

Toll-like Receptors and Thrombopoiesis
 , ,
, ,
Abstract
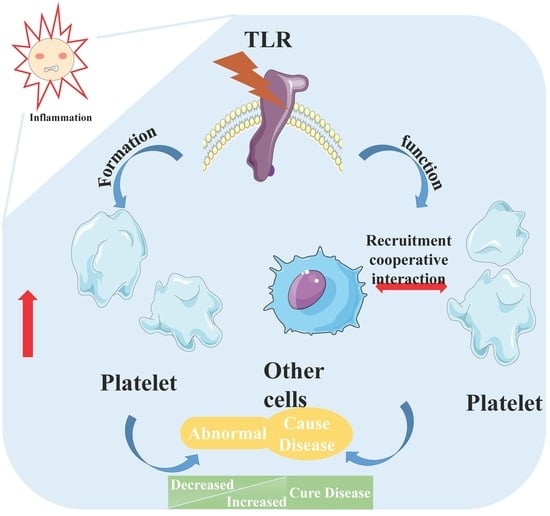
1. Introduction
2. Inflammation and Platelets
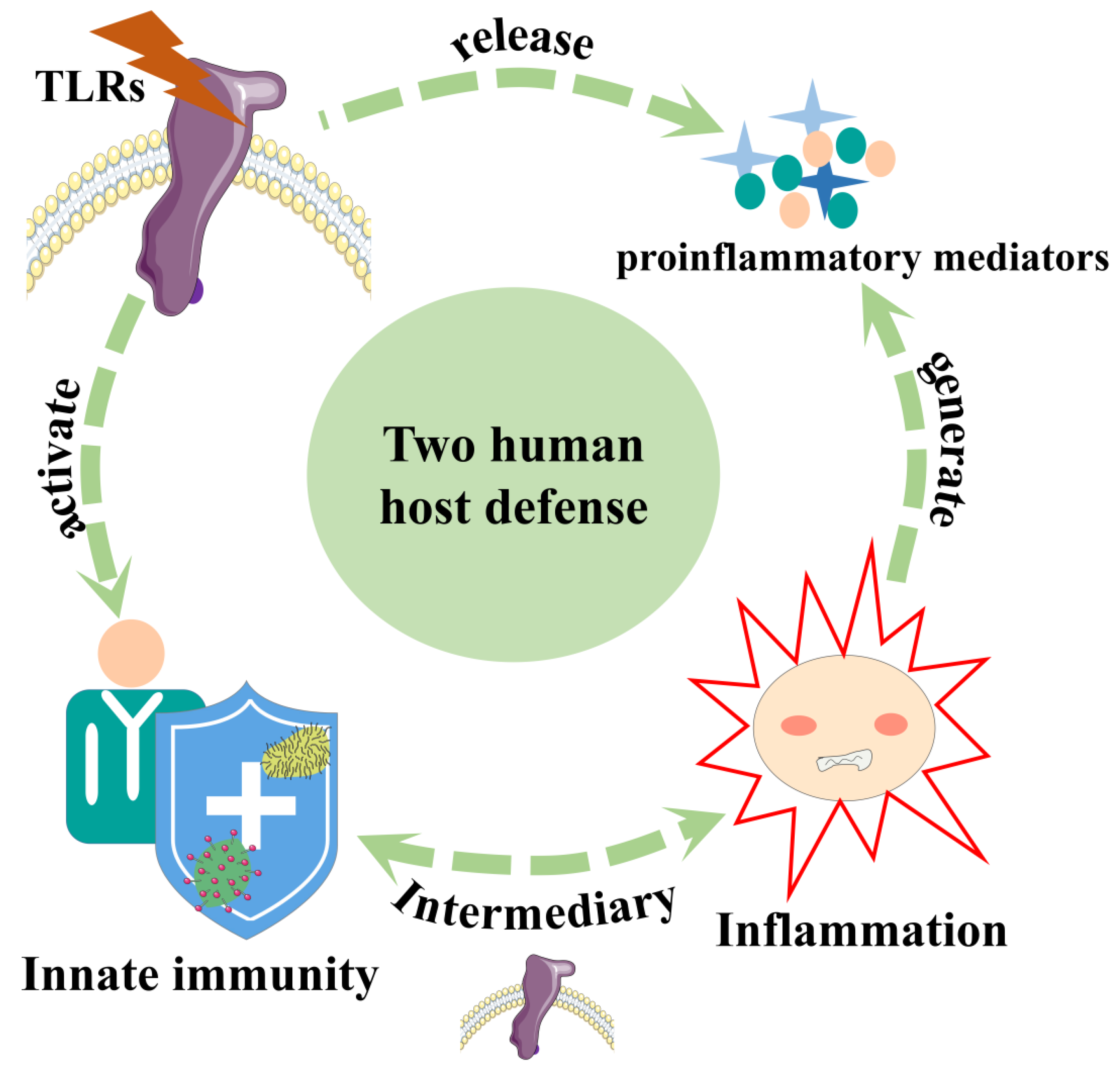
3. Inflammation and TLRs
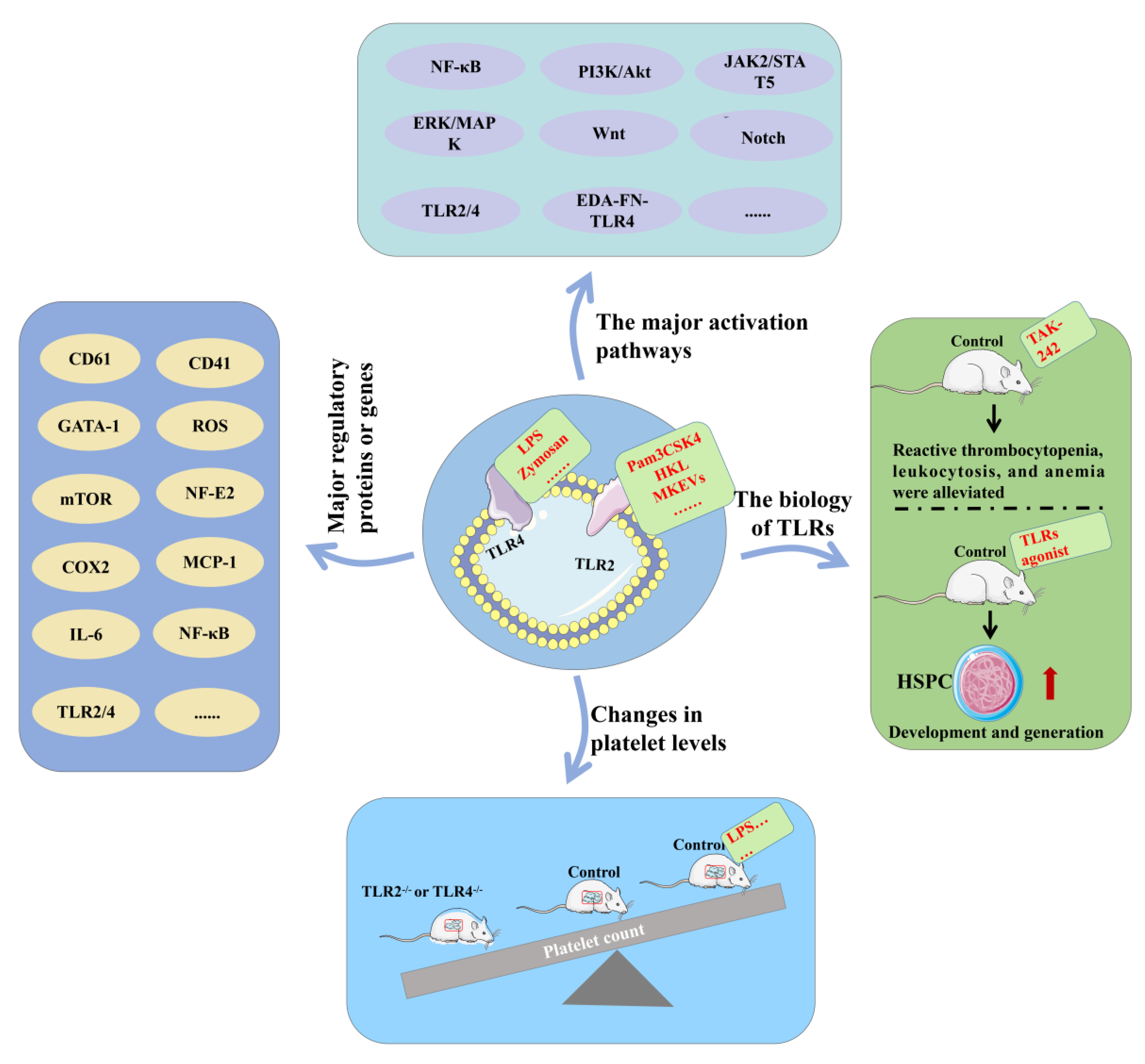
4. TLRs and Platelets
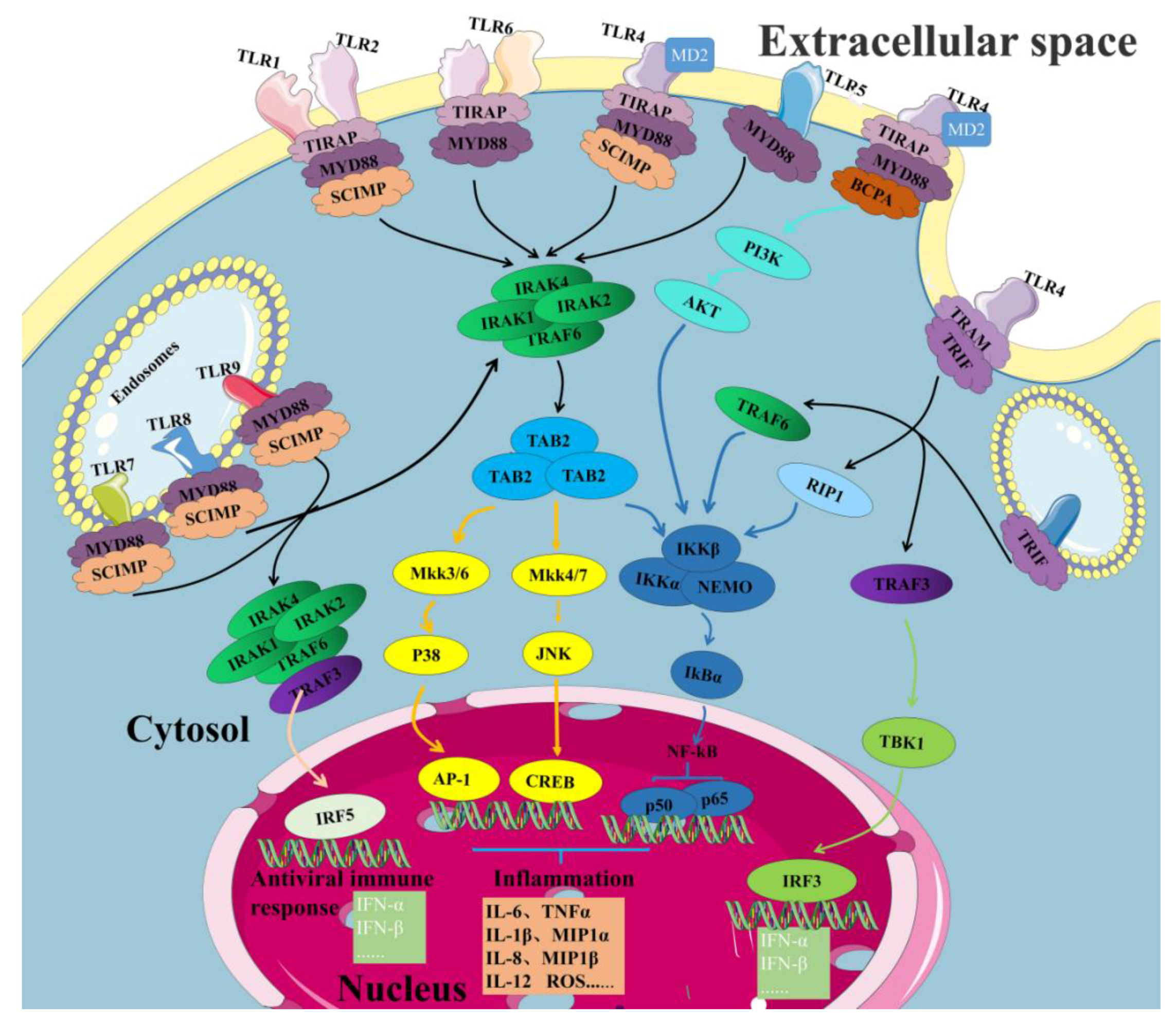
4.1. TLR2 and TLR4 Signaling Pathways
4.2. TLR2 and TLR4 Promote Platelet Formation
4.3. TLR2 and TLR4 Increase Platelet Function
4.4. Balanced TLR2 and TLR4 Pathways Are Critical
4.5. Other TLRs and Platelets
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thon, J.N.; Italiano, J.E. Platelets: Production, morphology and ultrastructure. In Antiplatelet Agents; Springer: Berlin/Heidelberg, Germany, 2012; pp. 3–22. [Google Scholar] [CrossRef]
- Eto, K.; Kunishima, S. Linkage between the mechanisms of thrombocytopenia and thrombopoiesis. Blood 2016, 127, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Long, M.W.; Williams, N.; Ebbe, S. Immature megakaryocytes in the mouse: Physical characteristics, cell cycle status, and in vitro responsiveness to thrombopoietic stimulatory factor. Blood 1982, 59, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E. The fine structure of the megakaryocyte in the mouse spleen. Cells Tissues Organs 1957, 29, 267–290. [Google Scholar] [CrossRef] [PubMed]
- Nakao, K.; Angrist, A.A. Membrane surface specialization of blood platelet and megakaryocyte. Nature 1968, 217, 960–961. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, L.J.; French, S.L.; Machlus, K.R. New Insights Into the Differentiation of Megakaryocytes From Hematopoietic Progenitors. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.; Lawrence, M.; Guerrero, J.A.; Stritt, S.; Waller, A.K.; Yan, Y.; Mifsud, R.W.; Ballester-Beltrán, J.; Baig, A.; Mueller, A.; et al. CRLF3 plays a key role in the final stage of platelet genesis and is a potential therapeutic target for thrombocythemia. Blood 2022, 139, 2227–2239. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef]
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67. [Google Scholar] [CrossRef]
- McMorran, B.J.; Marshall, V.M.; de Graaf, C.; Drysdale, K.E.; Shabbar, M.; Smyth, G.K.; Corbin, J.E.; Alexander, W.S.; Foote, S.J. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science 2009, 323, 797–800. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- Garraud, O.; Cognasse, F. Are Platelets Cells? And if Yes, are They Immune Cells? Front. Immunol. 2015, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Massberg, S. Platelets as key players in inflammation and infection. Curr. Opin. Hematol. 2020, 27, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Dib, P.R.B.; Quirino-Teixeira, A.C.; Merij, L.B.; Pinheiro, M.B.M.; Rozini, S.V.; Andrade, F.B.; Hottz, E.D. Innate immune receptors in platelets and platelet-leukocyte interactions. J. Leukoc. Biol. 2020, 108, 1157–1182. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Ohishi, H.M.; Aki, D.; Hanada, T. Regulation of TLR signaling and inflammation by SOCS family proteins. J. Leukoc. Biol. 2004, 75, 422–427. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Asami, J.; Shimizu, T. Structural and functional understanding of the toll-like receptors. Protein Sci. 2021, 30, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Lai, Y.; Gallo, R.L. Toll-like receptors in skin infections and inflammatory diseases. Infect. Disord. Drug Targets 2008, 8, 144–155. [Google Scholar] [CrossRef]
- Ospelt, C.; Gay, S. TLRs and chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 495–505. [Google Scholar] [CrossRef]
- Joosten, L.A.; Abdollahi-Roodsaz, S.; Dinarello, C.A.; O’Neill, L.; Netea, M.G. Toll-like receptors and chronic inflammation in rheumatic diseases: New developments. Nat. Rev. Rheumatol. 2016, 12, 344–357. [Google Scholar] [CrossRef]
- Baldridge, M.T.; King, K.Y.; Goodell, M.A. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011, 32, 57–65. [Google Scholar] [CrossRef]
- Dorner, M.; Brandt, S.; Tinguely, M.; Zucol, F.; Bourquin, J.P.; Zauner, L.; Berger, C.; Bernasconi, M.; Speck, R.F.; Nadal, D. Plasma cell toll-like receptor (TLR) expression differs from that of B cells, and plasma cell TLR triggering enhances immunoglobulin production. Immunology 2009, 128, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Kim, N.; Rho, J.; Choi, Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J. Immunol. 2002, 169, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Yu, Z.; Ding, N.; Yang, M.; Zhang, L.; Fan, X.; Zhou, Y.; Zou, Q.; Hou, J.; Zheng, J.; et al. The role of AGK in thrombocytopoiesis and possible therapeutic strategies. Blood 2020, 136, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, K.; Broudy, V.C.; Lin, N.; Jorgensen, M.J.; McCarty, J.; Fox, N.; Zucker-Franklin, D.; Lofton-Day, C. Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc. Natl. Acad. Sci. USA 1995, 92, 3234–3238. [Google Scholar] [CrossRef]
- Panuganti, S.; Schlinker, A.C.; Lindholm, P.F.; Papoutsakis, E.T.; Miller, W.M. Three-stage ex vivo expansion of high-ploidy megakaryocytic cells: Toward large-scale platelet production. Tissue Eng. Part A 2013, 19, 998–1014. [Google Scholar] [CrossRef]
- Clark, D.A.; Dessypris, E.N.; Koury, M.J. Induction of megakaryocytic colony-stimulating activity in mouse skin by inflammatory agents and tumor promoters. Proc. Soc. Exp. Biol. Med. 1987, 184, 245–249. [Google Scholar] [CrossRef]
