
Cpt1c Downregulation Causes Plasma Membrane Remodelling and Anthracycline Resistance in Breast Cancer

 , , and
, , and 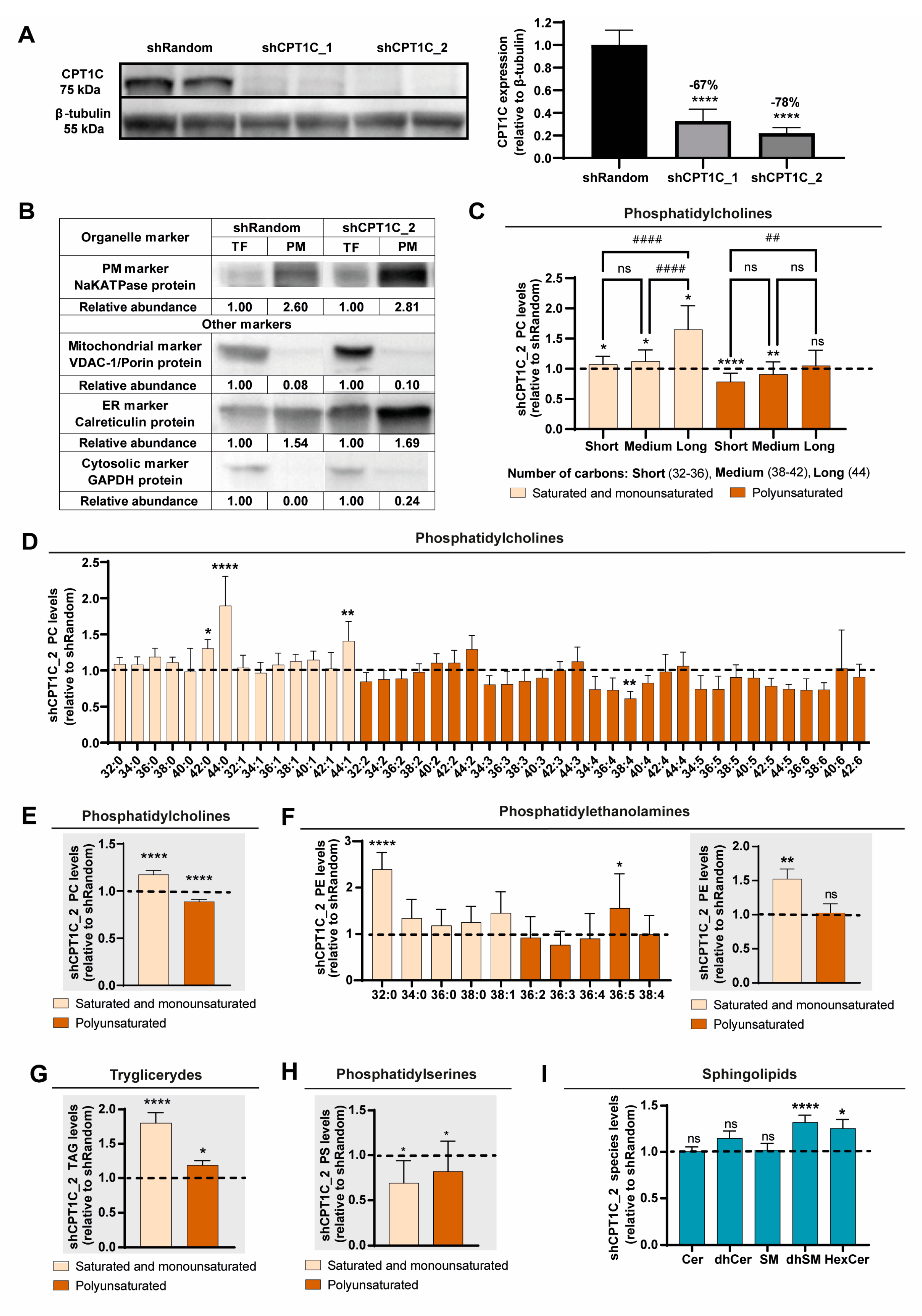{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. CPT1C Silencing Increases Phospholipid Saturation and Chain Length in the PM of MDA-MB-231 Cells
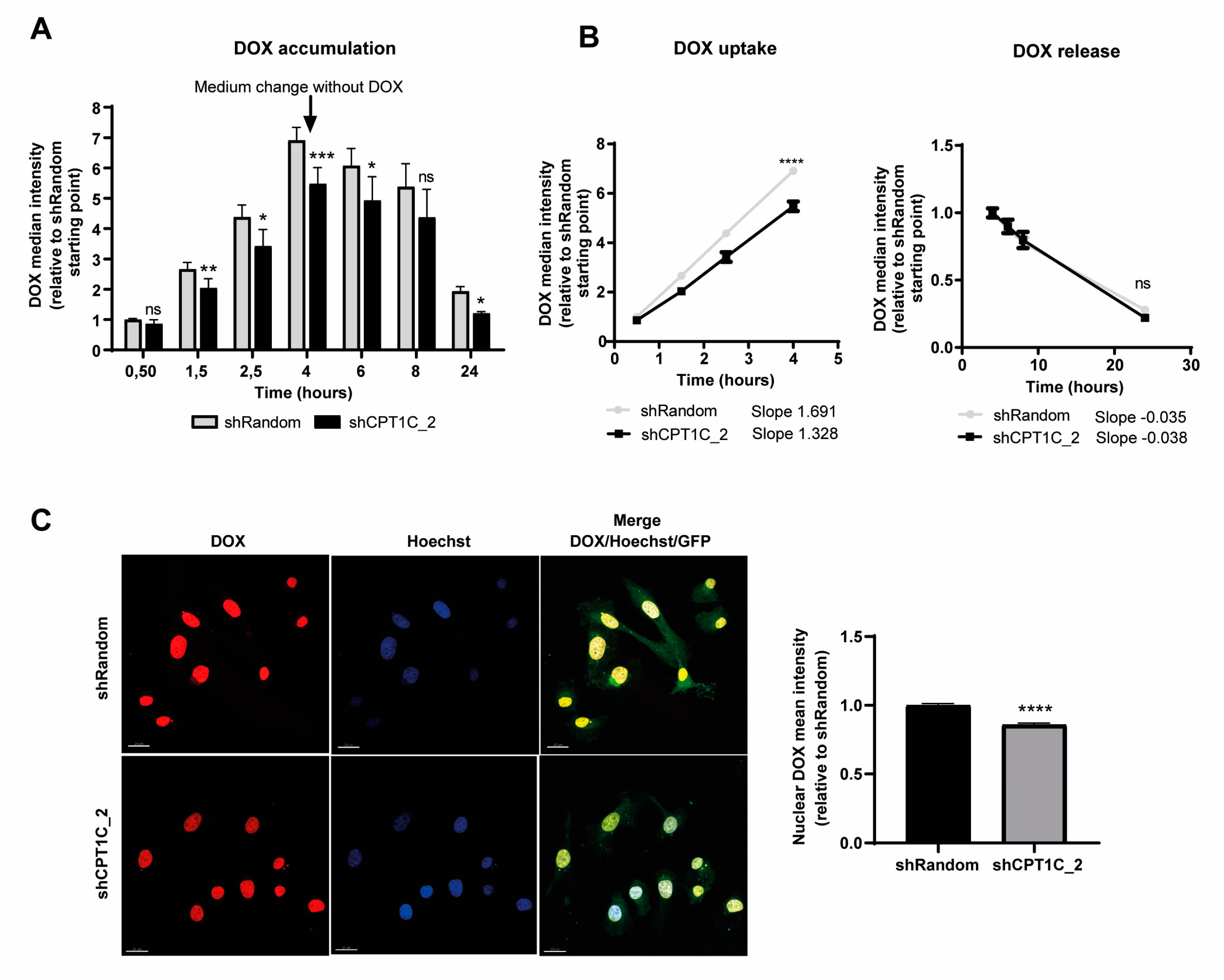
2.2. CPT1C Silencing Impairs DOX Uptake in MDA-MB-231 Cells
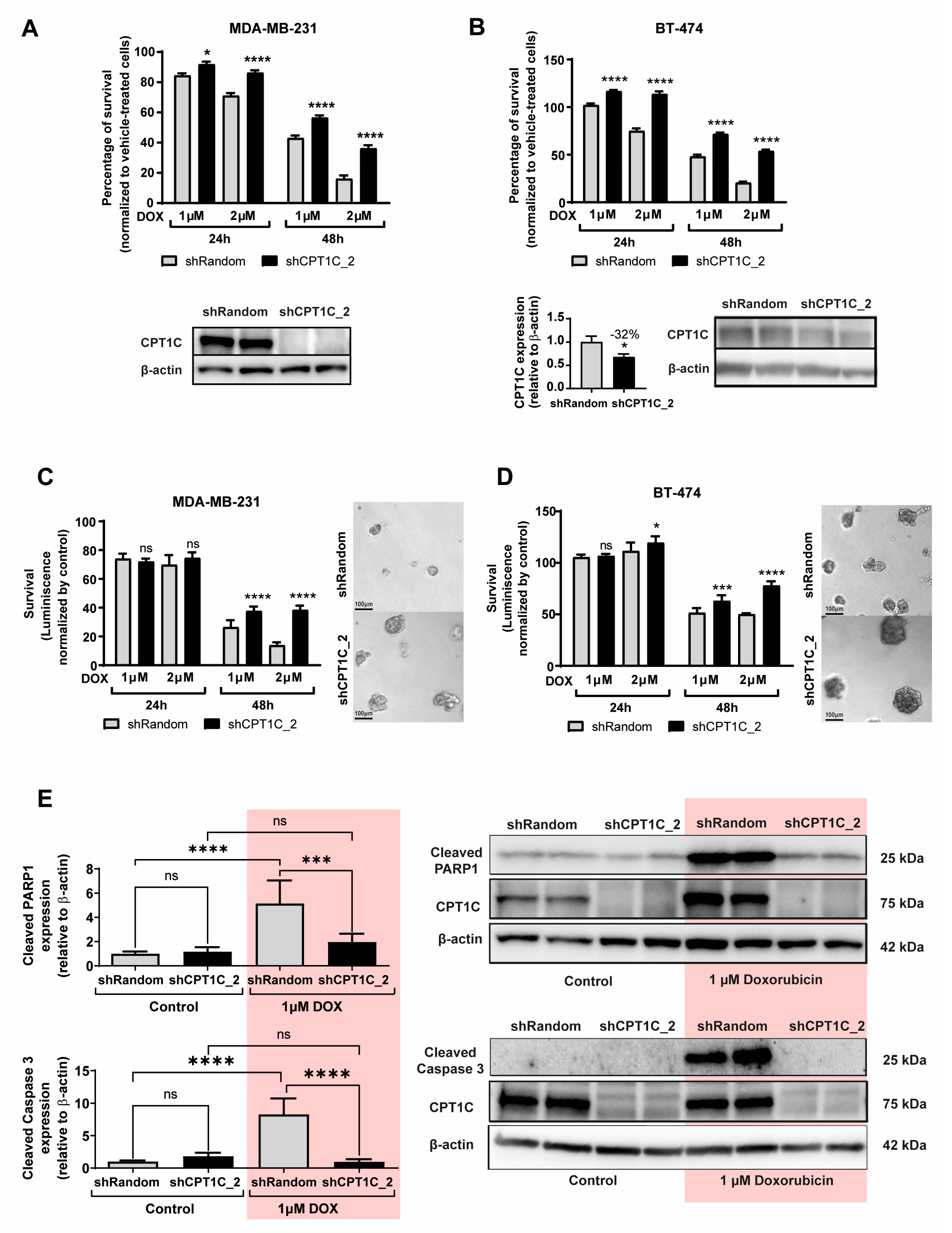
2.3. CPT1C Silencing Promotes Resistance to DOX in BC Cells
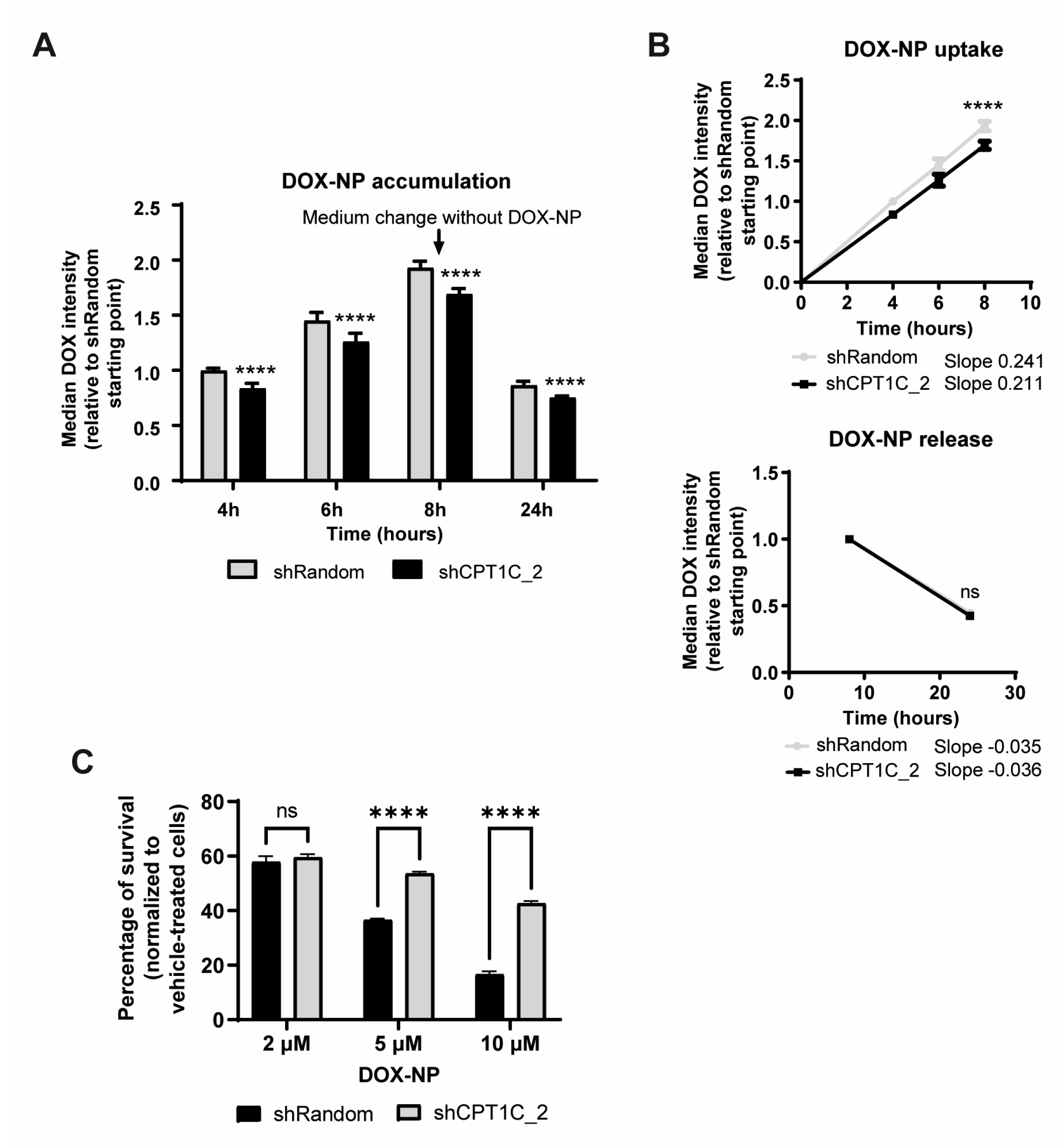
2.4. CPT1C Silencing Impairs Liposomal-DOX Uptake and Sensitivity in MDA-MB-231 Cells
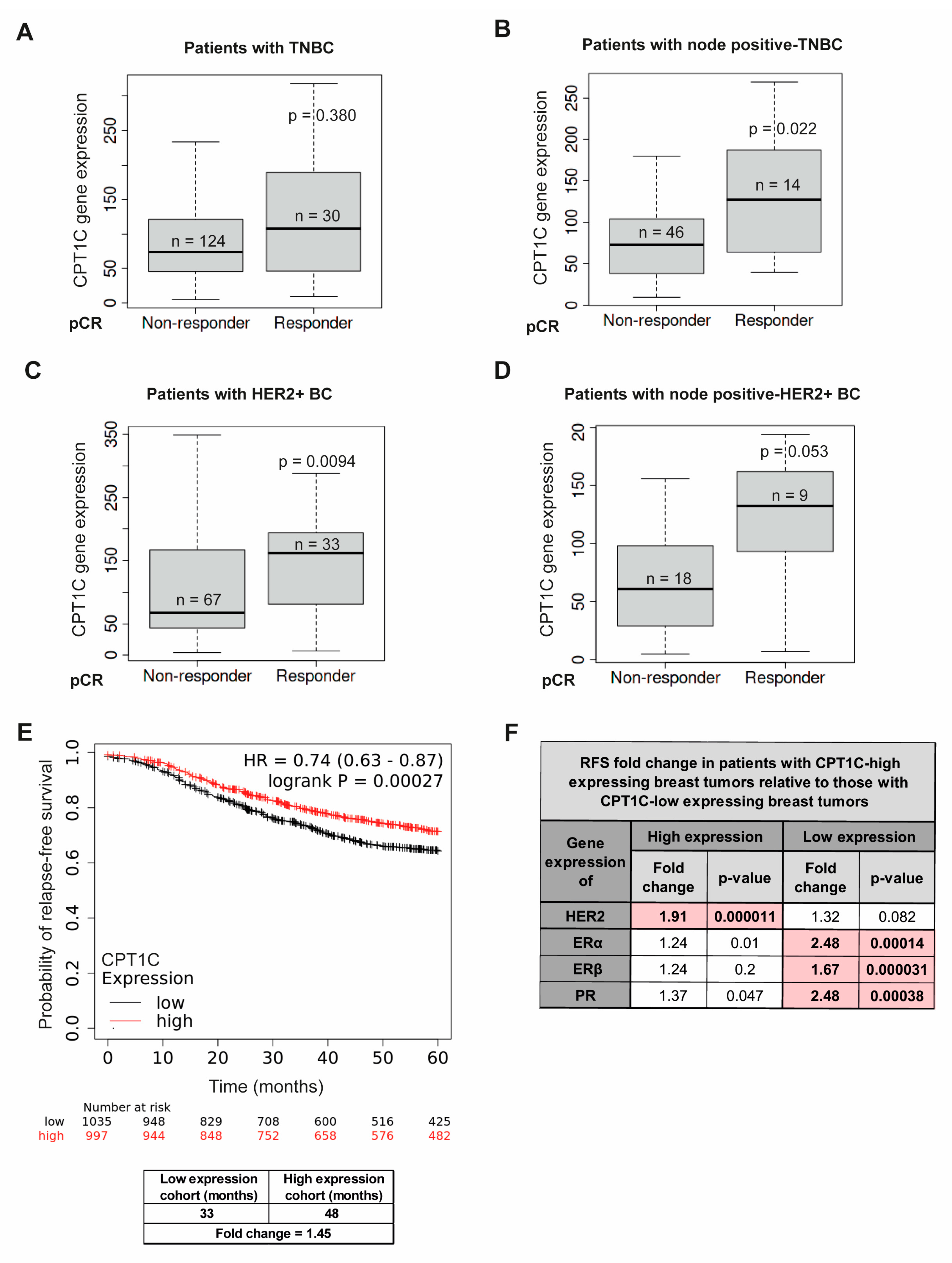
2.5. CPT1C as a Predictive Biomarker of Anthracycline Response Is of Prognostic Value for Patients with the TNBC and HER2+ BC Subtypes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids and Lentiviral Infection
4.3. 3D Culture
4.4. Cell Cytotoxicity Assays
4.4.1. MTT Cell Viability Assay
4.4.2. Cell Viability Assay
4.5. DOX Intracellular Accumulation Analysis
4.6. Isolation of PM-Enriched Fractions
4.7. Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS) Analysis
4.7.1. Glycerophospholipids and Neutral Lipids
4.7.2. Sphingolipids
4.7.3. LC-HRMS
4.8. Immunoblotting
4.9. Quantitative Real-Time Polymerase Chain Reaction (RT-PCR) Assays
4.10. Kaplan–Meier Plotter Analysis
4.11. ROC Plotter Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. C. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 24 September 2022).
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA J. Am. Med. Assoc. 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Efferth, T. Tumor Heterogeneity, Single-Cell Sequencing, and Drug Resistance. Pharmaceuticals 2016, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.N.; Gradishar, W.J. Adjuvant Anthracyclines in Breast Cancer: What Is Their Role? Oncologist 2018, 23, 1153. [Google Scholar] [CrossRef]
