
Seeing Neurodegeneration in a New Light Using Genetically Encoded Fluorescent Biosensors and iPSCs


Abstract
1. Introduction
2. Genetically Encoded Fluorescent Biosensor Advantages
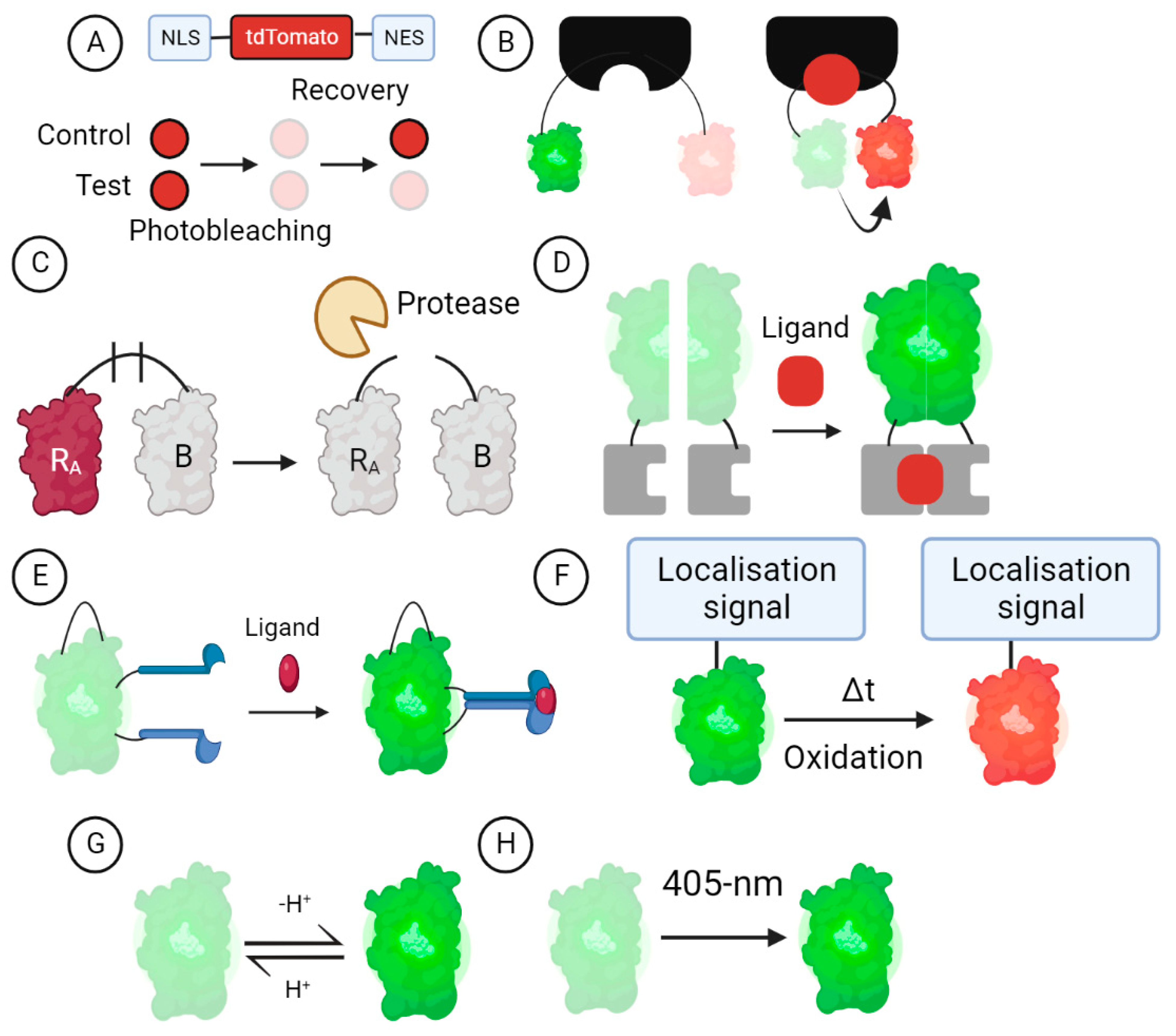
3. GEFB Designs
3.1. Turnover and Translocation-Based GEFBs
3.2. FRET-Based GEFBs
3.3. Dimerisation-Dependent GEFBs
3.4. cpFP GEFBs
3.5. Oxidation-Dependent GEFBs
3.6. Ion-Sensitive GEFBs
3.7. Photo-Transformable GEFBs
4. Leveraging GEFB and iPSC Technologies for Pre-Clinical Applications
4.1. Industrial and Lifestyle Exposures
4.1.1. Pesticides
4.1.2. Chemotherapy-Induced Peripheral Neuropathy
4.1.3. Pathogens and Pathogen-Derived Toxins
4.2. Genetics
4.2.1. Isogenic Disease Models
4.2.2. CRISPR-Based Genetic Screens
4.3. Cell–Cell and Cell–Environment Interactions
4.3.1. Neuroinflammation
4.3.2. Neurotransmitter Clearance, Hyperexcitability, and Excitotoxicity
4.3.3. Amyloid and Amyloid Plaques
4.4. Development and Testing of Novel Therapeutics
5. Challenges and Caveats of Using GEFBs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, M.L.; Cooper, B.A.; Paul, S.M.; Abrams, G.; Topp, K.; Kober, K.M.; Chesney, M.A.; Mazor, M.; Schumacher, M.A.; Conley, Y.P.; et al. Age-related differences in patient-reported and objective measures of chemotherapy-induced peripheral neuropathy among cancer survivors. Support. Care Cancer 2019, 27, 3905–3912. [Google Scholar] [CrossRef] [PubMed]
- Maihöfner, C.; Diel, I.; Tesch, H.; Quandel, T.; Baron, R. Chemotherapy-induced peripheral neuropathy (CIPN): Current therapies and topical treatment option with high-concentration capsaicin. Support. Care Cancer 2021, 29, 4223–4238. [Google Scholar] [CrossRef]
- Staff, N.P.; Grisold, A.; Grisold, W.; Windebank, A.J. Chemotherapy-induced peripheral neuropathy: A current review. Ann. Neurol. 2017, 81, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Grande, G.; Qiu, C.; Fratiglioni, L. Prevention of dementia in an ageing world: Evidence and biological rationale. Ageing Res. Rev. 2020, 64, 101045. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Ghosh, R.; Tabrizi, S.J. Clinical Features of Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 1–28. [Google Scholar] [CrossRef]
- Mo, C.; Hannan, A.J.; Renoir, T. Environmental factors as modulators of neurodegeneration: Insights from gene-environment interactions in Huntington’s disease. Neurosci. Biobehav. Rev. 2015, 52, 178–192. [Google Scholar] [CrossRef]
- Graham, N.S.N.; Jolly, A.; Zimmerman, K.; Bourke, N.J.; Scott, G.; Cole, J.H.; Schott, J.M.; Sharp, D.J. Diffuse axonal injury predicts neurodegeneration after moderate-severe traumatic brain injury. Brain A J. Neurol. 2020, 143, 3685–3698. [Google Scholar] [CrossRef]
- Linsley, J.W.; Shah, K.; Castello, N.; Chan, M.; Haddad, D.; Doric, Z.; Wang, S.; Leks, W.; Mancini, J.; Oza, V.; et al. Genetically encoded cell-death indicators (GEDI) to detect an early irreversible commitment to neurodegeneration. Nat. Commun. 2021, 12, 5284. [Google Scholar] [CrossRef]
- Greenwald, E.C.; Mehta, S.; Zhang, J. Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks. Chem. Rev. 2018, 118, 11707–11794. [Google Scholar] [CrossRef]
- Kim, H.; Ju, J.; Lee, H.N.; Chun, H.; Seong, J. Genetically Encoded Biosensors Based on Fluorescent Proteins. Sensors 2021, 21, 795. [Google Scholar] [CrossRef]
- Conley, J.M.; Radhakrishnan, S.; Valentino, S.A.; Tantama, M. Imaging extracellular ATP with a genetically-encoded, ratiometric fluorescent sensor. PLoS ONE 2017, 12, e0187481. [Google Scholar] [CrossRef]
- Ollivier, M.; Beudez, J.; Linck, N.; Grutter, T.; Compan, V.; Rassendren, F. P2X-GCaMPs as Versatile Tools for Imaging Extracellular ATP Signaling. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Goryashchenko, A.S.; Pakhomov, A.A.; Ryabova, A.V.; Romanishkin, I.D.; Maksimov, E.G.; Orsa, A.N.; Serova, O.V.; Mozhaev, A.A.; Maksimova, M.A.; Martynov, V.I.; et al. FLIM-Based Intracellular and Extracellular pH Measurements Using Genetically Encoded pH Sensor. Biosensors 2021, 11, 340. [Google Scholar] [CrossRef]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- Tantama, M.; Martínez-François, J.R.; Mongeon, R.; Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat. Commun. 2013, 4, 2550. [Google Scholar] [CrossRef]
- Hung, Y.P.; Albeck, J.G.; Tantama, M.; Yellen, G. Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab. 2011, 14, 545–554. [Google Scholar] [CrossRef]
- Bi, X.; Beck, C.; Gong, Y. Genetically Encoded Fluorescent Indicators for Imaging Brain Chemistry. Biosensors 2021, 11, 116. [Google Scholar] [CrossRef]
- Mehta, S.; Zhang, J. Biochemical Activity Architectures Visualized-Using Genetically Encoded Fluorescent Biosensors to Map the Spatial Boundaries of Signaling Compartments. Acc. Chem. Res. 2021, 54, 2409–2420. [Google Scholar] [CrossRef]
- Hong, S.G.; Yada, R.C.; Choi, K.; Carpentier, A.; Liang, T.J.; Merling, R.K.; Sweeney, C.L.; Malech, H.L.; Jung, M.; Corat, M.A.F.; et al. Rhesus iPSC Safe Harbor Gene-Editing Platform for Stable Expression of Transgenes in Differentiated Cells of All Germ Layers. Mol. Ther. 2017, 25, 44–53. [Google Scholar] [CrossRef]
- Sun, Y.-H.; Kao, H.K.J.; Chang, C.-W.; Merleev, A.; Overton, J.L.; Pretto, D.; Yechikov, S.; Maverakis, E.; Chiamvimonvat, N.; Chan, J.W.; et al. Human induced pluripotent stem cell line with genetically encoded fluorescent voltage indicator generated via CRISPR for action potential assessment post-cardiogenesis. Stem Cells 2020, 38, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Weisheit, I.; Kroeger, J.A.; Malik, R.; Klimmt, J.; Crusius, D.; Dannert, A.; Dichgans, M.; Paquet, D. Detection of Deleterious On-Target Effects after HDR-Mediated CRISPR Editing. Cell Rep. 2020, 31, 107689. [Google Scholar] [CrossRef] [PubMed]
- Stellon, D.; Tran, M.T.N.; Talbot, J.; Chear, S.; Khalid, M.K.N.M.; Pébay, A.; Vickers, J.C.; King, A.E.; Hewitt, A.W.; Cook, A.L. CRISPR/Cas-Mediated Knock-in of Genetically Encoded Fluorescent Biosensors into the AAVS1 Locus of Human-Induced Pluripotent Stem Cells. Methods Mol. Biol. 2021, 2549, 379–398. [Google Scholar] [CrossRef]
- Erapaneedi, R.; Belousov, V.V.; Schäfers, M.; Kiefer, F. A novel family of fluorescent hypoxia sensors reveal strong heterogeneity in tumor hypoxia at the cellular level. EMBO J. 2016, 35, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Kelly, C.J.; Colgan, S.P. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C350–C360. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Monitoring Autophagy Flux and Activity: Principles and Applications. BioEssays News Rev. Mol. Cell. Dev. Biol. 2020, 42, e2000122. [Google Scholar] [CrossRef]
- Gómez-Sánchez, R.; Pizarro-Estrella, E.; Yakhine-Diop, S.M.; Rodríguez-Arribas, M.; Bravo-San Pedro, J.M.; Fuentes, J.M.; González-Polo, R.A. Routine Western blot to check autophagic flux: Cautions and recommendations. Anal. Biochem. 2015, 477, 13–20. [Google Scholar] [CrossRef]