- Dan, K.; Gomi, S.; Inokuchi, K.; Ogata, K.; Yamada, T.; Ohki, I.; Hasegawa, S.; Nomura, T. Effects of interleukin-1 and tumor necrosis factor on megakaryocytopoiesis: Mechanism of reactive thrombocytosis. Acta Haematol. 1995, 93, 67–72. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, H.; Wang, H.; Zhang, T.; Hutchinson, M.R.; Yin, H.; Wang, X. Small-Molecule Modulators of Toll-like Receptors. Acc. Chem. Res. 2020, 53, 1046–1055. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Vincenzo, B.; Asif, I.J.; Nikolaos, P.; Francesco, M. Adaptive immunity and inflammation. Int. J. Inflam. 2015, 2015, 575406. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- Maskrey, B.H.; Megson, I.L.; Whitfield, P.D.; Rossi, A.G. Mechanisms of resolution of inflammation: A focus on cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Clancy, L.; Beaulieu, L.M.; Tanriverdi, K.; Freedman, J.E. The role of RNA uptake in platelet heterogeneity. Thromb. Haemost. 2017, 117, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Wienkamp, A.K.; Erpenbeck, L.; Rossaint, J. Platelets in the NETworks interweaving inflammation and thrombosis. Front. Immunol. 2022, 13, 953129. [Google Scholar] [CrossRef]
- Rayes, J.; Watson, S.P. Platelet GPVI repairs its own damage. Blood 2015, 126, 933–934. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Rosowski, E.E.; Huttenlocher, A. Motile Collectors: Platelets Promote Innate Immunity. Immunity 2018, 48, 16–18. [Google Scholar] [CrossRef]
- Bambach, S.K.; Lämmermann, T. Platelets, On Your Marks, Get Set, Migrate! Cell 2017, 171, 1256–1258. [Google Scholar] [CrossRef]
- Sexton, T.R.; Zhang, G.; Macaulay, T.E.; Callahan, L.A.; Charnigo, R.; Vsevolozhskaya, O.A.; Li, Z.; Smyth, S. Ticagrelor Reduces Thromboinflammatory Markers in Patients with Pneumonia. JACC Basic Transl. Sci. 2018, 3, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Kor, D.J.; Carter, R.E.; Park, P.K.; Festic, E.; Banner-Goodspeed, V.M.; Hinds, R.; Talmor, D.; Gajic, O.; Ware, L.B.; Gong, M.N. Effect of Aspirin on Development of ARDS in At-Risk Patients Presenting to the Emergency Department: The LIPS-A Randomized Clinical Trial. JAMA 2016, 315, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, K.; Lok, S.; Holly, R.D.; Broudy, V.C.; Lin, N.; Bailey, M.C.; Forstrom, J.W.; Buddle, M.M.; Oort, P.J.; Hagen, F.S.; et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature 1994, 369, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Trinh, B.Q.; Barengo, N.; Kim, S.B.; Lee, J.S.; Zweidler-McKay, P.A.; Naora, H. The homeobox gene DLX4 regulates erythro-megakaryocytic differentiation by stimulating IL-1β and NF-κB signaling. J. Cell Sci. 2015, 128, 3055–3067. [Google Scholar] [CrossRef]
- Wickenhauser, C.; Lorenzen, J.; Thiele, J.; Hillienhof, A.; Jungheim, K.; Schmitz, B.; Hansmann, M.L.; Fischer, R. Secretion of cytokines (interleukins-1 alpha, -3, and -6 and granulocyte-macrophage colony-stimulating factor) by normal human bone marrow megakaryocytes. Blood 1995, 85, 685–691. [Google Scholar] [CrossRef]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef]
- Behrens, K.; Alexander, W.S. Cytokine control of megakaryopoiesis. Growth Factors 2018, 36, 89–103. [Google Scholar] [CrossRef]
- Wolber, E.M.; Fandrey, J.; Frackowski, U.; Jelkmann, W. Hepatic thrombopoietin mRNA is increased in acute inflammation. Thromb. Haemost. 2001, 86, 1421–1424. [Google Scholar]
- Burmester, H.; Wolber, E.M.; Freitag, P.; Fandrey, J.; Jelkmann, W. Thrombopoietin production in wild-type and interleukin-6 knockout mice with acute inflammation. J. Interferon Cytokine Res. 2005, 25, 407–413. [Google Scholar] [CrossRef]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, Á.M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef]
- Ricciardi, S.; Miluzio, A.; Brina, D.; Clarke, K.; Bonomo, M.; Aiolfi, R.; Guidotti, L.G.; Falciani, F.; Biffo, S. Eukaryotic translation initiation factor 6 is a novel regulator of reactive oxygen species-dependent megakaryocyte maturation. J. Thromb. Haemost. 2015, 13, 2108–2118. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Nagasaki, M.; Kunishima, S.; Sawaguchi, A.; Sakata, A.; Sakaguchi, H.; Ohmori, T.; Manabe, I.; Italiano, J.E., Jr.; Ryu, T.; et al. IL-1α induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J. Cell Biol. 2015, 209, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Undi, R.B.; Sarvothaman, S.; Narasaiah, K.; Gutti, U.; Gutti, R.K. Toll-like receptor 2 signalling: Significance in megakaryocyte development through wnt signalling cross-talk and cytokine induction. Cytokine 2016, 83, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Tacchini-Cottier, F.; Vesin, C.; Redard, M.; Buurman, W.; Piguet, P.F. Role of TNFR1 and TNFR2 in TNF-induced platelet consumption in mice. J. Immunol. 1998, 160, 6182–6186. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, L.M.; Lin, E.; Mick, E.; Koupenova, M.; Weinberg, E.O.; Kramer, C.D.; Genco, C.A.; Tanriverdi, K.; Larson, M.G.; Benjamin, E.J.; et al. Interleukin 1 receptor 1 and interleukin 1β regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 552–564. [Google Scholar] [CrossRef]
- Nakayama, K. Expression of IL-6, IL-6 receptor and its signal transducer gp130 mRNAs in megakaryocytic cell lines. Leuk. Lymphoma 1998, 29, 399–405. [Google Scholar] [CrossRef]