- Rivera, E.; Gomez, H. Chemotherapy Resistance in Metastatic Breast Cancer: The Evolving Role of Ixabepilone. Breast Cancer Res. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vuger, A.T.; Tiscoski, K.; Apolinario, T.; Cardoso, F. Anthracyclines in the Treatment of Early Breast Cancer Friend or Foe? Breast 2022, 65, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? HHS Public Access. Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Szlasa, W.; Zendran, I.; Zalesińska, A.; Tarek, M.; Kulbacka, J. Lipid Composition of the Cancer Cell Membrane. J. Bioenerg. Biomembr. 2020, 52, 321–342. [Google Scholar] [CrossRef]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine Palmitoyltransferase 1C Promotes Cell Survival and Tumor Growth under Conditions of Metabolic Stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef]
- Reilly, P.T.; Mak, T.W. Molecular Pathways: Tumor Cells Co-Opt the Brain-Specific Metabolism Gene CPT1C to Promote Survival. Clin. Cancer Res. 2012, 18, 5850–5855. [Google Scholar] [CrossRef]
- Guan, L.; Chen, Y.; Wang, Y.; Zhang, H.; Fan, S.; Gao, Y.; Jiao, T.; Fu, K.; Sun, J.; Yu, A.; et al. Effects of Carnitine Palmitoyltransferases on Cancer Cellular Senescence. J. Cell. Physiol. 2019, 234, 1707–1719. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Macedo, N.; Feng, J.; Faubert, B.; Chang, N.; Elia, A.; Rushing, E.J.; Tsuchihara, K.; Bungard, D.; Berger, S.L.; Jones, R.G.; et al. Depletion of the Novel P53-Target Gene Carnitine Palmitoyltransferase 1C Delays Tumor Growth in the Neurofibromatosis Type i Tumor Model. Cell Death Differ. 2013, 20, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, T.; Zhou, Y.; Wang, S.; Zhou, X.; Wang, L.; Ou, T.; Chen, Y.; Zhou, Y.; Zhang, H.; et al. Carnitine Palmitoyltransferase 1C Contributes to Progressive Cellular Senescence. Aging 2020, 12, 6733–6755. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Y.; Han, F.; Zhao, Y.; Tu, M.; Wang, Y.; Huang, C.; Fan, S.; Chen, P.; Yao, X.; et al. A Novel MiR-1291-ERRα-CPT1C Axis Modulates Tumor Cell Proliferation, Metabolism and Tumorigenesis. Theranostics 2020, 10, 7193–7210. [Google Scholar] [CrossRef]