- Zhong, C.; Schleifenbaum, J. Genetically Encoded Calcium Indicators: A New Tool in Renal Hypertension Research. Front. Med. 2019, 6, 128. [Google Scholar] [CrossRef]
- Davidson, S.M.; Duchen, M.R. Imaging mitochondrial calcium signalling with fluorescent probes and single or two photon confocal microscopy. Methods Mol. Biol. 2012, 810, 219–234. [Google Scholar] [CrossRef]
- Martynov, V.I.; Pakhomov, A.A.; Deyev, I.E.; Petrenko, A.G. Genetically encoded fluorescent indicators for live cell pH imaging. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2924–2939. [Google Scholar] [CrossRef]
- Liu, Z.; Pei, H.; Zhang, L.; Tian, Y. Mitochondria-Targeted DNA Nanoprobe for Real-Time Imaging and Simultaneous Quantification of Ca(2+) and pH in Neurons. ACS Nano 2018, 12, 12357–12368. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Nagel, M.; Müßig, S.; Höflinger, P.; Schöls, L.; Hauser, S.; Schüle, R. Generation of the CRISPR/Cas9-mediated KIF1C knock-out human iPSC line HIHRSi003-A-1. Stem Cell Res. 2020, 49, 102059. [Google Scholar] [CrossRef]
- Ortiz-Virumbrales, M.; Moreno, C.L.; Kruglikov, I.; Marazuela, P.; Sproul, A.; Jacob, S.; Zimmer, M.; Paull, D.; Zhang, B.; Schadt, E.E.; et al. CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer’s PSEN2 N141I neurons. Acta Neuropathol. Commun. 2017, 5, 77. [Google Scholar] [CrossRef]
- Chang, K.-H.; Huang, C.-Y.; Ou-Yang, C.-H.; Ho, C.-H.; Lin, H.-Y.; Hsu, C.-L.; Chen, Y.-T.; Chou, Y.-C.; Chen, Y.-J.; Chen, Y.; et al. In vitro genome editing rescues parkinsonism phenotypes in induced pluripotent stem cells-derived dopaminergic neurons carrying LRRK2 p.G2019S mutation. Stem Cell Res. Ther. 2021, 12, 508. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral Vectors in Gene Therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhi, S.; Chen, Y.; Wu, G.; Wen, J.; Wu, J.; Liu, Q.; Li, Y.; Kang, R.; Hu, S.; Wang, J.; et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2022, 30, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, M. Modern approaches in gene therapy of motor neuron diseases. Med. Res. Rev. 2021, 41, 2634–2655. [Google Scholar] [CrossRef] [PubMed]
- Mo, G.C.H.; Ross, B.; Hertel, F.; Manna, P.; Yang, X.; Greenwald, E.; Booth, C.; Plummer, A.M.; Tenner, B.; Chen, Z.; et al. Genetically encoded biosensors for visualizing live-cell biochemical activity at super-resolution. Nat. Methods 2017, 14, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.-L.; Zhang, C.; Zhang, C.; Tang, Y.; Ye, B.-C. A genetically encoded biosensor for in vitro and in vivo detection of NADP(.). Biosens. Bioelectron. 2016, 77, 901–906. [Google Scholar] [CrossRef]
- Palmer, A.E.; Qin, Y.; Park, J.G.; McCombs, J.E. Design and application of genetically encoded biosensors. Trends Biotechnol. 2011, 29, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An Autophagic Flux Probe that Releases an Internal Control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, D.M.; Hink, M.A.; Joosen, L.; Gadella, T.W.J.; Verkhusha, V.V. An orange fluorescent protein with a large Stokes shift for single-excitation multicolor FCCS and FRET imaging. J. Am. Chem. Soc. 2012, 134, 7913–7923. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, O.; Soleja, N.; Khan, P.; Hassan, I.; Mohsin, M. Visualization of thiamine in living cells using genetically encoded fluorescent nanosensor. Biochem. Eng. J. 2019, 146, 170–178. [Google Scholar] [CrossRef]
- Tak, H.; Haque, M.M.; Kim, M.J.; Lee, J.H.; Baik, J.-H.; Kim, Y.; Kim, D.J.; Grailhe, R.; Kim, Y.K. Bimolecular fluorescence complementation; lighting-up tau-tau interaction in living cells. PLoS ONE 2013, 8, e81682. [Google Scholar] [CrossRef]
- Jing, M.; Zhang, P.; Wang, G.; Feng, J.; Mesik, L.; Zeng, J.; Jiang, H.; Wang, S.; Looby, J.C.; Guagliardo, N.A.; et al. A genetically encoded fluorescent acetylcholine indicator for in vitro and in vivo studies. Nat. Biotechnol. 2018, 36, 726–737. [Google Scholar] [CrossRef]
- Ast, C.; Foret, J.; Oltrogge, L.M.; De Michele, R.; Kleist, T.J.; Ho, C.-H.; Frommer, W.B. Ratiometric Matryoshka biosensors from a nested cassette of green- and orange-emitting fluorescent proteins. Nat. Commun. 2017, 8, 431. [Google Scholar] [CrossRef]
- Sun, F.; Zeng, J.; Jing, M.; Zhou, J.; Feng, J.; Owen, S.F.; Luo, Y.; Li, F.; Wang, H.; Yamaguchi, T.; et al. A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice. Cell 2018, 174, 481–496.e419. [Google Scholar] [CrossRef]
- Marvin, J.S.; Shimoda, Y.; Malgoire, V.; Leite, M.; Kawashima, T.; Jensen, T.P.; Knott, E.L.; Novak, O.; Podgorski, K.; Leidenheimer, N.J.; et al. A genetically encoded fluorescent sensor for in vivo imaging of GABA. bioRxiv 2018. [Google Scholar] [CrossRef]
- Marvin, J.S.; Borghuis, B.G.; Tian, L.; Cichon, J.; Harnett, M.T.; Akerboom, J.; Gordus, A.; Renninger, S.L.; Chen, T.-W.; Bargmann, C.I.; et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 2013, 10, 162–170. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, C.; Lischinsky, J.E.; Jing, M.; Zhou, J.; Wang, H.; Zhang, Y.; Dong, A.; Wu, Z.; Wu, H.; et al. A Genetically Encoded Fluorescent Sensor for Rapid and Specific In Vivo Detection of Norepinephrine. Neuron 2019, 102, 745–761.e748. [Google Scholar] [CrossRef]
- Unger, E.K.; Keller, J.P.; Altermatt, M.; Liang, R.; Matsui, A.; Dong, C.; Hon, O.J.; Yao, Z.; Sun, J.; Banala, S.; et al. Directed Evolution of a Selective and Sensitive Serotonin Sensor via Machine Learning. Cell 2020, 183, 1986–2002.e1926. [Google Scholar] [CrossRef]
- Laker, R.C.; Xu, P.; Ryall, K.A.; Sujkowski, A.; Kenwood, B.M.; Chain, K.H.; Zhang, M.; Royal, M.A.; Hoehn, K.L.; Driscoll, M.; et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 2014, 289, 12005–12015. [Google Scholar] [CrossRef]
- Ponsford, A.H.; Ryan, T.A.; Raimondi, A.; Cocucci, E.; Wycislo, S.A.; Fröhlich, F.; Swan, L.E.; Stagi, M. Live imaging of intra-lysosome pH in cell lines and primary neuronal culture using a novel genetically encoded biosensor. Autophagy 2021, 17, 1500–1518. [Google Scholar] [CrossRef]
- Chu, C.T.; Plowey, E.D.; Wang, Y.; Patel, V.; Jordan-Sciutto, K.L. Location, location, location: Altered transcription factor trafficking in neurodegeneration. J. Neuropathol. Exp. Neurol. 2007, 66, 873–883. [Google Scholar] [CrossRef]
- Patel, V.P.; Defranco, D.B.; Chu, C.T. Altered transcription factor trafficking in oxidatively-stressed neuronal cells. Biochim. Biophys. Acta 2012, 1822, 1773–1782. [Google Scholar] [CrossRef]
- van Royen, M.E.; Farla, P.; Mattern, K.A.; Geverts, B.; Trapman, J.; Houtsmuller, A.B. Fluorescence recovery after photobleaching (FRAP) to study nuclear protein dynamics in living cells. Methods Mol. Biol. 2009, 464, 363–385. [Google Scholar] [CrossRef]
