 2019, 142, 33–37. [Google Scholar] [CrossRef]
- Luo, S.; Zheng, N.; Lang, B. ULK4 in Neurodevelopmental and Neuropsychiatric Disorders. Front. Cell Dev. Biol. 2022, 10, 821. [Google Scholar] [CrossRef]
- Edelbusch, C.; Cindrić, S.; Dougherty, G.W.; Loges, N.T.; Olbrich, H.; Rivlin, J.; Wallmeier, J.; Pennekamp, P.; Amirav, I.; Omran, H. Mutation of serine/threonine protein kinase 36 (STK36) causes primary ciliary dyskinesia with a central pair defect. Hum. Mutat. 2017, 38, 964–969. [Google Scholar] [CrossRef]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Ren, H.; Bakas, N.A.; Vamos, M.; Chaikuad, A.; Limpert, A.S.; Wimer, C.D.; Brun, S.N.; Lambert, L.J.; Tautz, L.; Celeridad, M.; et al. Design, Synthesis, and Characterization of an Orally Active Dual-Specific ULK1/2 Autophagy Inhibitor that Synergizes with the PARP Inhibitor Olaparib for the Treatment of Triple-Negative Breast Cancer. J. Med. Chem. 2020, 63, 14609–14625. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Shokat, K.M. Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorg. Med. Chem. 2015, 23, 5483–5488. [Google Scholar] [CrossRef]
- Zachari, M.; Rainard, J.M.; Pandarakalam, G.C.; Robinson, L.; Gillespie, J.; Rajamanickam, M.; Hamon, V.; Morrison, A.; Ganley, I.G.; McElroy, S.P. The identification and characterisation of autophagy inhibitors from the published kinase inhibitor sets. Biochem. J. 2020, 477, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Yang, Z.; Zhen, Y.; Yang, Y.; Chen, Y.; Yuan, Y.; Zhang, L.; Zeng, X.; Chen, L. Discovery of 5-bromo-4-phenoxy-N-phenylpyrimidin-2-amine derivatives as novel ULK1 inhibitors that block autophagy and induce apoptosis in non-small cell lung cancer. Eur. J. Med. Chem. 2020, 208, 112782. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.D.; Grant, W.; Adrados, I.; Choi, J.Y.; Alburger, J.M.; Duckett, D.R.; Roush, W.R. In Silico HTS and Structure Based Optimization of Indazole-Derived ULK1 Inhibitors. ACS Med. Chem. Lett. 2017, 8, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.T.; Li, K.; Gao, Y.; Zhao, L.Y.; Gao, Y.; Yi, H.; Jiang, J.D.; Li, Z.R. The role of the key autophagy kinase ULK1 in hepa-tocellular carcinoma and its validation as a treatment target. Autophagy 2020, 16, 1823–1837. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, Y.; Li, Q.; Lyu, W.; Feng, F.; Guo, Q.; Zhao, L.; Sun, H. In silico approaches using pharmacophore model combined with molecular docking for discovery of novel ULK1 inhibitors. Future Med. Chem. 2021, 13, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Dower, C.M.; Bhat, N.; Gebru, M.T.; Chen, L.; Wills, C.A.; Miller, B.A.; Wang, H.-G. Targeted Inhibition of ULK1 Promotes Apoptosis and Suppresses Tumor Growth and Metastasis in Neuroblastoma. Mol. Cancer Ther. 2018, 17, 2365–2376. [Google Scholar] [CrossRef]
- Lu, J.; Zhu, L.; Zheng, L.P.; Cui, Q.; Zhu, H.H.; Zhao, H.; Shen, Z.J.; Dong, H.Y.; Chen, S.S.; Wu, W.Z.; et al. Overexpression of ULK1 Represents a Potential Diagnostic Marker for Clear Cell Renal Carcinoma and the Antitumor Effects of SBI-0206965. EBioMedicine 2018, 34, 85–93. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, X.; Wang, C.; Hu, Y.; Zhang, H.; Zhang, L.; Tu, S.; He, Y.; Li, Y. Dual targeting of NUAK1 and ULK1 using the multitargeted inhibitor MRT68921 exerts potent antitumor activities. Cell Death Dis. 2020, 11, 712. [Google Scholar] [CrossRef]
- Skah, S.; Richartz, N.; Duthil, E.; Gilljam, K.M.; Bindesbøll, C.; Naderi, E.H.; Eriksen, A.B.; Ruud, E.; Dirdal, M.M.; Simonsen, A.; et al. cAMP-mediated autophagy inhibits DNA damage-induced death of leukemia cells independent of p53. Oncotarget 2018, 9, 30434–30449. [Google Scholar] [CrossRef][Green Version]
- Schöffski, P.; Jones, S.F.; Dumez, H.; Infante, J.R.; Van Mieghem, E.; Fowst, C.; Gerletti, P.; Xu, H.; Jakubczak, J.L.; English, P.A.; et al. Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase in-hibitor PF-03814735 in advanced solid tumours. Eur. J. Cancer 2011, 47, 2256–2264. [Google Scholar] [CrossRef]