- Gao, X.F.; Chen, W.; Kong, X.P.; Xu, A.M.; Wang, Z.G.; Sweeney, G.; Wu, D. Enhanced Susceptibility of Cpt1c Knockout Mice to Glucose Intolerance Induced by a High-Fat Diet Involves Elevated Hepatic Gluconeogenesis and Decreased Skeletal Muscle Glucose Uptake. Diabetologia 2009, 52, 912–920. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, Q.; Zhang, H.; Gao, Y.; Zhou, Y.; Chen, Y.; Guan, L.; Jiao, T.; Zhao, Y.; Huang, M.; et al. Carnitine Palmitoyltransferase 1C Reverses Cellular Senescence of MRC-5 Fibroblasts via Regulating Lipid Accumulation and Mitochondrial Function. J. Cell. Physiol. 2021, 236, 958–970. [Google Scholar] [CrossRef]
- Price, N.T.; Van Der Leij, F.R.; Jackson, V.N.; Corstorphine, C.G.; Thomson, R.; Sorensen, A.; Zammit, V.A. A Novel Brain-Expressed Protein Related to Carnitine Palmitoyltransferase I. Genomics 2002, 80, 433–442. [Google Scholar] [CrossRef]
- Roa-Mansergas, X.; Fadó, R.; Atari, M.; Mir, J.F.; Muley, H.; Serra, D.; Casals, N. CPT1C Promotes Human Mesenchymal Stem Cells Survival under Glucose Deprivation through the Modulation of Autophagy. Sci. Rep. 2018, 8, 6997. [Google Scholar] [CrossRef]
- Carrasco, P.; Sahún, I.; McDonald, J.; Ramírez, S.; Jacas, J.; Gratacós, E.; Sierra, A.Y.; Serra, D.; Herrero, L.; Acker-Palmer, A.; et al. Ceramide Levels Regulated by Carnitine Palmitoyltransferase 1C Control Dendritic Spine Maturation and Cognition. J. Biol. Chem. 2012, 287, 21224–21232. [Google Scholar] [CrossRef]