- Barton, G.M. A calculated response: Control of inflammation by the innate immune system. J. Clin. Investig. 2008, 118, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.V.; Bokla, L.; Nüsslein-Volhard, C. Establishment of dorsal-ventral polarity in the Drosophila embryo: The induction of polarity by the Toll gene product. Cell 1985, 42, 791–798. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Barreiro, L.B.; Ben-Ali, M.; Quach, H.; Laval, G.; Patin, E.; Pickrell, J.K.; Bouchier, C.; Tichit, M.; Neyrolles, O.; Gicquel, B.; et al. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 2009, 5, e1000562. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int. Immunopharmacol. 2020, 89, 107087. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, L.M.; Lin, E.; Morin, K.M.; Tanriverdi, K.; Freedman, J.E. Regulatory effects of TLR2 on megakaryocytic cell function. Blood 2011, 117, 5963–5974. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, R.; Inoue, N.; Kawasaki, S.; Takei, A.; Kadotani, M.; Ohnishi, Y.; Ejiri, J.; Kobayashi, S.; Hirata, K.; Kawashima, S.; et al. Expression of Toll-like receptors on human platelets. Thromb. Res. 2004, 113, 379–385. [Google Scholar] [CrossRef]
- Cognasse, F.; Hamzeh, H.; Chavarin, P.; Acquart, S.; Genin, C.; Garraud, O. Evidence of Toll-like receptor molecules on human platelets. Immunol. Cell Biol. 2005, 83, 196–198. [Google Scholar] [CrossRef]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 2014, 124, 791–802. [Google Scholar] [CrossRef]
- Thon, J.N.; Peters, C.G.; Machlus, K.R.; Aslam, R.; Rowley, J.; Macleod, H.; Devine, M.T.; Fuchs, T.A.; Weyrich, A.S.; Semple, J.W.; et al. T granules in human platelets function in TLR9 organization and signaling. J. Cell Biol. 2012, 198, 561–574. [Google Scholar] [CrossRef]
- Sioud, M.; Fløisand, Y.; Forfang, L.; Lund-Johansen, F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J. Mol. Biol. 2006, 364, 945–954. [Google Scholar] [CrossRef]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, E.I.; Qureshi, S.T.; Schnare, M. The role of toll-like receptors in acute and chronic lung inflammation. J. Inflamm. 2010, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Botos, I.; Segal, D.M.; Davies, D.R. The structural biology of Toll-like receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lucas, R.M.; Liu, L.; Stow, J.L. Signalling, sorting and scaffolding adaptors for Toll-like receptors. J. Cell Sci. 2019, 133, jcs239194. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Adachi, O.; Ogawa, T.; Takeda, K.; Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 1999, 11, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Loiarro, M.; Gallo, G.; Fantò, N.; De Santis, R.; Carminati, P.; Ruggiero, V.; Sette, C. Identification of critical residues of the MyD88 death domain involved in the recruitment of downstream kinases. J. Biol. Chem. 2009, 284, 28093–28103. [Google Scholar] [CrossRef]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef]
- Gay, N.J.; Gangloff, M.; O’Neill, L.A. What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 2011, 32, 104–109. [Google Scholar] [CrossRef]
- Latty, S.L.; Sakai, J.; Hopkins, L.; Verstak, B.; Paramo, T.; Berglund, N.A.; Cammarota, E.; Cicuta, P.; Gay, N.J.; Bond, P.J.; et al. Activation of Toll-like receptors nucleates assembly of the MyDDosome signaling hub. eLife 2018, 7, e31377. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S.M.; Fitzgerald, K.A.; Rosains, J.; Rowe, D.C.; Golenbock, D.T.; Maniatis, T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2004, 101, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Berthet, J.; Damien, P.; Hamzeh-Cognasse, H.; Pozzetto, B.; Garraud, O.; Cognasse, F. Toll-like receptor 4 signal transduction in platelets: Novel pathways. Br. J. Haematol. 2010, 151, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef]
- Kang, J.Y.; Nan, X.; Jin, M.S.; Youn, S.J.; Ryu, Y.H.; Mah, S.; Han, S.H.; Lee, H.; Paik, S.G.; Lee, J.O. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 2009, 31, 873–884. [Google Scholar] [CrossRef]
- Molteni, M.; Gemma, S.; Rossetti, C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediat. Inflamm. 2016, 2016, 6978936. [Google Scholar] [CrossRef]
- Tanimura, N.; Saitoh, S.; Matsumoto, F.; Akashi-Takamura, S.; Miyake, K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 2008, 368, 94–99. [Google Scholar] [CrossRef]
- Latz, E.; Visintin, A.; Lien, E.; Fitzgerald, K.A.; Espevik, T.; Golenbock, D.T. The LPS receptor generates inflammatory signals from the cell surface. J. Endotoxin Res. 2003, 9, 375–380. [Google Scholar] [CrossRef]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef]
- Lim, K.H.; Staudt, L.M. Toll-like receptor signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a011247. [Google Scholar] [CrossRef] [PubMed]
- Häcker, H.; Tseng, P.H.; Karin, M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat. Rev. Immunol. 2011, 11, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Akashi-Takamura, S.; Miyake, K. TLR accessory molecules. Curr. Opin. Immunol. 2008, 20, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef]
- Troutman, T.D.; Hu, W.; Fulenchek, S.; Yamazaki, T.; Kurosaki, T.; Bazan, J.F.; Pasare, C. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc. Natl. Acad. Sci. USA 2012, 109, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Tukhvatulin, A.I.; Logunov, D.Y.; Shcherbinin, D.N.; Shmarov, M.M.; Naroditsky, B.S.; Gudkov, A.V.; Gintsburg, A.L. Toll-like receptors and their adapter molecules. Biochemistry 2010, 75, 1098–1114. [Google Scholar] [CrossRef]
- Estruch, M.; Bancells, C.; Beloki, L.; Sanchez-Quesada, J.L.; Ordóñez-Llanos, J.; Benitez, S. CD14 and TLR4 mediate cytokine release promoted by electronegative LDL in monocytes. Atherosclerosis 2013, 229, 356–362. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Senger, D.R.; Dvorak, A.M.; Harvey, V.S.; McDonagh, J. Regulation of extravascular coagulation by microvascular permeability. Science 1985, 227, 1059–1061. [Google Scholar] [CrossRef]