- Bizzarri, R.; Cardarelli, F.; Serresi, M.; Beltram, F. Fluorescence recovery after photobleaching reveals the biochemistry of nucleocytoplasmic exchange. Anal. Bioanal. Chem. 2012, 403, 2339–2351. [Google Scholar] [CrossRef]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Kumar, M.S.; Ramesh, N.; Anderson, E.N.; Nguyen, A.T.; Kim, B.; Cheung, S.; McDonough, J.A.; Skarnes, W.C.; Lopez-Gonzalez, R.; et al. Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway. Nat. Neurosci. 2021, 24, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.C.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 2017, 94, 93–107.e106. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e927. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.J.; Pluciennik, A.; Merry, D.E. Impaired Nuclear Export of Polyglutamine-Expanded Androgen Receptor in Spinal and Bulbar Muscular Atrophy. Sci. Rep. 2019, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Youssef, S.; Zhang, S.; Ai, H.-W. A Genetically Encoded, Ratiometric Fluorescent Biosensor for Hydrogen Sulfide. ACS Sens. 2019, 4, 1626–1632. [Google Scholar] [CrossRef]
- Liu, L.; He, F.; Yu, Y.; Wang, Y. Application of FRET Biosensors in Mechanobiology and Mechanopharmacological Screening. Front. Bioeng. Biotechnol. 2020, 8, 595497. [Google Scholar] [CrossRef]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef]
- Picó, S.; Parras, A.; Santos-Galindo, M.; Pose-Utrilla, J.; Castro, M.; Fraga, E.; Hernández, I.H.; Elorza, A.; Anta, H.; Wang, N.; et al. CPEB alteration and aberrant transcriptome-polyadenylation lead to a treatable SLC19A3 deficiency in Huntington’s disease. Sci. Transl. Med. 2021, 13, eabe7104. [Google Scholar] [CrossRef]
- Manzetti, S.; Zhang, J.; van der Spoel, D. Thiamin function, metabolism, uptake, and transport. Biochemistry 2014, 53, 821–835. [Google Scholar] [CrossRef]
- Liu, D.; Ke, Z.; Luo, J. Thiamine Deficiency and Neurodegeneration: The Interplay Among Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy. Mol. Neurobiol. 2017, 54, 5440–5448. [Google Scholar] [CrossRef]
- Tomasulo, P.A.; Kater, R.M.; Iber, F.L. Impairment of thiamine absorption in alcoholism. Am. J. Clin. Nutr. 1968, 21, 1341–1344. [Google Scholar] [CrossRef]
- Yeh, W.-Y.; Lian, L.-M.; Chang, A.; Cheng, C.-K. Thiamine-deficient optic neuropathy associated with Wernicke’s encephalopathy in patients with chronic diarrhea. J. Formos. Med. Assoc. 2013, 112, 165–170. [Google Scholar] [CrossRef]
- Abdou, E.; Hazell, A.S. Thiamine deficiency: An update of pathophysiologic mechanisms and future therapeutic considerations. Neurochem. Res. 2015, 40, 353–361. [Google Scholar] [CrossRef]
- Wesół-Kucharska, D.; Greczan, M.; Kaczor, M.; Pajdowska, M.; Piekutowska-Abramczuk, D.; Ciara, E.; Halat-Wolska, P.; Kowalski, P.; Jurkiewicz, E.; Rokicki, D. Early treatment of biotin-thiamine-responsive basal ganglia disease improves the prognosis. Mol. Genet. Metab. Rep. 2021, 29, 100801. [Google Scholar] [CrossRef]
- Hazell, A.S. Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem. Int. 2009, 55, 129–135. [Google Scholar] [CrossRef]
- Yu, Q.; Liu, H.; Sang, S.; Chen, L.; Zhao, Y.; Wang, Y.; Zhong, C. Thiamine deficiency contributes to synapse and neural circuit defects. Biol. Res. 2018, 51, 35. [Google Scholar] [CrossRef]
- Jhala, S.S.; Hazell, A.S. Modeling neurodegenerative disease pathophysiology in thiamine deficiency: Consequences of impaired oxidative metabolism. Neurochem. Int. 2011, 58, 248–260. [Google Scholar] [CrossRef]
- Ding, Y.; Li, J.; Enterina, J.R.; Shen, Y.; Zhang, I.; Tewson, P.H.; Mo, G.C.H.; Zhang, J.; Quinn, A.M.; Hughes, T.E.; et al. Ratiometric biosensors based on dimerization-dependent fluorescent protein exchange. Nat. Methods 2015, 12, 195–198. [Google Scholar] [CrossRef]
- Mitchell, A.C.; Alford, S.C.; Hunter, S.A.; Kannan, D.; Parra Sperberg, R.A.; Chang, C.H.; Cochran, J.R. Development of a Protease Biosensor Based on a Dimerization-Dependent Red Fluorescent Protein. ACS Chem. Biol. 2018, 13, 66–72. [Google Scholar] [CrossRef]
- Hertel, F.; Li, S.; Chen, M.; Pott, L.; Mehta, S.; Zhang, J. Fluorescent Biosensors for Multiplexed Imaging of Phosphoinositide Dynamics. ACS Chem. Biol. 2020, 15, 33–38. [Google Scholar] [CrossRef]
- McCrea, H.J.; Camilli, P.D. Mutations in Phosphoinositide Metabolizing Enzymes and Human Disease. Physiology 2009, 24, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Voronov, S.V.; Frere, S.G.; Giovedi, S.; Pollina, E.A.; Borel, C.; Zhang, H.; Schmidt, C.; Akeson, E.C.; Wenk, M.R.; Cimasoni, L.; et al. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 9415–9420. [Google Scholar] [CrossRef] [PubMed]
- Tebo, A.G.; Gautier, A. A split fluorescent reporter with rapid and reversible complementation. Nat. Commun. 2019, 10, 2822. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.-J.; Lee, H.-J.; Lee, S.-J. Cell Models to Study Cell-to-Cell Transmission of α-Synuclein. Methods Mol. Biol. 2016, 1345, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; AlOkda, A.; Jackson, M.P.; Riguet, N.; Duce, J.A.; Lashuel, H.A. Monitoring alpha-synuclein oligomerization and aggregation using bimolecular fluorescence complementation assays: What you see is not always what you get. J. Neurochem. 2021, 157, 872–888. [Google Scholar] [CrossRef]
- Kostyuk, A.I.; Demidovich, A.D.; Kotova, D.A.; Belousov, V.V.; Bilan, D.S. Circularly Permuted Fluorescent Protein-Based Indicators: History, Principles, and Classification. Int. J. Mol. Sci. 2019, 20, 4200. [Google Scholar] [CrossRef]
- Pédelacq, J.D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef]
- Marvin, J.S.; Scholl, B.; Wilson, D.E.; Podgorski, K.; Kazemipour, A.; Müller, J.A.; Schoch, S.; Quiroz, F.J.U.; Rebola, N.; Bao, H.; et al. Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat. Methods 2018, 15, 936–939. [Google Scholar] [CrossRef]
- Leopold, A.V.; Shcherbakova, D.M.; Verkhusha, V.V. Fluorescent Biosensors for Neurotransmission and Neuromodulation: Engineering and Applications. Front. Cell. Neurosci. 2019, 13, 474. [Google Scholar] [CrossRef]
- Shen, Y.; Dana, H.; Abdelfattah, A.S.; Patel, R.; Shea, J.; Molina, R.S.; Rawal, B.; Rancic, V.; Chang, Y.-F.; Wu, L.; et al. A genetically encoded Ca2+ indicator based on circularly permutated sea anemone red fluorescent protein eqFP578. BMC Biol. 2018, 16, 9. [Google Scholar] [CrossRef]
- Czapski, G.A.; Strosznajder, J.B. Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11677. [Google Scholar] [CrossRef]
- Chiodi, V.; Uchigashima, M.; Beggiato, S.; Ferrante, A.; Armida, M.; Martire, A.; Potenza, R.L.; Ferraro, L.; Tanganelli, S.; Watanabe, M.; et al. Unbalance of CB1 receptors expressed in GABAergic and glutamatergic neurons in a transgenic mouse model of Huntington’s disease. Neurobiol. Dis. 2012, 45, 983–991. [Google Scholar] [CrossRef]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Arena, J.D.; Smith, D.H. Traumatic Brain Injury as a Trigger of Neurodegeneration. Adv. Neurobiol. 2017, 15, 383–400. [Google Scholar] [CrossRef]
- Terskikh, A.; Fradkov, A.; Ermakova, G.; Zaraisky, A.; Tan, P.; Kajava, A.V.; Zhao, X.; Lukyanov, S.; Matz, M.; Kim, S.; et al. “Fluorescent timer”: Protein that changes color with time. Science 2000, 290, 1585–1588. [Google Scholar] [CrossRef]