- Sierra, A.Y.; Gratacós, E.; Carrasco, P.; Clotet, J.; Ureña, J.; Serra, D.; Asins, G.; Hegardt, F.G.; Casals, N. CPT1c Is Localized in Endoplasmic Reticulum of Neurons and Has Carnitine Palmitoyltransferase Activity. J. Biol. Chem. 2008, 283, 6878–6885. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Guan, L.; Chen, Y.; Chen, P.; Sun, J.; Gonzalez, F.J.; Huang, M.; Bi, H. Lipidomics Reveals Carnitine Palmitoyltransferase 1C Protects Cancer Cells from Lipotoxicity and Senescence. J. Pharm. Anal. 2020, 11, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Huang, Y.; Zeng, H.; Hu, B.; Guan, L.; Zhang, H.; Yu, A.M.; Johnson, C.H.; Gonzalez, F.J.; et al. PPARα Regulates Tumor Cell Proliferation and Senescence via a Novel Target Gene Carnitine Palmitoyltransferase 1C. Carcinogenesis 2017, 38, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, G.; Hu, H.; Wu, C. Enhanced Fatty Acid Oxidation Mediated by CPT1C Promotes Gastric Cancer Progression. J. Gastrointest. Oncol. 2020, 11, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cheng, Y.; Su, D.; Gong, B.; He, X.; Zhou, X.; Pang, Z.; Cheng, L.; Chen, Y.; Yao, Z. Cpt1c Regulated by AMPK Promotes Papillary Thyroid Carcinomas Cells Survival under Metabolic Stress Conditions. J. Cancer 2017, 8, 3675–3681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, Y.; Liu, J.; Ma, Y.; Ye, Q.; Yan, X.; Ding, L. MicroRNA-377-3p Inhibits Hepatocellular Carcinoma Growth and Metastasis through Negative Regulation of CPT1C-Mediated Fatty Acid Oxidation. Cancer Metab. 2022, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Hiraide, T.; Morita, Y.; Horikawa, M.; Sugiyama, E.; Sato, T.; Kahyo, T.; Furuhashi, S.; Takeda, M.; Kikuchi, H.; Hiramatsu, Y.; et al. Saturated Fatty Acids in Cell Membrane Lipids Induce Resistance to 5-Fluorouracil in Colorectal Cancer Cells. Anticancer Res. 2022, 42, 3313–3324. [Google Scholar] [CrossRef]
- Peetla, C.; Bhave, R.; Vijayaraghavalu, S.; Stine, A.; Kooijman, E.; Labhasetwar, V. Drug Resistance in Breast Cancer Cells: Biophysical Characterization of and Doxorubicin Interactions with Membrane Lipids. Mol. Pharm. 2010, 7, 2334–2348. [Google Scholar] [CrossRef]