- Singha, B.; Laski, J.; Valdés, Y.R.; Liu, E.; DiMattia, G.E.; Shepherd, T.G. Inhibiting ULK1 kinase decreases autophagy and cell viability in high-grade serous ovarian cancer spheroids. Am. J. Cancer Res. 2020, 10, 1384–1399. [Google Scholar] [PubMed]
- Sankhe, K.; Prabhu, A.; Khan, T. Design strategies, SAR, and mechanistic insight of Aurora kinase inhibitors in cancer. Chem. Biol. Drug Des. 2021, 98, 73–93. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Thakral, F.; Singhal, P.; Aggarwal, D.; Srivastava, S.; Pandey, A.; Sak, K.; Varol, M.; Khan, A.; et al. Molecular mechanisms of action of hesperidin in cancer: Recent trends and advancements. Exp. Biol. Med. 2020, 245, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Zhou, G.; Cao, S. Targeted inhibition of ULK1 enhances daunorubicin sensitivity in acute myeloid leukemia. Life Sci. 2019, 243, 117234. [Google Scholar] [CrossRef]
- Yu, L.; Shi, Q.; Jin, Y.; Liu, Z.; Li, J.; Sun, W. Blockage of AMPK-ULK1 pathway mediated autophagy promotes cell apoptosis to increase doxorubicin sensitivity in breast cancer (BC) cells: An in vitro study. BMC Cancer 2021, 21, 195. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Liu, S.; Sun, W.; Huang, H.; Zhang, Q.; Yan, J. Inhibition of ULK1 promotes the death of leukemia cell in an autophagy irrelevant manner and exerts the antileukemia effect. Clin. Transl. Med. 2021, 11, e282. [Google Scholar] [CrossRef]
- Follo, C.; Cheng, Y.; Richards, W.G.; Bueno, R.; Broaddus, V.C. Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol. Carcinog. 2017, 57, 319–332. [Google Scholar] [CrossRef]
- Dite, T.A.; Langendorf, C.; Hoque, A.; Galic, S.; Rebello, R.J.; Ovens, A.J.; Lindqvist, L.M.; Ngoei, K.R.; Ling, N.; Furic, L.; et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J. Biol. Chem. 2018, 293, 8874–8885. [Google Scholar] [CrossRef]
- Ahwazi, D.; Neopane, K.; Markby, G.R.; Kopietz, F.; Ovens, A.J.; Dall, M.; Hassing, A.S.; Grasle, P.; Alshuweishi, Y.; Treebak, J.T.; et al. Investigation of the specificity and mechanism of action of the ULK1/AMPK inhibitor SBI-0206965. Biochem. J. 2021, 478, 2977–2997. [Google Scholar] [CrossRef]
- Knudsen, J.R.; Madsen, A.B.; Persson, K.W.; Henriquez-Olguin, C.; Li, Z.; Jensen, T.E. The ULK1/2 and AMPK Inhibitor SBI-0206965 Blocks AICAR and Insulin-Stimulated Glucose Transport. Int. J. Mol. Sci. 2020, 21, 2344. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhang, X.; Li, Z. ULK1 inhibitor induces spindle microtubule disorganization and inhibits phosphorylation of Ser10 of histone H3. FEBS Open Bio 2020, 10, 2452–2463. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Longo, M.; Ganley, I.G. Aberrant autophagosome formation occurs upon small molecule inhibition of ULK1 kinase activity. Life Sci. Alliance 2020, 3, e202000815. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karmacharya, U.; Jung, J.-W. Small Molecule Inhibitors for Unc-51-like Autophagy-Activating Kinase Targeting Autophagy in Cancer. Int. J. Mol. Sci. 2023, 24, 953. https://doi.org/10.3390/ijms24020953
Karmacharya U, Jung J-W. Small Molecule Inhibitors for Unc-51-like Autophagy-Activating Kinase Targeting Autophagy in Cancer. International Journal of Molecular Sciences. 2023; 24(2):953. https://doi.org/10.3390/ijms24020953
Chicago/Turabian StyleKarmacharya, Ujjwala, and Jong-Wha Jung. 2023. "Small Molecule Inhibitors for Unc-51-like Autophagy-Activating Kinase Targeting Autophagy in Cancer" International Journal of Molecular Sciences 24, no. 2: 953. https://doi.org/10.3390/ijms24020953
APA StyleKarmacharya, U., & Jung, J.-W. (2023). Small Molecule Inhibitors for Unc-51-like Autophagy-Activating Kinase Targeting Autophagy in Cancer. International Journal of Molecular Sciences, 24(2), 953. https://doi.org/10.3390/ijms24020953