- Yarbrough, D.; Wachter, R.M.; Kallio, K.; Matz, M.V.; Remington, S.J. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-A resolution. Proc. Natl. Acad. Sci. USA 2001, 98, 462–467. [Google Scholar] [CrossRef]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.-J. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016, 1637, 34–55. [Google Scholar] [CrossRef]
- Krzystek, T.J.; Banerjee, R.; Thurston, L.; Huang, J.; Swinter, K.; Rahman, S.N.; Falzone, T.L.; Gunawardena, S. Differential mitochondrial roles for α-synuclein in DRP1-dependent fission and PINK1/Parkin-mediated oxidation. Cell Death Dis. 2021, 12, 796. [Google Scholar] [CrossRef]
- Mahon, M.J. pHluorin2: An enhanced, ratiometric, pH-sensitive green florescent protein. Adv. Biosci. Biotechnol. 2011, 2, 132–137. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Bagh, M.B.; Peng, S.; Chandra, G.; Zhang, Z.; Singh, S.P.; Pattabiraman, N.; Liu, A.; Mukherjee, A.B. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat. Commun. 2017, 8, 14612. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef]
- Bourdenx, M.; Daniel, J.; Genin, E.; Soria, F.N.; Blanchard-Desce, M.; Bezard, E.; Dehay, B. Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases. Autophagy 2016, 12, 472–483. [Google Scholar] [CrossRef]
- Liu, A.; Huang, X.; He, W.; Xue, F.; Yang, Y.; Liu, J.; Chen, L.; Yuan, L.; Xu, P. pHmScarlet is a pH-sensitive red fluorescent protein to monitor exocytosis docking and fusion steps. Nat. Commun. 2021, 12, 1413. [Google Scholar] [CrossRef]
- Zha, X.-M. Acid-sensing ion channels: Trafficking and synaptic function. Mol. Brain 2013, 6, 1. [Google Scholar] [CrossRef]
- Mango, D.; Nisticò, R. Neurodegenerative Disease: What Potential Therapeutic Role of Acid-Sensing Ion Channels? Front. Cell. Neurosci. 2021, 15, 730641. [Google Scholar] [CrossRef]
- Decker, Y.; Németh, E.; Schomburg, R.; Chemla, A.; Fülöp, L.; Menger, M.D.; Liu, Y.; Fassbender, K. Decreased pH in the aging brain and Alzheimer’s disease. Neurobiol. Aging 2021, 101, 40–49. [Google Scholar] [CrossRef]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar] [CrossRef]
- Wiedenmann, J.; Ivanchenko, S.; Oswald, F.; Schmitt, F.; Röcker, C.; Salih, A.; Spindler, K.-D.; Nienhaus, G.U. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15905–15910. [Google Scholar] [CrossRef]
- Ando, R.; Mizuno, H.; Miyawaki, A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 2004, 306, 1370–1373. [Google Scholar] [CrossRef]
- Heinrich, R.; Hussein, W.; Berlin, S. Photo-transformable genetically-encoded optical probes for functional highlighting in vivo. J. Neurosci. Methods 2021, 355, 109129. [Google Scholar] [CrossRef]
- Ruta, V.; Datta, S.R.; Vasconcelos, M.L.; Freeland, J.; Looger, L.L.; Axel, R. A dimorphic pheromone circuit in Drosophila from sensory input to descending output. Nature 2010, 468, 686–690. [Google Scholar] [CrossRef]
- Chen, Y.; MacDonald, P.J.; Skinner, J.P.; Patterson, G.H.; Müller, J.D. Probing nucleocytoplasmic transport by two-photon activation of PA-GFP. Microsc. Res. Tech. 2006, 69, 220–226. [Google Scholar] [CrossRef]
- Lovy, A.; Molina, A.J.A.; Cerqueira, F.M.; Trudeau, K.; Shirihai, O.S. A faster, high resolution, mtPA-GFP-based mitochondrial fusion assay acquiring kinetic data of multiple cells in parallel using confocal microscopy. J. Vis. Exp. 2012, e3991. [Google Scholar] [CrossRef]
- Yao, J.; Kaberniuk, A.A.; Li, L.; Shcherbakova, D.M.; Zhang, R.; Wang, L.; Li, G.; Verkhusha, V.V.; Wang, L.V. Multiscale photoacoustic tomography using reversibly switchable bacterial phytochrome as a near-infrared photochromic probe. Nat. Methods 2016, 13, 67–73. [Google Scholar] [CrossRef]
- Adam, Y.; Kim, J.J.; Lou, S.; Zhao, Y.; Xie, M.E.; Brinks, D.; Wu, H.; Mostajo-Radji, M.A.; Kheifets, S.; Parot, V.; et al. Voltage imaging and optogenetics reveal behaviour-dependent changes in hippocampal dynamics. Nature 2019, 569, 413–417. [Google Scholar] [CrossRef]
- Zhang, C.; Quan, R.; Wang, J. Development and application of CRISPR/Cas9 technologies in genomic editing. Hum. Mol. Genet. 2018, 27, R79–R88. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chear, S.; Wing, K.; Stellon, D.; Nguyen Tran, M.T.; Talbot, J.; Pébay, A.; Hewitt, A.W.; Cook, A.L. Use of CRISPR/Cas ribonucleoproteins for high throughput gene editing of induced pluripotent stem cells. Methods 2021, 194, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Ahmed, M.; Ahmad, S.; Sahoo, C.R.; Iqubal, M.K.; Haque, S.E. Environmental neurotoxic pollutants: Review. Environ. Sci. Pollut. Res. Int. 2020, 27, 41175–41198. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.Y.; Wang, P.; Sun, Y.J.; Jiang, L.; Xu, M.Y.; Wu, Y.J. Autophagy in Tri-o-cresyl Phosphate-Induced Delayed Neurotoxicity. J. Neuropathol. Exp. Neurol. 2017, 76, 52–60. [Google Scholar] [CrossRef]
- Andrew, A.; Zhou, J.; Gui, J.; Harrison, A.; Shi, X.; Li, M.; Guetti, B.; Nathan, R.; Tischbein, M.; Pioro, E.P.; et al. Pesticides applied to crops and amyotrophic lateral sclerosis risk in the U.S. Neurotoxicology 2021, 87, 128–135. [Google Scholar] [CrossRef]
- Konno, N.; Katoh, K.; Yamauchi, T.; Fukushima, M. Delayed neurotoxicity of triphenyl phosphite in hens: Pharmacokinetic and biochemical studies. Toxicol. Appl. Pharmacol. 1989, 100, 440–450. [Google Scholar] [CrossRef]
- Burke, R.D.; Todd, S.W.; Lumsden, E.; Mullins, R.J.; Mamczarz, J.; Fawcett, W.P.; Gullapalli, R.P.; Randall, W.R.; Pereira, E.F.R.; Albuquerque, E.X. Developmental neurotoxicity of the organophosphorus insecticide chlorpyrifos: From clinical findings to preclinical models and potential mechanisms. J. Neurochem. 2017, 142 (Suppl. S2), 162–177. [Google Scholar] [CrossRef]
- Pannu, A.K.; Bhalla, A.; Vishnu, R.I.; Dhibar, D.P.; Sharma, N.; Vijayvergiya, R. Organophosphate induced delayed neuropathy after an acute cholinergic crisis in self-poisoning. Clin. Toxicol. 2021, 59, 488–492. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Wallace Hayes, A.; Najafi, N.; Karimi, G. The protective effect of natural compounds against rotenone-induced neurotoxicity. J. Biochem. Mol. Toxicol. 2020, 34, e22605. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.X.; Chen, A.D.; Wang, Q.J.; Xin, Y.Y.; Yin, J.; Jing, Y.H. Protective effect of metformin against rotenone-induced parkinsonism in mice. Toxicol. Mech. Methods 2020, 30, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236. [Google Scholar] [CrossRef]
- Richardson, J.R.; Fitsanakis, V.; Westerink, R.H.S.; Kanthasamy, A.G. Neurotoxicity of pesticides. Acta Neuropathol. 2019, 138, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Ruan, Z.; Zhang, D.; Liu, X.; Hou, L.; Wang, Q. Rotenone impairs learning and memory in mice through microglia-mediated blood brain barrier disruption and neuronal apoptosis. Chemosphere 2022, 291, 132982. [Google Scholar] [CrossRef]