- Corsetto, P.A.; Colombo, I.; Kopecka, J.; Rizzo, A.M.; Riganti, C. ω-3 Long Chain Polyunsaturated Fatty Acids as Sensitizing Agents and Multidrug Resistance Revertants in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2770. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Kinoshita, M.; Kyo, T.; Matsumori, N. Assembly Formation of Minor Dihydrosphingomyelin in Sphingomyelin-Rich Ordered Membrane Domains. Sci. Rep. 2020, 10, 11794. [Google Scholar] [CrossRef]
- Pike, L.J. The Challenge of Lipid Rafts. J. Lipid Res. 2009, 50, S323. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, Y.S.; Choi, K.M.; Yoo, K.S.; Sin, D.M.; Kim, W.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; Yoo, H.S. Quantitative Analysis of Sphingomyelin by High-Performance Liquid Chromatography after Enzymatic Hydrolysis. Evid. Based. Complement. Alternat. Med. 2012, 2012, 396218. [Google Scholar] [CrossRef] [PubMed]
- Muley, H.; Fadó, R.; Rodríguez-Rodríguez, R.; Casals, N. Drug Uptake-Based Chemoresistance in Breast Cancer Treatment. Biochem. Pharmacol. 2020, 177, 113959. [Google Scholar] [CrossRef] [PubMed]
- Fekete, J.T.; Győrffy, B. ROCplot.Org: Validating Predictive Biomarkers of Chemotherapy/Hormonal Therapy/Anti-HER2 Therapy Using Transcriptomic Data of 3,104 Breast Cancer Patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological Complete Response and Long-Term Clinical Benefit in Breast Cancer: The CTNeoBC Pooled Analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- Spring, L.M.; Fell, G.; Arfe, A.; Sharma, C.; Greenup, R.; Reynolds, K.L.; Smith, B.L.; Alexander, B.; Moy, B.; Isakoff, S.J.; et al. Pathological Complete Response after Neoadjuvant Chemotherapy and Impact on Breast Cancer Recurrence and Survival: A Comprehensive Meta-Analysis. Clin. Cancer Res. 2020, 26, 2838–2848. [Google Scholar] [CrossRef]