- Schett, G.; Redlich, K.; Xu, Q.; Bizan, P.; Gröger, M.; Tohidast-Akrad, M.; Kiener, H.; Smolen, J.; Steiner, G. Enhanced expression of heat shock protein 70 (hsp70) and heat shock factor 1 (HSF1) activation in rheumatoid arthritis synovial tissue. Differential regulation of hsp70 expression and hsf1 activation in synovial fibroblasts by proinflammatory cytokines, shear stress, and antiinflammatory drugs. J. Clin. Investig. 1998, 102, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Kawahara, K.; Yone, K.; Hashiguchi, T.; Yamakuchi, M.; Goto, M.; Inoue, K.; Yamada, S.; Ijiri, K.; Matsunaga, S.; et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003, 48, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.L.; Ma, C.; O’Connell, R.M.; Mehta, A.; DiLoreto, R.; Heath, J.R.; Baltimore, D. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 2014, 14, 445–459. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef]
- Pignatelli, P.; Carnevale, R. Megakaryocyte TLR2: Immunity bullet? Blood 2011, 117, 5791–5792. [Google Scholar] [CrossRef][Green Version]
- Kimura, A.; Naka, T.; Muta, T.; Takeuchi, O.; Akira, S.; Kawase, I.; Kishimoto, T. Suppressor of cytokine signaling-1 selectively inhibits LPS-induced IL-6 production by regulating JAK-STAT. Proc. Natl. Acad. Sci. USA 2005, 102, 17089–17094. [Google Scholar] [CrossRef]
- Wu, D.; Xie, J.; Wang, X.; Zou, B.; Yu, Y.; Jing, T.; Zhang, S.; Zhang, Q. Micro-concentration Lipopolysaccharide as a Novel Stimulator of Megakaryocytopoiesis that Synergizes with IL-6 for Platelet Production. Sci. Rep. 2015, 5, 13748. [Google Scholar] [CrossRef]
- Zhiyi, H.; Wenshan, L.; Wenze, Z.; Ning, D.; Chi, Z.; Kenan, Y.; Ping, W.; Qianqian, W.; Qing, Z. Secretion expression and activity assay of a novel fusion protein of thrombopoietin and interleukin-6 in Pichia pastoris. J. Biochem. 2007, 142, 17–24. [Google Scholar] [CrossRef]
- Malara, A.; Gruppi, C.; Abbonante, V.; Cattaneo, D.; De Marco, L.; Massa, M.; Iurlo, A.; Gianelli, U.; Balduini, C.L.; Tira, M.E.; et al. EDA fibronectin-TLR4 axis sustains megakaryocyte expansion and inflammation in bone marrow fibrosis. J. Exp. Med. 2019, 216, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Kovuru, N.; Raghuwanshi, S.; Gutti, R.K. Exosome mediated differentiation of megakaryocytes: A study on TLR mediated effects. J. Thromb. Thrombolysis 2019, 48, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Kovuru, N.; Raghuwanshi, S.; Sangeeth, A.; Malleswarapu, M.; Sharma, D.S.; Dahariya, S.; Pallepati, A.; Gutti, R.K. Co-stimulatory effect of TLR2 and TLR4 stimulation on megakaryocytic development is mediated through PI3K/NF-ĸB and XBP-1 loop. Cell. Signal. 2021, 80, 109924. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, L.P.; Rodríguez, C.S.; Miguel, C.P.; Pozner, R.G.; Ortiz Wilczyñski, J.M.; Negrotto, S.; Carrera-Silva, E.A.; Heller, P.G.; Schattner, M. Activation of toll-like receptors 2 and 4 on CD34+ cells increases human megakaryo/thrombopoiesis induced by thrombopoietin. J. Thromb. Haemost. 2019, 17, 2196–2210. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, M.; Brunn, G.J.; Karnicki, K.; Miller, R.S.; Owen, W.G.; Miller, V.M. In vivo effects of lipopolysaccharide and TLR4 on platelet production and activity: Implications for thrombotic risk. J. Appl. Physiol. 2007, 102, 429–433. [Google Scholar] [CrossRef]
- Espín-Palazón, R.; Stachura, D.L.; Campbell, C.A.; García-Moreno, D.; Del Cid, N.; Kim, A.D.; Candel, S.; Meseguer, J.; Mulero, V.; Traver, D. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell 2014, 159, 1070–1085. [Google Scholar] [CrossRef]
- He, Q.; Zhang, C.; Wang, L.; Zhang, P.; Ma, D.; Lv, J.; Liu, F. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 2015, 125, 1098–1106. [Google Scholar] [CrossRef]
- Vieira-de-Abreu, A.; Campbell, R.A.; Weyrich, A.S.; Zimmerman, G.A. Platelets: Versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin. Immunopathol. 2012, 34, 5–30. [Google Scholar] [CrossRef]
- Anabel, A.S.; Eduardo, P.C.; Pedro Antonio, H.C.; Carlos, S.M.; Juana, N.M.; Honorio, T.A.; Nicolás, V.S.; Sergio Roberto, A.R. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum. Immunol. 2014, 75, 1244–1251. [Google Scholar] [CrossRef]
- Zakeri, A.; Russo, M. Dual Role of Toll-like Receptors in Human and Experimental Asthma Models. Front. Immunol. 2018, 9, 1027. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Shashkin, P.N.; Brown, G.T.; Ghosh, A.; Marathe, G.K.; McIntyre, T.M. Lipopolysaccharide is a direct agonist for platelet RNA splicing. J. Immunol. 2008, 181, 3495–3502. [Google Scholar] [CrossRef]
- Ståhl, A.L.; Svensson, M.; Mörgelin, M.; Svanborg, C.; Tarr, P.I.; Mooney, J.C.; Watkins, S.L.; Johnson, R.; Karpman, D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 2006, 108, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Hamzeh-Cognasse, H.; Lafarge, S.; Delezay, O.; Pozzetto, B.; McNicol, A.; Garraud, O. Toll-like receptor 4 ligand can differentially modulate the release of cytokines by human platelets. Br. J. Haematol. 2008, 141, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Hally, K.E.; Bird, G.K.; La Flamme, A.C.; Harding, S.A.; Larsen, P.D. Platelets modulate multiple markers of neutrophil function in response to in vitro Toll-like receptor stimulation. PLoS ONE 2019, 14, e0223444. [Google Scholar] [CrossRef]
- Assinger, A.; Laky, M.; Schabbauer, G.; Hirschl, A.M.; Buchberger, E.; Binder, B.R.; Volf, I. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J. Thromb. Haemost. 2011, 9, 799–809. [Google Scholar] [CrossRef]