- Ruan, Z.; Zhang, D.; Huang, R.; Sun, W.; Hou, L.; Zhao, J.; Wang, Q. Microglial Activation Damages Dopaminergic Neurons through MMP-2/-9-Mediated Increase of Blood-Brain Barrier Permeability in a Parkinson’s Disease Mouse Model. Int. J. Mol. Sci. 2022, 23, 2793. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Bruna, J.; Marmiroli, P.; Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit. Rev. Oncol. /Hematol. 2012, 82, 51–77. [Google Scholar] [CrossRef]
- Calls, A.; Torres-Espin, A.; Navarro, X.; Yuste, V.J.; Udina, E.; Bruna, J. Cisplatin-induced peripheral neuropathy is associated with neuronal senescence-like response. Neuro-oncology 2021, 23, 88–99. [Google Scholar] [CrossRef]
- Klein, I.; Lehmann, H.C. Pathomechanisms of Paclitaxel-Induced Peripheral Neuropathy. Toxics 2021, 9, 229. [Google Scholar] [CrossRef]
- Zajączkowska, R.; Kocot-Kępska, M.; Leppert, W.; Wrzosek, A.; Mika, J.; Wordliczek, J. Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. Int. J. Mol. Sci. 2019, 20, 1451. [Google Scholar] [CrossRef]
- Brinkmann, V.; Fritz, G. Prevention of anticancer therapy-induced neurotoxicity: Putting DNA damage in perspective. Neurotoxicology 2022, 91, 1–10. [Google Scholar] [CrossRef]
- Nielsen, S.W.; Hasselsteen, S.D.; Dominiak, H.S.H.; Labudovic, D.; Reiter, L.; Dalton, S.O.; Herrstedt, J. Oral cannabidiol for prevention of acute and transient chemotherapy-induced peripheral neuropathy. Support. Care Cancer Off. J. Multinatl. Assoc. Support. Care Cancer 2022, 30, 9441–9451. [Google Scholar] [CrossRef]
- Huehnchen, P.; Bangemann, N.; Lischewski, S.; Märschenz, S.; Paul, F.; Schmitz-Hübsch, T.; Blohmer, J.U.; Eberhardt, C.; Rauch, G.; Flöel, A.; et al. Rationale and design of the prevention of paclitaxel-related neurological side effects with lithium trial—Protocol of a multicenter, randomized, double-blind, placebo- controlled proof-of-concept phase-2 clinical trial. Front. Med. 2022, 9, 967964. [Google Scholar] [CrossRef]
- Haji Gholami, A.; Ansari, H.; Fardani, F. Investigating the Effect of Zinc on the Prevention of Acute Peripheral Neuropathy in Cancer Patients Treated with Taxanes. Adv. Biomed. Res. 2022, 11, 61. [Google Scholar] [CrossRef]
- Gérard, H.C.; Dreses-Werringloer, U.; Wildt, K.S.; Deka, S.; Oszust, C.; Balin, B.J.; Frey, W.H., 2nd; Bordayo, E.Z.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol. Med. Microbiol. 2006, 48, 355–366. [Google Scholar] [CrossRef]
- Gérard, H.C.; Wildt, K.L.; Whittum-Hudson, J.A.; Lai, Z.; Ager, J.; Hudson, A.P. The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb. Pathog. 2005, 39, 19–26. [Google Scholar] [CrossRef]
- Sochocka, M.; Zwolińska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef]
- Caputi, V.; Giron, M.C. Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 1689. [Google Scholar] [CrossRef]
- Vigasova, D.; Nemergut, M.; Liskova, B.; Damborsky, J. Multi-pathogen infections and Alzheimer’s disease. Microb. Cell Fact. 2021, 20, 25. [Google Scholar] [CrossRef]
- Alonso, R.; Pisa, D.; Marina, A.I.; Morato, E.; Rábano, A.; Carrasco, L. Fungal infection in patients with Alzheimer’s disease. J. Alzheimers. Dis. 2014, 41, 301–311. [Google Scholar] [CrossRef]
- Bando, H.; Fukuda, Y.; Watanabe, N.; Olawale, J.T.; Kato, K. Depletion of Intracellular Glutamine Pools Triggers Toxoplasma gondii Stage Conversion in Human Glutamatergic Neurons. Front. Cell. Infect. Microbiol. 2021, 11, 788303. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.; Robinson, S.-A.; Kim, D.-G.; Yan, A.; Cleland, T.A.; Bynoe, M.S. Toxoplasma gondii alters NMDAR signaling and induces signs of Alzheimer’s disease in wild-type, C57BL/6 mice. J. Neuroinflamm. 2018, 15, 57. [Google Scholar] [CrossRef] [PubMed]
- David, C.N.; Frias, E.S.; Szu, J.I.; Vieira, P.A.; Hubbard, J.A.; Lovelace, J.; Michael, M.; Worth, D.; McGovern, K.E.; Ethell, I.M.; et al. GLT-1-Dependent Disruption of CNS Glutamate Homeostasis and Neuronal Function by the Protozoan Parasite Toxoplasma gondii. PLoS Pathog. 2016, 12, e1005643. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, J.; Zhao, J.; Wen, H.; Pan, J.; Zhang, S.; Fang, Y.; Li, X.; Cai, Y.; Wang, X.; et al. Autophagy activated by Toxoplasma gondii infection in turn facilitates Toxoplasma gondii proliferation. Parasitol. Res. 2014, 113, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- Dincel, G.C.; Atmaca, H.T. Role of oxidative stress in the pathophysiology of Toxoplasma gondii infection. Int. J. Immunopathol. Pharmacol. 2016, 29, 226–240. [Google Scholar] [CrossRef]
- Shin, D.-W.; Cha, D.-Y.; Hua, Q.J.; Cha, G.-H.; Lee, Y.-H. Seroprevalence of Toxoplasma gondii infection and characteristics of seropositive patients in general hospitals in Daejeon, Korea. Korean J. Parasitol. 2009, 47, 125–130. [Google Scholar] [CrossRef]
- Xiao, Y.; Yin, J.; Jiang, N.; Xiang, M.; Hao, L.; Lu, H.; Sang, H.; Liu, X.; Xu, H.; Ankarklev, J.; et al. Seroepidemiology of human Toxoplasma gondii infection in China. BMC Infect. Dis. 2010, 10, 4. [Google Scholar] [CrossRef]
- Kamani, J.; Mani, A.U.; Egwu, G.O.; Kumshe, H.A. Seroprevalence of human infection with Toxoplasma gondii and the associated risk factors, in Maiduguri, Borno state, Nigeria. Ann. Trop. Med. Parasitol. 2009, 103, 317–321. [Google Scholar] [CrossRef]
- Swai, E.S.; Schoonman, L. Seroprevalence of Toxoplasma gondii infection amongst residents of Tanga district in north-east Tanzania. Tanzan. J. Health Res. 2009, 11, 205–209. [Google Scholar] [CrossRef]
- Molan, A.; Nosaka, K.; Hunter, M.; Wang, W. Global status of Toxoplasma gondii infection: Systematic review and prevalence snapshots. Trop. Biomed. 2019, 36, 898–925. [Google Scholar]
- Buratti, F.M.; Manganelli, M.; Vichi, S.; Stefanelli, M.; Scardala, S.; Testai, E.; Funari, E. Cyanotoxins: Producing organisms, occurrence, toxicity, mechanism of action and human health toxicological risk evaluation. Arch. Toxicol. 2017, 91, 1049–1130. [Google Scholar] [CrossRef]
- Bradley, W.G.; Andrew, A.S.; Traynor, B.J.; Chio, A.; Butt, T.H.; Stommel, E.W. Gene-Environment-Time Interactions in Neurodegenerative Diseases: Hypotheses and Research Approaches. Ann. Neurosci. 2018, 25, 261–267. [Google Scholar] [CrossRef]
- Martin, R.M.; Bereman, M.S.; Marsden, K.C. The Cyanotoxin 2,4-DAB Reduces Viability and Causes Behavioral and Molecular Dysfunctions Associated with Neurodegeneration in Larval Zebrafish. Neurotox. Res. 2022, 40, 347–364. [Google Scholar] [CrossRef]
- Main, B.J.; Rodgers, K.J. Assessing the Combined Toxicity of BMAA and Its Isomers 2,4-DAB and AEG In Vitro Using Human Neuroblastoma Cells. Neurotox. Res. 2018, 33, 33–42. [Google Scholar] [CrossRef]
- Ra, D.; Sa, B.; Sl, B.; Js, M.; Sj, M.; Da, D.; Ew, S.; Karlsson, O.; Eb, B.; Ad, C.; et al. Is Exposure to BMAA a Risk Factor for Neurodegenerative Diseases? A Response to a Critical Review of the BMAA Hypothesis. Neurotox. Res. 2021, 39, 81–106. [Google Scholar] [CrossRef]
- Rao, S.D.; Banack, S.A.; Cox, P.A.; Weiss, J.H. BMAA selectively injures motor neurons via AMPA/kainate receptor activation. Exp. Neurol. 2006, 201, 244–252. [Google Scholar] [CrossRef]