- Cao, J.; Zhang, M.; Wang, B.; Zhang, L.; Zhou, F.; Fang, M. Chemoresistance and Metastasis in Breast Cancer Molecular Mechanisms and Novel Clinical Strategies. Front. Oncol. 2021, 11, 658552. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, X.; Yuan, Y.; Jiang, J.; Zhang, P.; Zhang, B. ARTICLE Identification of a Novel Mechanism for Reversal of Doxorubicin-Induced Chemotherapy Resistance by TXNIP in Triple-Negative Breast Cancer via Promoting Reactive Oxygen-Mediated DNA Damage. Cell Death Dis. 2022, 13, 338. [Google Scholar] [CrossRef]
- Wu, Y.; Sarkissyan, M.; Mcghee, E.; Lee, S.; Vadgama, J.V. Combined Inhibition of Glycolysis and AMPK Induces Synergistic Breast Cancer Cell Killing. Breast Cancer Res. Treat. 2015, 151, 529–539. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Wang, C.-H.; Mai, R.-T.; Chen, T.-W.; Li, C.-W.; Chao, C.-H. Mutant P53-MicroRNA-200c-ZEB2-Axis-Induced CPT1C Elevation Contributes to Metabolic Reprogramming and Tumor Progression in Basal-Like Breast Cancers. Front. Oncol. 2022, 12, 940402. [Google Scholar] [CrossRef]
- Casals, N.; Zammit, V.; Herrero, L.; Fadó, R.; Rodríguez-Rodríguez, R.; Serra, D. Carnitine Palmitoyltransferase 1C: From Cognition to Cancer. Prog. Lipid Res. 2016, 61, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Fadó, R.; Rodríguez-Rodríguez, R.; Casals, N. NC-ND License The Return of Malonyl-CoA to the Brain: Cognition and Other Stories. Prog. Lipid Res. 2021, 81, 101071. [Google Scholar] [CrossRef]
- Casas, M.; Fadó, R.; Domínguez, J.L.; Roig, A.; Kaku, M.; Chohnan, S.; Solé, M.; Unzeta, M.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J.; et al. Sensing of Nutrients by CPT1C Controls SAC1 Activity to Regulate AMPA Receptor Trafficking. J. Cell Biol. 2020, 219, 201912045. [Google Scholar] [CrossRef] [PubMed]