- Kälvegren, H.; Skoglund, C.; Helldahl, C.; Lerm, M.; Grenegård, M.; Bengtsson, T. Toll-like receptor 2 stimulation of platelets is mediated by purinergic P2X1-dependent Ca2+ mobilisation, cyclooxygenase and purinergic P2Y1 and P2Y12 receptor activation. Thromb. Haemost. 2010, 103, 398–407. [Google Scholar] [CrossRef]
- Niklaus, M.; Klingler, P.; Weber, K.; Koessler, A.; Boeck, M.; Kobsar, A.; Koessler, J. The involvement of toll-like receptors 2 and 4 in human platelet signalling pathways. Cell. Signal. 2020, 76, 109817. [Google Scholar] [CrossRef]
- Jerez-Dolz, D.; Torramade-Moix, S.; Palomo, M.; Moreno-Castaño, A.; Lopez-Vilchez, I.; Hernandez, R.; Badimon, J.J.; Zafar, M.U.; Diaz-Ricart, M.; Escolar, G. Internalization of microparticles by platelets is partially mediated by toll-like receptor 4 and enhances platelet thrombogenicity. Atherosclerosis 2020, 294, 17–24. [Google Scholar] [CrossRef]
- Rex, S.; Beaulieu, L.M.; Perlman, D.H.; Vitseva, O.; Blair, P.S.; McComb, M.E.; Costello, C.E.; Freedman, J.E. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb. Haemost. 2009, 102, 97–110. [Google Scholar] [CrossRef] [PubMed]
- de Stoppelaar, S.F.; Claushuis, T.A.; Schaap, M.C.; Hou, B.; van der Poll, T.; Nieuwland, R.; van ‘t Veer, C. Toll-Like Receptor Signalling Is Not Involved in Platelet Response to Streptococcus pneumoniae In Vitro or In Vivo. PLoS ONE 2016, 11, e0156977. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.E.; La Flamme, A.C.; Larsen, P.D.; Harding, S.A. Platelet Toll-like receptor (TLR) expression and TLR-mediated platelet activation in acute myocardial infarction. Thromb. Res. 2017, 158, 8–15. [Google Scholar] [CrossRef]
- Hally, K.E.; La Flamme, A.C.; Harding, S.A.; Larsen, P.D. Platelets regulate leucocyte responses to Toll-like receptor stimulation. Clin. Transl. Immunol. 2018, 7, e1036. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Nalamolu, K.R.; Challa, S.R.; Fornal, C.A.; Grudzien, N.A.; Jorgenson, L.C.; Choudry, M.M.; Smith, N.J.; Palmer, C.J.; Pinson, D.M.; Klopfenstein, J.D.; et al. Attenuation of the Induction of TLRs 2 and 4 Mitigates Inflammation and Promotes Neurological Recovery After Focal Cerebral Ischemia. Transl. Stroke Res. 2021, 12, 923–936. [Google Scholar] [CrossRef]
- Branchford, B.R.; Carpenter, S.L. The Role of Inflammation in Venous Thromboembolism. Front. Pediatr. 2018, 6, 142. [Google Scholar] [CrossRef]
- Cheng, Z.; Jia, W.; Tian, X.; Jiang, P.; Zhang, Y.; Li, J.; Tian, C.; Liu, J. Cotinine inhibits TLR4/NF-κB signaling pathway and improves deep vein thrombosis in rats. Biosci. Rep. 2020, 40, BSR20201293. [Google Scholar] [CrossRef]
- He, Y.; Feng, D.; Li, M.; Gao, Y.; Ramirez, T.; Cao, H.; Kim, S.J.; Yang, Y.; Cai, Y.; Ju, C.; et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology 2017, 66, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Bezhaeva, T.; Karper, J.; Quax, P.H.A.; de Vries, M.R. The Intriguing Role of TLR Accessory Molecules in Cardiovascular Health and Disease. Front. Cardiovasc. Med. 2022, 9, 820962. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Yan, H.; Han, X.; Weng, L.; Wei, Q.; Sun, X.; Lu, W.; Wei, Q.; Ye, J.; Cai, X.; et al. Ginseng-derived nanoparticles alter macrophage polarization to inhibit melanoma growth. J. Immunother. Cancer 2019, 7, 326. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Ruvolo, G.; Lio, D.; Madonna, R. Toll-like receptor-4 signaling pathway in aorta aging and diseases: “its double nature”. J. Mol. Cell. Cardiol. 2017, 110, 38–53. [Google Scholar] [CrossRef]
- Sharma, S.; Garg, I.; Ashraf, M.Z. TLR signalling and association of TLR polymorphism with cardiovascular diseases. Vasc. Pharmacol. 2016, 87, 30–37. [Google Scholar] [CrossRef]
- Cen, X.; Liu, S.; Cheng, K. The Role of Toll-Like Receptor in Inflammation and Tumor Immunity. Front. Pharmacol. 2018, 9, 878. [Google Scholar] [CrossRef] [PubMed]
- Pozner, R.G.; Ure, A.E.; Jaquenod de Giusti, C.; D’Atri, L.P.; Italiano, J.E.; Torres, O.; Romanowski, V.; Schattner, M.; Gómez, R.M. Junín virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type I IFN signaling. PLoS Pathog. 2010, 6, e1000847. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, L.P.; Etulain, J.; Rivadeneyra, L.; Lapponi, M.J.; Centurion, M.; Cheng, K.; Yin, H.; Schattner, M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J. Thromb. Haemost. 2015, 13, 839–850. [Google Scholar] [CrossRef]
- Claushuis, T.A.M.; Van Der Veen, A.I.P.; Horn, J.; Schultz, M.J.; Houtkooper, R.H.; Van ‘t Veer, C.; Van Der Poll, T. Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets 2019, 30, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Bortolus, C.; Lecointe, K.; Parny, M.; Charlet, R.; Sendid, B.; Jawhara, S. Fungal Chitin Reduces Platelet Activation Mediated via TLR8 Stimulation. Front. Cell. Infect. Microbiol. 2019, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef]
- Abdollahi-Roodsaz, S.; Joosten, L.A.; Roelofs, M.F.; Radstake, T.R.; Matera, G.; Popa, C.; van der Meer, J.W.; Netea, M.G.; van den Berg, W.B. Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007, 56, 2957–2967. [Google Scholar] [CrossRef]