- Dunlop, R.A.; Guillemin, G.J. The Cyanotoxin and Non-protein Amino Acid β-Methylamino-L-Alanine (L-BMAA) in the Food Chain: Incorporation into Proteins and Its Impact on Human Health. Neurotox. Res. 2019, 36, 602–611. [Google Scholar] [CrossRef]
- Xie, X.; Basile, M.; Mash, D.C. Cerebral uptake and protein incorporation of cyanobacterial toxin β-N-methylamino-L-alanine. Neuroreport 2013, 24, 779–784. [Google Scholar] [CrossRef]
- Shematorova, E.K.; Shpakovski, G.V. Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease. Int. J. Mol. Sci. 2020, 21, 8055. [Google Scholar] [CrossRef]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Schmid, B.; Holst, B.; Clausen, C.; Bahnassawy, L.; Reinhardt, P.; Bakker, M.H.M.; Díaz-Guerra, E.; Vicario, C.; Martino-Adami, P.V.; Thoenes, M.; et al. Generation of a set of isogenic iPSC lines carrying all APOE genetic variants (ε2/ε3/ε4) and knock-out for the study of APOE biology in health and disease. Stem Cell Res. 2021, 52, 102180. [Google Scholar] [CrossRef] [PubMed]
- Weykopf, B.; Haupt, S.; Jungverdorben, J.; Flitsch, L.J.; Hebisch, M.; Liu, G.H.; Suzuki, K.; Belmonte, J.C.I.; Peitz, M.; Blaess, S.; et al. Induced pluripotent stem cell-based modeling of mutant LRRK2-associated Parkinson’s disease. Eur. J. Neurosci. 2019, 49, 561–589. [Google Scholar] [CrossRef]
- Pantazis, C.B.; Yang, A.; Lara, E.; McDonough, J.A.; Blauwendraat, C.; Peng, L.; Oguro, H.; Kanaujiya, J.; Zou, J.; Sebesta, D.; et al. A reference human induced pluripotent stem cell line for large-scale collaborative studies. Cell Stem Cell 2022, 29, 1685–1702.e1622. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.M.; Skarnes, W.C.; Singleton, A.B.; Cookson, M.R.; Ward, M.E. Tackling neurodegenerative diseases with genomic engineering: A new stem cell initiative from the NIH. Neuron 2021, 109, 1080–1083. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Bjerke, M.; Hens, E.; Sieben, A.; Timmers, M.; De Roeck, A.; Vandenberghe, R.; Sleegers, K.; Martin, J.J.; De Deyn, P.P.; et al. Amyloid-β(1-43) cerebrospinal fluid levels and the interpretation of APP, PSEN1 and PSEN2 mutations. Alzheimer’s Res. Ther. 2020, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Cacace, R.; van der Zee, J.; Van Broeckhoven, C. Emerging genetic complexity and rare genetic variants in neurodegenerative brain diseases. Genome Med. 2021, 13, 59. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.; Sauerbeck, A.D.; St-Pierre, M.K.; Xiong, M.; Kim, N.; Serrano, J.R.; Tremblay, M.; Kummer, T.T.; Colonna, M.; et al. Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Investig. 2020, 130, 4954–4968. [Google Scholar] [CrossRef]
- de Waard, D.M.; Bugiani, M. Astrocyte-Oligodendrocyte-Microglia Crosstalk in Astrocytopathies. Front. Cell. Neurosci. 2020, 14, 608073. [Google Scholar] [CrossRef]
- Bugiani, M.; Vuong, C.; Breur, M.; van der Knaap, M.S. Vanishing white matter: A leukodystrophy due to astrocytic dysfunction. Brain Pathol. 2018, 28, 408–421. [Google Scholar] [CrossRef]
- Saá, P.; Harris, D.A.; Cervenakova, L. Mechanisms of prion-induced neurodegeneration. Expert Rev. Mol. Med. 2016, 18, e5. [Google Scholar] [CrossRef]
- Kousi, M.; Lehesjoki, A.E.; Mole, S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef]
- Tang, C.; Han, J.; Dalvi, S.; Manian, K.; Winschel, L.; Volland, S.; Soto, C.A.; Galloway, C.A.; Spencer, W.; Roll, M.; et al. A human model of Batten disease shows role of CLN3 in phagocytosis at the photoreceptor-RPE interface. Commun. Biol. 2021, 4, 161. [Google Scholar] [CrossRef]
- Parviainen, L.; Dihanich, S.; Anderson, G.W.; Wong, A.M.; Brooks, H.R.; Abeti, R.; Rezaie, P.; Lalli, G.; Pope, S.; Heales, S.J.; et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol. Commun. 2017, 5, 74. [Google Scholar] [CrossRef]
- Chear, S.; Perry, S.; Wilson, R.; Bindoff, A.; Talbot, J.; Ware, T.L.; Grubman, A.; Vickers, J.C.; Pébay, A.; Ruddle, J.B.; et al. Lysosomal alterations and decreased electrophysiological activity in CLN3 disease patient-derived cortical neurons. Dis. Model. Mech. 2022, 15, dmm049651. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Mohammad, D.; Trépanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef]
- Sawada, M.; Imamura, K.; Nagatsu, T. Role of cytokines in inflammatory process in Parkinson’s disease. J. Neural Transm. Suppl. 2006, 373–381. [Google Scholar] [CrossRef]
- Brettschneider, J.; Libon, D.J.; Toledo, J.B.; Xie, S.X.; McCluskey, L.; Elman, L.; Geser, F.; Lee, V.M.Y.; Grossman, M.; Trojanowski, J.Q. Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 123, 395–407. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Gordon, R.; Singh, N.; Lawana, V.; Ghosh, A.; Harischandra, D.S.; Jin, H.; Hogan, C.; Sarkar, S.; Rokad, D.; Panicker, N.; et al. Protein kinase Cδ upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson’s disease. Neurobiol. Dis. 2016, 93, 96–114. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-J.; Hwang, Y.G.; Sharma, N.; Tran, H.-Q.; Dang, D.-K.; Jang, C.-G.; Jeong, J.H.; Nah, S.-Y.; Nabeshima, T.; Kim, H.-C. Role of protein kinase Cδ in dopaminergic neurotoxic events. Food Chem. Toxicol. 2018, 121, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, H.J.; Kim, D.K.; Lee, S.J. Non-cell-autonomous actions of α-synuclein: Implications in glial synucleinopathies. Prog. Neurobiol. 2018, 169, 158–171. [Google Scholar] [CrossRef]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambarè, D.; Bellini, E.; Ordazzo, G.; et al. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting Microglial α-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef]
- Chen, L.; Ding, Y.; Cagniard, B.; Van Laar, A.D.; Mortimer, A.; Chi, W.; Hastings, T.G.; Kang, U.J.; Zhuang, X. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 2008, 28, 425–433. [Google Scholar] [CrossRef]
- Mattson, M.P. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N. Y. Acad. Sci. USA 2008, 1144, 97–112. [Google Scholar] [CrossRef]
- Wilson, D.A.; Linster, C. Neurobiology of a simple memory. J. Neurophysiol. 2008, 100, 2–7. [Google Scholar] [CrossRef]
- Chantranupong, L.; Sabatini, B.L. Sunlight Brightens Learning and Memory. Cell 2018, 173, 1570–1572. [Google Scholar] [CrossRef]
- Santos, M.D.; Mohammadi, M.H.; Yang, S.; Liang, C.W.; Kao, J.P.Y.; Alger, B.E.; Thompson, S.M.; Tang, C.-M. Dendritic hold and read: A gated mechanism for short term information storage and retrieval. PLoS ONE 2012, 7, e37542. [Google Scholar] [CrossRef]
- Maragos, W.F.; Greenamyre, J.T.; Penney, J.B.; Young, A.B. Glutamate dysfunction in Alzheimer’s disease: An hypothesis. Trends Neurosci. 1987, 10, 65–68. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Stanika, R.I.; Pivovarova, N.B.; Brantner, C.A.; Watts, C.A.; Winters, C.A.; Andrews, S.B. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9854–9859. [Google Scholar] [CrossRef]
- Petralia, R.S.; Wang, Y.X.; Hua, F.; Yi, Z.; Zhou, A.; Ge, L.; Stephenson, F.A.; Wenthold, R.J. Organization of NMDA receptors at extrasynaptic locations. Neuroscience 2010, 167, 68–87. [Google Scholar] [CrossRef]