- Miralpeix, C.; Reguera, A.C.; Fosch, A.; Casas, M.; Lillo, J.; Navarro, G.; Franco, R.; Casas, J.; Alexander, S.P.H.; Casals, N.; et al. CPT1C Negatively Regulates the Endocannabinoid Hydrolase ABHD6 Depending on Nutritional Status. Br. J. Pharmacol. 2021, 178, 1507–1523. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in Cancer: The Relevance of Drug-Membrane Interaction Studies. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2231–2244. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Guan, L.; Zhang, H.; Huang, Y.; Johnson, C.H.; Wu, Z.; Gonzalez, F.J.; Yu, A.; Huang, P.; et al. Carnitine Palmitoyltransferase 1C Regulates Cancer Cell Senescence through Mitochondria-Associated Metabolic Reprograming. Cell Death Differ. 2018, 25, 733–746. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and Cancer—Role and Therapeutic Opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, H.; Sun, Y. Tumor Microenvironment and Cellular Senescence: Understanding Therapeutic Resistance and Harnessing Strategies. Semin. Cancer Biol. 2021, 86, 769–781. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The Fate of Chemoresistance in Triple Negative Breast Cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef]
- Mandapati, A.; Lukong, E. Triple Negative Breast Cancer: Approved Treatment Options and Their Mechanisms of Action. J. Cancer Res. Clin. Oncol. 2022, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Bae, J.S.; Kim, K.M.; Moon, Y.J.; Park, S.H.; Ha, S.H.; Hussein, U.K.; Zhang, Z.; Park, H.S.; Park, B.H.; et al. The PARP Inhibitor Olaparib Potentiates the Effect of the DNA Damaging Agent Doxorubicin in Osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 107. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, W.K.D.; Garcia-Chica, J.; Ariza, X.; Zagmutt, S.; Fukushima, S.; Garcia, J.; Mochida, Y.; Serra, D.; Herrero, L.; Kinoh, H.; et al. Poly-Ion Complex Micelles Effectively Deliver CoA-Conjugated CPT1A Inhibitors to Modulate Lipid Metabolism in Brain Cells. Biomater. Sci. 2021, 9, 7076–7091. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gage, F.H.; Tronot, D.; Verma, I.M. Genetic Engineering of Viruses and of Virus Vectors. Proc. Natl. Acad. Sci. USA 1996, 93, 11287. [Google Scholar] [CrossRef]