- Pierer, M.; Wagner, U.; Rossol, M.; Ibrahim, S. Toll-like receptor 4 is involved in inflammatory and joint destructive pathways in collagen-induced arthritis in DBA1J mice. PLoS ONE 2011, 6, e23539. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Stokes, J.A.; Corr, M.; Yaksh, T.L. Toll-like receptor signaling regulates cisplatin-induced mechanical allodynia in mice. Cancer Chemother. Pharmacol. 2014, 73, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, H.; Li, T.; Zhou, M.; Hao, S.; Yan, H.; Yu, Z.; Li, W.; Li, K.; Hang, C. TLR4 inhibitor resatorvid provides neuroprotection in experimental traumatic brain injury: Implication in the treatment of human brain injury. Neurochem. Int. 2014, 75, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Buisman-Pijlman, F.; Hutchinson, M.R. Toll-like receptor 4: Innate immune regulator of neuroimmune and neuroendocrine interactions in stress and major depressive disorder. Front. Neurosci. 2014, 8, 309. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Laterre, P.F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The ACCESS randomized trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef]
- Rice, T.W.; Wheeler, A.P.; Bernard, G.R.; Vincent, J.L.; Angus, D.C.; Aikawa, N.; Demeyer, I.; Sainati, S.; Amlot, N.; Cao, C.; et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med. 2010, 38, 1685–1694. [Google Scholar] [CrossRef]
- Tidswell, M.; Tillis, W.; Larosa, S.P.; Lynn, M.; Wittek, A.E.; Kao, R.; Wheeler, J.; Gogate, J.; Opal, S.M. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit. Care Med. 2010, 38, 72–83. [Google Scholar] [CrossRef]
- Bennett-Guerrero, E.; Grocott, H.P.; Levy, J.H.; Stierer, K.A.; Hogue, C.W.; Cheung, A.T.; Newman, M.F.; Carter, A.A.; Rossignol, D.P.; Collard, C.D. A phase II, double-blind, placebo-controlled, ascending-dose study of Eritoran (E5564), a lipid A antagonist, in patients undergoing cardiac surgery with cardiopulmonary bypass. Anesth. Analg. 2007, 104, 378–383. [Google Scholar] [CrossRef]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Manček-Keber, M.; Jerala, R. Postulates for validating TLR4 agonists. Eur. J. Immunol. 2015, 45, 356–370. [Google Scholar] [CrossRef]
- Cheng, K.; Gao, M.; Godfroy, J.I.; Brown, P.N.; Kastelowitz, N.; Yin, H. Specific activation of the TLR1-TLR2 heterodimer by small-molecule agonists. Sci. Adv. 2015, 1, e1400139. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Zhu, G.; Yang, J.; Yang, J.; Guo, J.; Jin, J.; Nandakumar, K.S.; Yang, W.; Yin, H.; Liu, S.; et al. TLR1/2 Specific Small-Molecule Agonist Suppresses Leukemia Cancer Cell Growth by Stimulating Cytotoxic T Lymphocytes. Adv. Sci. 2019, 6, 1802042. [Google Scholar] [CrossRef] [PubMed]
- Rose, W.A., 2nd; McGowin, C.L.; Pyles, R.B. FSL-1, a bacterial-derived toll-like receptor 2/6 agonist, enhances resistance to experimental HSV-2 infection. Virol. J. 2009, 6, 195. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Shihab, P.K.; Jasem, S.; Behbehani, K. FSL-1 induces MMP-9 production through TLR-2 and NF-κB /AP-1 signaling pathways in monocytic THP-1 cells. Cell. Physiol. Biochem. 2014, 34, 929–942. [Google Scholar] [CrossRef]
- Kurkjian, C.J.; Guo, H.; Montgomery, N.D.; Cheng, N.; Yuan, H.; Merrill, J.R.; Sempowski, G.D.; Brickey, W.J.; Ting, J.P. The Toll-Like Receptor 2/6 Agonist, FSL-1 Lipopeptide, Therapeutically Mitigates Acute Radiation Syndrome. Sci. Rep. 2017, 7, 17355. [Google Scholar] [CrossRef]
- Hug, B.A.; Matheny, C.J.; Burns, O.; Struemper, H.; Wang, X.; Washburn, M.L. Safety, Pharmacokinetics, and Pharmacodynamics of the TLR4 Agonist GSK1795091 in Healthy Individuals: Results from a Randomized, Double-blind, Placebo-controlled, Ascending Dose Study. Clin. Ther. 2020, 42, 1519–1534.e33. [Google Scholar] [CrossRef]
- Maroof, A.; Yorgensen, Y.M.; Li, Y.; Evans, J.T. Intranasal vaccination promotes detrimental Th17-mediated immunity against influenza infection. PLoS Pathog. 2014, 10, e1003875. [Google Scholar] [CrossRef]
- Shanmugam, A.; Rajoria, S.; George, A.L.; Mittelman, A.; Suriano, R.; Tiwari, R.K. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS ONE 2012, 7, e30839. [Google Scholar] [CrossRef]
- Das, N.; Dewan, V.; Grace, P.M.; Gunn, R.J.; Tamura, R.; Tzarum, N.; Watkins, L.R.; Wilson, I.A.; Yin, H. HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia. Cell Rep. 2016, 17, 1128–1140. [Google Scholar] [CrossRef]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Mullarkey, M.; Rose, J.R.; Bristol, J.; Kawata, T.; Kimura, A.; Kobayashi, S.; Przetak, M.; Chow, J.; Gusovsky, F.; Christ, W.J.; et al. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J. Pharmacol. Exp. Ther. 2003, 304, 1093–1102. [Google Scholar] [CrossRef]
- Reilly, M.; Miller, R.M.; Thomson, M.H.; Patris, V.; Ryle, P.; McLoughlin, L.; Mutch, P.; Gilboy, P.; Miller, C.; Broekema, M.; et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin. Pharmacol. Ther. 2013, 94, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Fort, M.M.; Mozaffarian, A.; Stöver, A.G.; Correia Jda, S.; Johnson, D.A.; Crane, R.T.; Ulevitch, R.J.; Persing, D.H.; Bielefeldt-Ohmann, H.; Probst, P.; et al. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J. Immunol. 2005, 174, 6416–6423. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Pearce, S.; Peri, F.; Neumann, F.; Cockerill, G.; Pirianov, G. A novel small molecule TLR4 antagonist (IAXO-102) negatively regulates non-hematopoietic toll like receptor 4 signalling and inhibits aortic aneurysms development. Atherosclerosis 2015, 242, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhang, X.; Zhang, T.; Grace, P.M.; Li, H.; Wang, Y.; Li, H.; Chen, H.; Watkins, L.R.; Hutchinson, M.R.; et al. Lovastatin inhibits Toll-like receptor 4 signaling in microglia by targeting its co-receptor myeloid differentiation protein 2 and attenuates neuropathic pain. Brain Behav. Immun. 2019, 82, 432–444. [Google Scholar] [CrossRef]
- Cheng, K.; Wang, X.; Zhang, S.; Yin, H. Discovery of small-molecule inhibitors of the TLR1/TLR2 complex. Angew. Chem. Int. Ed. Engl. 2012, 51, 12246–12249. [Google Scholar] [CrossRef]