- Jayakar, S.S.; Dikshit, M. AMPA receptor regulation mechanisms: Future target for safer neuroprotective drugs. Int. J. Neurosci. 2004, 114, 695–734. [Google Scholar] [CrossRef]
- Wu, J.; Abdelfattah, A.S.; Zhou, H.; Ruangkittisakul, A.; Qian, Y.; Ballanyi, K.; Campbell, R.E. Genetically Encoded Glutamate Indicators with Altered Color and Topology. ACS Chem. Biol. 2018, 13, 1832–1837. [Google Scholar] [CrossRef]
- Fang, Y.; Ding, X.; Zhang, Y.; Cai, L.; Ge, Y.; Ma, K.; Xu, R.; Li, S.; Song, M.; Zhu, H.; et al. Fluoxetine inhibited the activation of A1 reactive astrocyte in a mouse model of major depressive disorder through astrocytic 5-HT2BR/β-arrestin2 pathway. J. Neuroinflamm. 2022, 19, 23. [Google Scholar] [CrossRef]
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Piña-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. α-Synuclein Oligomers Induce Glutamate Release from Astrocytes and Excessive Extrasynaptic NMDAR Activity in Neurons, Thus Contributing to Synapse Loss. J. Neurosci. 2021, 41, 2264–2273. [Google Scholar] [CrossRef]
- Juźwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. MicroRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef]
- Wang, X.; Liu, D.; Huang, H.-Z.; Wang, Z.-H.; Hou, T.-Y.; Yang, X.; Pang, P.; Wei, N.; Zhou, Y.-F.; Dupras, M.-J.; et al. A Novel MicroRNA-124/PTPN1 Signal Pathway Mediates Synaptic and Memory Deficits in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 395–405. [Google Scholar] [CrossRef]
- Valera, E.; Spencer, B.; Mott, J.; Trejo, M.; Adame, A.; Mante, M.; Rockenstein, E.; Troncoso, J.C.; Beach, T.G.; Masliah, E.; et al. MicroRNA-101 Modulates Autophagy and Oligodendroglial Alpha-Synuclein Accumulation in Multiple System Atrophy. Front. Mol. Neurosci. 2017, 10, 329. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Gong, F.; Ge, X.; Lv, C.; Huang, C.; Feng, S.; Zhou, Z.; Rong, Y.; Wang, J.; Ji, C.; et al. Neuron-derived exosomes-transmitted miR-124-3p protect traumatically injured spinal cord by suppressing the activation of neurotoxic microglia and astrocytes. J. Nanobiotechnol. 2020, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Poggini, S.; Garofalo, S.; Milior, G.; El Hajj, H.; Lecours, C.; Girard, I.; Gagnon, S.; Boisjoly-Villeneuve, S.; Brunello, N.; et al. Fluoxetine treatment affects the inflammatory response and microglial function according to the quality of the living environment. Brain Behav. Immun. 2016, 58, 261–271. [Google Scholar] [CrossRef]
- Catlow, B.J.; Song, S.; Paredes, D.A.; Kirstein, C.L.; Sanchez-Ramos, J. Effects of psilocybin on hippocampal neurogenesis and extinction of trace fear conditioning. Exp. Brain Res. 2013, 228, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Castrén, E.; Kasper, S.; Lanzenberger, R. Serotonin and neuroplasticity—Links between molecular, functional and structural pathophysiology in depression. Neurosci. Biobehav. Rev. 2017, 77, 317–326. [Google Scholar] [CrossRef]
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn errors of coenzyme A metabolism and neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef]
- Di Meo, I.; Tiranti, V. Classification and molecular pathogenesis of NBIA syndromes. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2018, 22, 272–284. [Google Scholar] [CrossRef]
- Santillo, A.F.; Skoglund, L.; Lindau, M.; Eeg-Olofsson, K.E.; Tovi, M.; Engler, H.; Brundin, R.M.; Ingvast, S.; Lannfelt, L.; Glaser, A.; et al. Frontotemporal dementia-amyotrophic lateral sclerosis complex is simulated by neurodegeneration with brain iron accumulation. Alzheimer Dis. Assoc. Disord. 2009, 23, 298–300. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, Z.-H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef]
- Kim, J.; Kim, T.Y.; Hwang, J.J.; Lee, J.Y.; Shin, J.H.; Gwag, B.J.; Koh, J.Y. Accumulation of labile zinc in neurons and astrocytes in the spinal cords of G93A SOD-1 transgenic mice. Neurobiol. Dis. 2009, 34, 221–229. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Dhawan, U.; Boudeau, S.; Lei, S.; Carlomagno, Y.; Knobel, M.; Al Mohanna, L.F.A.; Boomhower, S.R.; Newland, M.C.; Sherr, D.H.; et al. Heavy Metal Neurotoxicants Induce ALS-Linked TDP-43 Pathology. Toxicol. Sci. Off. J. Soc. Toxicol. 2019, 167, 105–115. [Google Scholar] [CrossRef]
- McDade, E.; Cummings, J.L.; Dhadda, S.; Swanson, C.J.; Reyderman, L.; Kanekiyo, M.; Koyama, A.; Irizarry, M.; Kramer, L.D.; Bateman, R.J. Lecanemab in patients with early Alzheimer’s disease: Detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study. Alzheimer’s Res. Ther. 2022, 14, 191. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Ooi, L.; Dottori, M.; Cook, A.L.; Engel, M.; Gautam, V.; Grubman, A.; Hernandez, D.; King, A.E.; Maksour, S.; Targa Dias Anastacio, H.; et al. If Human Brain Organoids Are the Answer to Understanding Dementia, What Are the Questions? Neuroscientist 2020, 26, 438–454. [Google Scholar] [CrossRef]
- Armijo, E.; Gonzalez, C.; Shahnawaz, M.; Flores, A.; Davis, B.; Soto, C. Increased susceptibility to Abeta toxicity in neuronal cultures derived from familial Alzheimer’s disease (PSEN1-A246E) induced pluripotent stem cells. Neurosci. Lett. 2017, 639, 74–81. [Google Scholar] [CrossRef]
- Robbins, J.P.; Perfect, L.; Ribe, E.M.; Maresca, M.; Dangla-Valls, A.; Foster, E.M.; Killick, R.; Nowosiad, P.; Reid, M.J.; Polit, L.D.; et al. Clusterin Is Required for beta-Amyloid Toxicity in Human iPSC-Derived Neurons. Front. Neurosci. 2018, 12, 504. [Google Scholar] [CrossRef]
- Sackmann, C.; Hallbeck, M. Oligomeric amyloid-beta induces early and widespread changes to the proteome in human iPSC-derived neurons. Sci. Rep. 2020, 10, 6538. [Google Scholar] [CrossRef]
- Berry, B.J.; Smith, A.S.T.; Long, C.J.; Martin, C.C.; Hickman, J.J. Physiological Abeta Concentrations Produce a More Biomimetic Representation of the Alzheimer’s Disease Phenotype in iPSC Derived Human Neurons. ACS Chem. Neurosci. 2018, 9, 1693–1701. [Google Scholar] [CrossRef]
- Xiang, X.; Werner, G.; Bohrmann, B.; Liesz, A.; Mazaheri, F.; Capell, A.; Feederle, R.; Knuesel, I.; Kleinberger, G.; Haass, C. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol. Med. 2016, 8, 992–1004. [Google Scholar] [CrossRef]
- Ovechkina, V.S.; Zakian, S.M.; Medvedev, S.P.; Valetdinova, K.R. Genetically Encoded Fluorescent Biosensors for Biomedical Applications. Biomedicines 2021, 9, 1528. [Google Scholar] [CrossRef] [PubMed]
- Potekhina, E.S.; Bass, D.Y.; Kelmanson, I.V.; Fetisova, E.S.; Ivanenko, A.V.; Belousov, V.V.; Bilan, D.S. Drug Screening with Genetically Encoded Fluorescent Sensors: Today and Tomorrow. Int. J. Mol. Sci. 2020, 22, 148. [Google Scholar] [CrossRef] [PubMed]
- Combes, R.D.; Balls, M. The Three Rs--opportunities for improving animal welfare and the quality of scientific research. Altern. Lab. Anim. 2014, 42, 245–259. [Google Scholar] [CrossRef]