- Suski, J.M.; Lebiedzinska, M.; Wojtala, A.; Duszynski, J.; Giorgi, C.; Pinton, P.; Wieckowski, M.R. Isolation of Plasma Membrane-Associated Membranes from Rat Liver. Nat. Protoc. 2014, 9, 312–322. [Google Scholar] [CrossRef]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An Online Survival Analysis Tool to Rapidly Assess the Effect of 22,277 Genes on Breast Cancer Prognosis Using Microarray Data of 1,809 Patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muley, H.; Valencia, K.; Casas, J.; Moreno, B.; Botella, L.; Lecanda, F.; Fadó, R.; Casals, N. Cpt1c Downregulation Causes Plasma Membrane Remodelling and Anthracycline Resistance in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 946. https://doi.org/10.3390/ijms24020946
Muley H, Valencia K, Casas J, Moreno B, Botella L, Lecanda F, Fadó R, Casals N. Cpt1c Downregulation Causes Plasma Membrane Remodelling and Anthracycline Resistance in Breast Cancer. International Journal of Molecular Sciences. 2023; 24(2):946. https://doi.org/10.3390/ijms24020946
Chicago/Turabian StyleMuley, Helena, Karmele Valencia, Josefina Casas, Bea Moreno, Luis Botella, Fernando Lecanda, Rut Fadó, and Núria Casals. 2023. "Cpt1c Downregulation Causes Plasma Membrane Remodelling and Anthracycline Resistance in Breast Cancer" International Journal of Molecular Sciences 24, no. 2: 946. https://doi.org/10.3390/ijms24020946
APA StyleMuley, H., Valencia, K., Casas, J., Moreno, B., Botella, L., Lecanda, F., Fadó, R., & Casals, N. (2023). Cpt1c Downregulation Causes Plasma Membrane Remodelling and Anthracycline Resistance in Breast Cancer. International Journal of Molecular Sciences, 24(2), 946. https://doi.org/10.3390/ijms24020946