- Piazza, M.; Rossini, C.; Della Fiorentina, S.; Pozzi, C.; Comelli, F.; Bettoni, I.; Fusi, P.; Costa, B.; Peri, F. Glycolipids and benzylammonium lipids as novel antisepsis agents: Synthesis and biological characterization. J. Med. Chem. 2009, 52, 1209–1213. [Google Scholar] [CrossRef]
- Calvo-Rodríguez, M.; de la Fuente, C.; García-Durillo, M.; García-Rodríguez, C.; Villalobos, C.; Núñez, L. Aging and amyloid β oligomers enhance TLR4 expression, LPS-induced Ca2+ responses, and neuron cell death in cultured rat hippocampal neurons. J. Neuroinflamm. 2017, 14, 24. [Google Scholar] [CrossRef]
- Zhang, X.; Peng, Y.; Grace, P.M.; Metcalf, M.D.; Kwilasz, A.J.; Wang, Y.; Zhang, T.; Wu, S.; Selfridge, B.R.; Portoghese, P.S.; et al. Stereochemistry and innate immune recognition: (+)-norbinaltorphimine targets myeloid differentiation protein 2 and inhibits toll-like receptor 4 signaling. FASEB J. 2019, 33, 9577–9587. [Google Scholar] [CrossRef]
- Monnet, E.; Choy, E.H.; McInnes, I.; Kobakhidze, T.; de Graaf, K.; Jacqmin, P.; Lapeyre, G.; de Min, C. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: A phase II study. Ann. Rheum. Dis. 2020, 79, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Laird, M.H.; Schwarz, R.S.; Greene, S.; Dyson, T.; Snyder, G.A.; Xiao, T.S.; Chauhan, J.; Fletcher, S.; Toshchakov, V.Y.; et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc. Natl. Acad. Sci. USA 2015, 112, 5455–5460. [Google Scholar] [CrossRef] [PubMed]
- Lamphier, M.; Zheng, W.; Latz, E.; Spyvee, M.; Hansen, H.; Rose, J.; Genest, M.; Yang, H.; Shaffer, C.; Zhao, Y.; et al. Novel small molecule inhibitors of TLR7 and TLR9: Mechanism of action and efficacy in vivo. Mol. Pharmacol. 2014, 85, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Koymans, K.J.; Feitsma, L.J.; Brondijk, T.H.; Aerts, P.C.; Lukkien, E.; Lössl, P.; van Kessel, K.P.; de Haas, C.J.; van Strijp, J.A.; Huizinga, E.G. Structural basis for inhibition of TLR2 by staphylococcal superantigen-like protein 3 (SSL3). Proc. Natl. Acad. Sci. USA 2015, 112, 11018–11023. [Google Scholar] [CrossRef]
- Wang, Y.; Tu, Q.; Yan, W.; Xiao, D.; Zeng, Z.; Ouyang, Y.; Huang, L.; Cai, J.; Zeng, X.; Chen, Y.J.; et al. CXC195 suppresses proliferation and inflammatory response in LPS-induced human hepatocellular carcinoma cells via regulating TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem. Biophys. Res. Commun. 2015, 456, 373–379. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, G.; Huang, J.; Ma, S.; Mi, K.; Cheng, J.; Zhu, Y.; Zha, X.; Huang, W. Atractylenolide I modulates ovarian cancer cell-mediated immunosuppression by blocking MD-2/TLR4 complex-mediated MyD88/NF-κB signaling in vitro. J. Transl. Med. 2016, 14, 104. [Google Scholar] [CrossRef]
- Premkumar, V.; Dey, M.; Dorn, R.; Raskin, I. MyD88-dependent and independent pathways of Toll-Like Receptors are engaged in biological activity of Triptolide in ligand-stimulated macrophages. BMC Chem. Biol. 2010, 10, 3. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, Q.; Qian, R.; Liu, S.; Hu, W.; Liu, Z. Paeonol antagonizes oncogenesis of osteosarcoma by inhibiting the function of TLR4/MAPK/NF-κB pathway. Acta Histochem. 2020, 122, 151455. [Google Scholar] [CrossRef]
- Grabowski, M.; Murgueitio, M.S.; Bermudez, M.; Rademann, J.; Wolber, G.; Weindl, G. Identification of a pyrogallol derivative as a potent and selective human TLR2 antagonist by structure-based virtual screening. Biochem. Pharmacol. 2018, 154, 148–160. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Typical Aptamer | Regulatory Aptamer |
|---|---|
| myeloid differentiation primary-response protein 88 (MyD88) | Sterile α and armadillo motif-containing protein (SARM) |
| TIR-domain-containing adaptor protein (TIRAP), also known as MyD88 adapter-like protein (MAL) | B-cell adaptor for phosphoinositide 3-kinase (BCAP), also known as Phosphoinositide 3-kinase adapter protein 1 (PIK3AP1) |
| TIR-domain-containing adaptor protein inducing interferon-β (TRIF), also known as TIR domain-containing adapter molecule 1 (TICAM1) | SLP adapter and Csk-interacting membrane protein(SCIMP) |
| TRIF-related adaptor molecule (TRAM) also known as TIR domain-containing adapter molecule 2 (TICAM2) |
| Tagonist | Target TLR | Biological Activity | References |
|---|---|---|---|
| LPS/lipid A | TLR4 | It is a compound of lipids and polysaccharides. This product is lipopolypaccharide purified from E. coli O111:B4, which can specifically activate TLR4 but not TLR2. | |
| Paclitaxel | TLR4 | Paclitaxel is one of the confirmed direct mouse TLR4/MD-2 agonists and is an antagonist of the human TLR4 receptor complex | [161] |
| Ni2+ ions | TLR4 | Specific activation of human TLR4/MD-2 by Ni2+, but not of the mouse receptor | [161] |
| CU-T12-9 | TLR2 | CU-T12-9 is a specific TLR1/2 agonist with EC50 of 52.9 nM in HEK-Blue hTLR2 SEAP assay. CU-T12-9 activates both the innate and the adaptive immune systems. CU-T12-9 selectively activates the TLR1/2 heterodimer, not TLR2/6. CU-T12-9 signals through NF-κB and invokes an elevation of the downstream effectors TNF-α, IL-10, and iNOS | [162] |
| SMU-Z1 | TLR2 | SMU-Z1 exhibited specific activation of TLR1/TLR2 signaling and showed antitumor immunity against leukemia in a murine leukemia model. | [163] |
| FSL-1 | TLR2 | FSL-1, a bacterial-derived toll-like receptor 2/6 (TLR2/6) agonist, enhances resistance to experimental HSV-2 infection. | [164,165] |
| FSL-1 TFA | TLR2 | FSL-1 TFA, a bacterial-derived toll-like receptor 2/6 (TLR2/6) agonist, enhances resistance to experimental HSV-2 infection. FSL-1 TFA induces MMP-9 production through TLR2 and NF-κB/AP-1 signaling pathways in monocytic THP-1 cells | [164,166] |