- Honarnejad, K.; Daschner, A.; Giese, A.; Zall, A.; Schmidt, B.; Szybinska, A.; Kuznicki, J.; Herms, J. Development and implementation of a high-throughput compound screening assay for targeting disrupted ER calcium homeostasis in Alzheimer’s disease. PLoS ONE 2013, 8, e80645. [Google Scholar] [CrossRef] [PubMed]
- Boivin, B.; Roet, K.C.D.; Huang, X.; Karhohs, K.W.; Rohban, M.H.; Sandoe, J.; Wiskow, O.; Maeda, R.; Grantham, A.; Dornon, M.K.; et al. A Multiparametric Activity Profiling Platform for Neuron Disease Phenotyping and Drug Screening. Mol. Biol. Cell 2021, 33, ar54. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Roet, K.C.D.; Zhang, L.; Brault, A.; Berg, A.P.; Jefferson, A.B.; Klug-McLeod, J.; Leach, K.L.; Vincent, F.; Yang, H.; et al. Human amyotrophic lateral sclerosis excitability phenotype screen: Target discovery and validation. Cell Rep. 2021, 35, 109224. [Google Scholar] [CrossRef]
- Depry, C.; Mehta, S.; Zhang, J. Multiplexed visualization of dynamic signaling networks using genetically encoded fluorescent protein-based biosensors. Pflug. Arch. 2013, 465, 373–381. [Google Scholar] [CrossRef][Green Version]
- Shcherbakova, D.M.; Stepanenko, O.V.; Turoverov, K.K.; Verkhusha, V.V. Near-Infrared Fluorescent Proteins: Multiplexing and Optogenetics across Scales. Trends Biotechnol. 2018, 36, 1230–1243. [Google Scholar] [CrossRef]
- Bhalla, R.M.; Hülsemann, M.; Verkhusha, P.V.; Walker, M.G.; Shcherbakova, D.M.; Hodgson, L. Multiplex Imaging of Rho GTPase Activities in Living Cells. Methods Mol. Biol. 2021, 2350, 43–68. [Google Scholar] [CrossRef]
- Ast, C.; De Michele, R.; Kumke, M.U.; Frommer, W.B. Single-fluorophore membrane transport activity sensors with dual-emission read-out. Elife 2015, 4, e07113. [Google Scholar] [CrossRef]
- Yang, J.-M.; Chi, W.-Y.; Liang, J.; Takayanagi, S.; Iglesias, P.A.; Huang, C.-H. Deciphering cell signaling networks with massively multiplexed biosensor barcoding. Cell 2021, 184, 6193–6206.e6114. [Google Scholar] [CrossRef]
- Cuomo, A.S.E.; Seaton, D.D.; McCarthy, D.J.; Martinez, I.; Bonder, M.J.; Garcia-Bernardo, J.; Amatya, S.; Madrigal, P.; Isaacson, A.; Buettner, F.; et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun. 2020, 11, 810. [Google Scholar] [CrossRef]
- Neavin, D.R.; Steinmann, A.M.; Chiu, H.S.; Daniszewski, M.S.; Moutinho, C.; Chan, C.-L.; Tyebally, M.; Gnanasambandapillai, V.; Lam, C.E.; Nguyen, U.; et al. Village in a dish: A model system for population-scale hiPSC studies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Senabouth, A.; Daniszewski, M.; Lidgerwood, G.E.; Liang, H.H.; Hernandez, D.; Mirzaei, M.; Keenan, S.N.; Zhang, R.; Han, X.; Neavin, D.; et al. Transcriptomic and proteomic retinal pigment epithelium signatures of age-related macular degeneration. Nat. Commun. 2022, 13, 4233. [Google Scholar] [CrossRef]
- Benmoyal-Segal, L.; Vander, T.; Shifman, S.; Bryk, B.; Ebstein, R.P.; Marcus, E.-L.; Stessman, J.; Darvasi, A.; Herishanu, Y.; Friedman, A.; et al. Acetylcholinesterase/paraoxonase interactions increase the risk of insecticide-induced Parkinson’s disease. FASEB J. 2005, 19, 452–454. [Google Scholar] [CrossRef]
- Benmoyal-Segal, L.; Soreq, H. Gene-environment interactions in sporadic Parkinson’s disease. J. Neurochem. 2006, 97, 1740–1755. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Example(s) | Advantages | Disadvantages Compared to GEFBs | Reference |
|---|---|---|---|---|
| Specialised analytical devices | Seahorse XF HS Mini Analyzer (Agilent); Multi-electrode arrays | Allows for real-time data acquisition and continuous monitoring. | Usually, samples small areas near the sensor and measurements are indirect calculations. Does not allow for visualisation of cellular compartments. | [24,25] |
| Endpoint assay | Many cell-based assay kits from various companies | Quantitative measurements, high-throughput, with convenient and economic kits available. | Does not allow for continuous high spatiotemporal visualisation in living cells or tracking at single-cell resolution. | [26,27] |
| Organic dyes | Calcium indicators (e.g., FURA-2) | Quick to use; little preparation needed. | Relatively more invasive, long-exposure can lead to accumulated toxicity. Extended excitation can lead to more photobleaching. Short-term imaging. Dye leakage significantly contributes to accuracy. Difficulty monitoring activity in specific cell types and specific subcellular compartments. | [28,29] |
| Fluorescent pH probes | HPTS, SNARF-1, Lysotracker | High spatiotemporal resolution, long-term fluorescent and structural stability. | Difficulty in penetrating the cell membrane, and targeting methods to subcellular locations can perturb the cell and affect pH in the long run. May exhibit rapid photobleaching. | [30,31] |
| Design | GEFB | Sensing | FP | Reference | Addgene Plasmid Number |
|---|---|---|---|---|---|
| Turnover and translocation of FP | GFP-LC3-RFP-LC3ΔF | Autophagy | GFP and RFP | [42] | 84572 |
| Turnover and translocation of FP | NLS-tdTomato-NES | Nucleocytoplasmic transport defects | tdTomato | [43] | 112579 |
| FRET | LSSmOrange-DEVD-mKate2 | Caspase 3 | LSSmOrange and mKate2 | [44] | 37132 |
| FRET | FLIPT | Thiamine | CFP and YFP | [45] | N.A. * |
| BiFC | Tau-BiFC | Tau–tau interaction | Venus | [46] | N.A. * |
| cpFP | GACh2.0 | Acetylcholine | cpGFP | [47] | 106073 |
| cpFP | MatryoshCaMP6s | Calcium signalling | LSSmOrange and cpEGFP | [48] | 100025 |
| cpFP | GRABDA | Dopamine | cpEGFP | [49] | 113050 and 113049 |
| cpFP | iGABASnFR | GABA | cpSFGFP | [50] | 112176 |
| cpFP | iGluSnFR | Glutamate | cpGFP | [51] | 41732 |
| cpFP | GRABNE1M | Norepinephrine | cpEGFP | [52] | 123309 and 123308 |
| cpFP | iSeroSnFR | Serotonin | cpSFGFP | [53] | 128484 |
| Oxidation-dependent | MitoTimer | Mitochondrial health | GFP and DsRed1 | [54] | 52659 |
| Ion-sensitive | RpH-LAMP1-3xFLAG | Lysosomal pH | pHlourin and mCherry | [55] | 163018 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stellon, D.; Talbot, J.; Hewitt, A.W.; King, A.E.; Cook, A.L. Seeing Neurodegeneration in a New Light Using Genetically Encoded Fluorescent Biosensors and iPSCs. Int. J. Mol. Sci. 2023, 24, 1766. https://doi.org/10.3390/ijms24021766
Stellon D, Talbot J, Hewitt AW, King AE, Cook AL. Seeing Neurodegeneration in a New Light Using Genetically Encoded Fluorescent Biosensors and iPSCs. International Journal of Molecular Sciences. 2023; 24(2):1766. https://doi.org/10.3390/ijms24021766
Chicago/Turabian StyleStellon, David, Jana Talbot, Alex W. Hewitt, Anna E. King, and Anthony L. Cook. 2023. "Seeing Neurodegeneration in a New Light Using Genetically Encoded Fluorescent Biosensors and iPSCs" International Journal of Molecular Sciences 24, no. 2: 1766. https://doi.org/10.3390/ijms24021766
APA StyleStellon, D., Talbot, J., Hewitt, A. W., King, A. E., & Cook, A. L. (2023). Seeing Neurodegeneration in a New Light Using Genetically Encoded Fluorescent Biosensors and iPSCs. International Journal of Molecular Sciences, 24(2), 1766. https://doi.org/10.3390/ijms24021766