| GSK1795091 (CRX-601) | TLR4 | GSK1795091 (CRX-601), an immunologic stimulator, is a synthetic TLR4 agonist. Antitumor activity. GSK1795091 can be used as a vaccine adjuvant to enhance both mucosal and systemic immunity to influenza virus vaccines | [167,168] |
| RS 09 TFA | TLR4 | RS 09 TFA is a TLR4 agonist. RS 09 TFA promotes NF-κB nuclear translocation and induces inflammatory cytokine secretion in RAW264.7 macrophages in vitro. RS 09 TFA acts as an adjuvant in vivo; RS 09 TFA enhances X-15 specific antibody serum concentrations, when administered with X-15-KLH in mice | [169,170] |
| HMGB1 | TLR5 | HMGB1 can bind to TLR5 to initiate its downstream NF-κB signaling pathway activation and induce proinflammatory cytokine | [170] |
| Imiquimod | TLR7 | Imiquimod is the first FDA-approved agonist targeting TLR7 for the treatment of external genital warts. | [171] |
| Antagonist | Target TLR | Mechanism | References |
|---|---|---|---|
| TAK-242 (Resatorvid) | TLR4 | TAK-242 binds selectively to TLR4 (Cys747) and subsequently disrupts the interaction of TLR4 with adaptor molecules, thereby inhibiting TLR4 signal transduction and its downstream signaling events | [172] |
| E5564 (Eritoran) | TLR4 | E5564 (a novel Toll-like receptor 4-directed endotoxin antagonist) can block TLR4 activation through prevention of LPS binding to the TLR4-MD2 complex and lack agonistic activity in human and animal model systems, making it a potentially effective therapeutic agent for treatment of disease states caused by endotoxin. | [173] |
| OPN-305 | TLR2 | OPN-305 is the first humanized IgG4 monoclonal antibody against TLR2 in development and is intended for the prevention of reperfusion injury following renal transplantation and other indications. | [174] |
| CRX-526 | TLR4 | CRX-526, which has antagonistic activity for TLR4 and can block TLR4 activation through prevention of LPS binding to the TLR4-MD2 complex. CRX-526 can prevent the expression of proinflammatory genes stimulated by LPS in vitro | [175] |
| IAXO-102 | TLR4 | IAXO-102 is a TLR4 antagonist which negatively regulates TLR4 signaling. IAXO-102 inhibits MAPK and p65 NF-κB phosphorylation and expression of TLR4 dependent proinflammatory protein. IAXO-102 also prevents experimental abdominal aortic aneurysm development | [176] |
| lovastatin | TLR4 | lovastatin may be a potential drug to be repurposed for treating chronic pain | [177] |
| CU-CPT22 | TLR2 | CU-CPT22 is a potent protein complex of toll-like receptor 1 and 2 (TLR1/2) inhibitor, and competes with the synthetic triacylated lipoprotein (Pam3CSK4) binding to TLR1/2. | [178] |
| CAY10614 | TLR4 | CAY10614 is a potent TLR4 antagonist. CAY10614 inhibits the lipid A-induced activation of TLR4. | [179,180] |
| (+)-norbinaltorphimine | TLR4 | the TLR4 antagonistic activity of (+)-norbinaltorphimine increased to 4.7 ± 1.8 μM in the NO assay and significantly enhanced and prolonged morphine analgesia in vivo. | [181] |
| NI-0101 | TLR4 | Primarily blocking THE dimerization of TLR4 and blocking the production of pro-inflammatory cytokines in synovial stimulated monocytes in RA patients, it has been tested in clinical trials in RA patients but unfortunately has not shown any benefit | [182] |
| C16H15NO4 (C29) | TLR2 | C29, and its derivative, ortho-vanillin (o-vanillin), inhibited TLR2/1 and TLR2/6 signaling induced by synthetic and bacterial TLR2 agonists in human HEK-TLR2 and THP-1 cells, but only TLR2/1 signaling in murine macrophages. | [183] |
| Hydroxychloroquine | TLR7/9 | Hydroxychloroquine is an autophagy inhibitor, which may target TLR7 and TLR9. | [184] |
| SSL3 | TLR2 | SSL3 inhibits binding of bacterial lipopeptides, and, second, if a lipopeptide has already been engaged by TLR2, SSL3 prevents the formation of TLR2–TLR1 and TLR2–TLR6 heterodimers. | [185] |
| CXC195 | TLR4 | CXC195 Exerts anti-proliferative effects through TLR4-mediated suppression of inflammatory cytokines. | [186] |
| Atractylenolide | TLR4 | Atractylenolide can make TLR4 and MyD88/NF-κB in ovarian cancer cells downregulatie | [187] |
| Triptolide | TLR4 | Triptolide can inducie suppressive effect on the TLR4/NF-κB axis. | [188] |
| Paeonol | TLR4 | Paeonol can abolish the propagation of the TLR4/MAPK/NF-κB signaling axis. | [189] |
| MMG-11 | TLR2 | MMG-11 is a potent and selective human TLR2 antagonist with low cytotoxicity. MMG-11 inhibits both TLR2/1 and TLR2/6 signaling. | [190] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, X.; Xu, Q.; Yang, S.; Huang, X.; Wang, L.; Huang, F.; Luo, J.; Zhou, X.; Wu, A.; Mei, Q.; et al. Toll-like Receptors and Thrombopoiesis. Int. J. Mol. Sci. 2023, 24, 1010. https://doi.org/10.3390/ijms24021010
Tang X, Xu Q, Yang S, Huang X, Wang L, Huang F, Luo J, Zhou X, Wu A, Mei Q, et al. Toll-like Receptors and Thrombopoiesis. International Journal of Molecular Sciences. 2023; 24(2):1010. https://doi.org/10.3390/ijms24021010
Chicago/Turabian StyleTang, Xiaoqin, Qian Xu, Shuo Yang, Xinwu Huang, Long Wang, Feihong Huang, Jiesi Luo, Xiaogang Zhou, Anguo Wu, Qibing Mei, and et al. 2023. "Toll-like Receptors and Thrombopoiesis" International Journal of Molecular Sciences 24, no. 2: 1010. https://doi.org/10.3390/ijms24021010
APA StyleTang, X., Xu, Q., Yang, S., Huang, X., Wang, L., Huang, F., Luo, J., Zhou, X., Wu, A., Mei, Q., Zhao, C., & Wu, J. (2023). Toll-like Receptors and Thrombopoiesis. International Journal of Molecular Sciences, 24(2), 1010. https://doi.org/10.3390/ijms24021010