

Characterization of Lipopolysaccharide Effects on LRRK2 Signaling in RAW Macrophages
 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. LPS Increases the Kinase Activity of LRRK2
2.2. LRRK2 Deletion Changes the Cell Morphology after LPS Stimulation
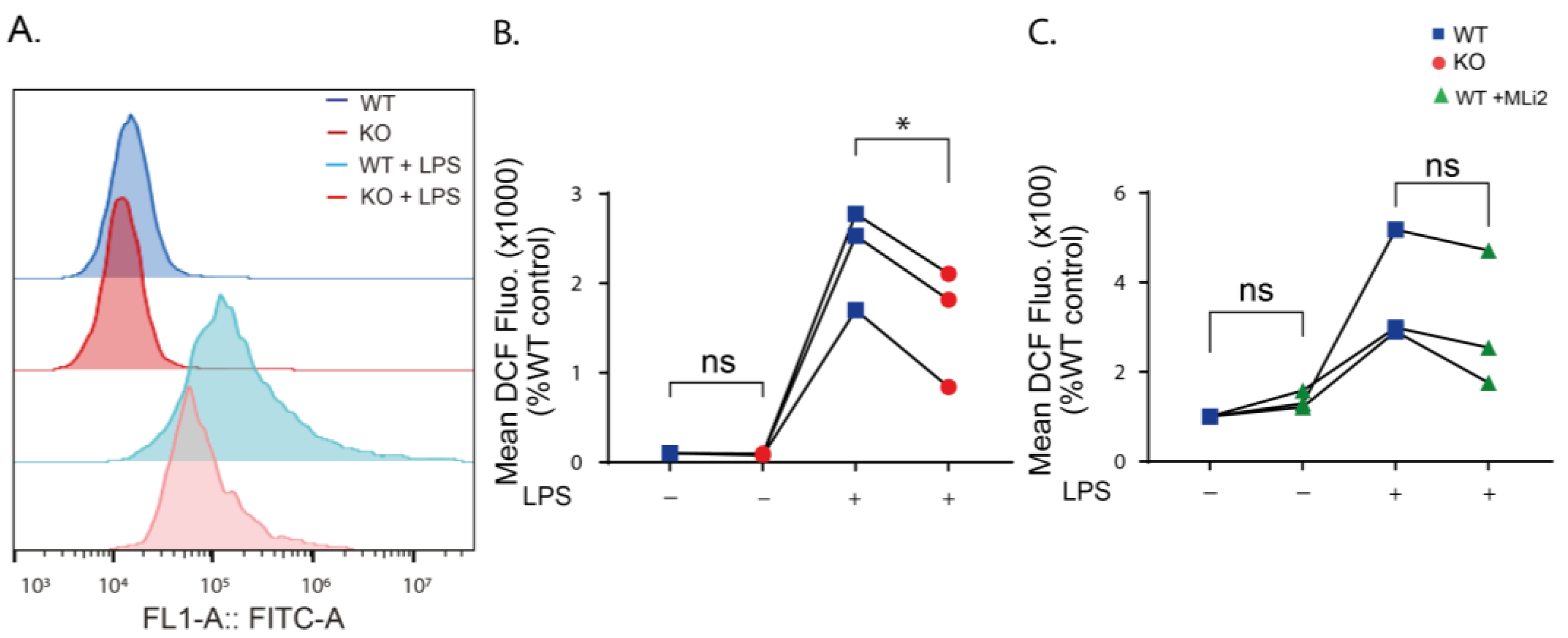
2.3. LRRK2 Mediates LPS-Stimulated ROS Production
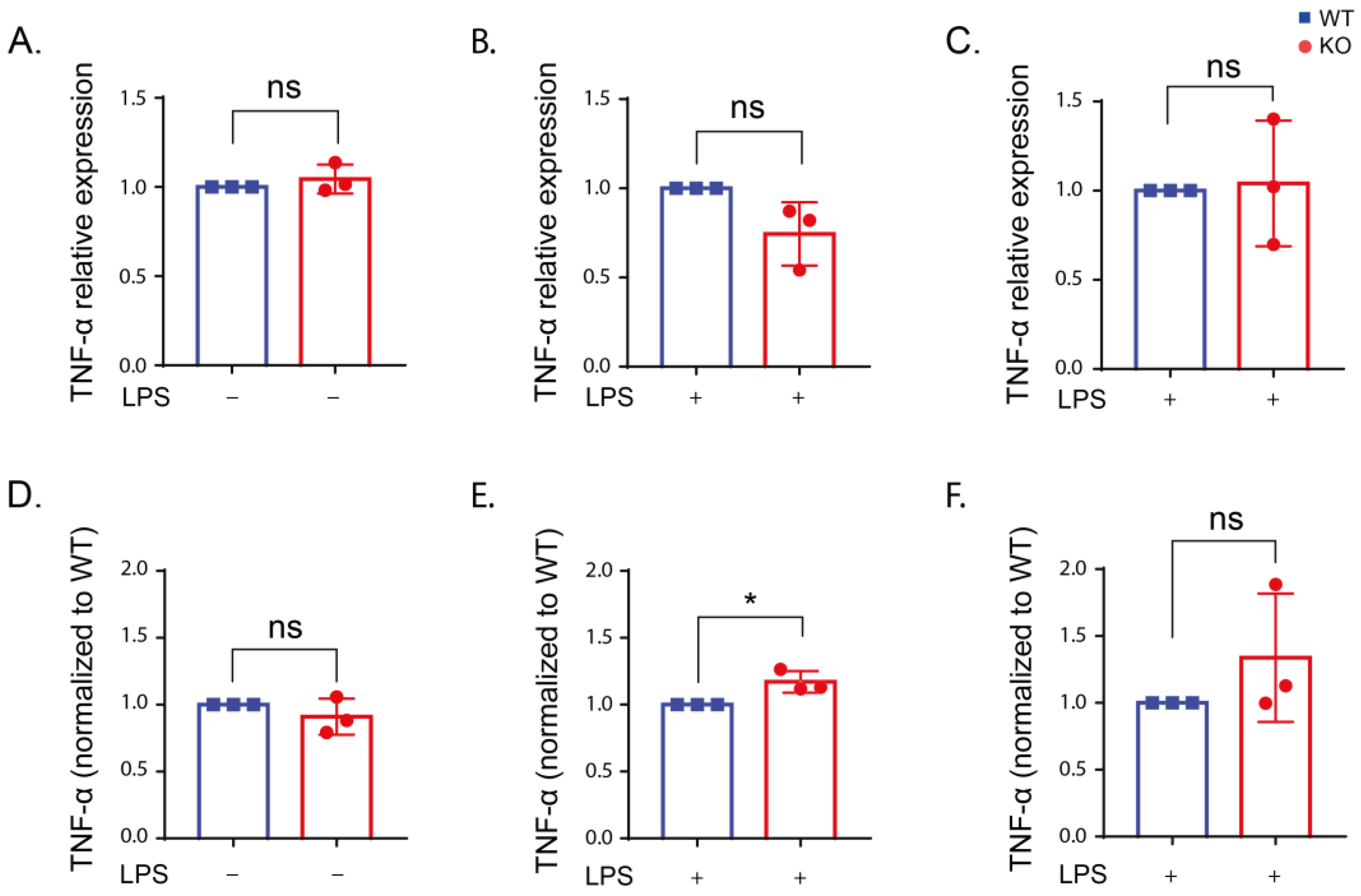
2.4. Inflammatory Cytokine Production in Stimulated RAW Macrophages
2.5. LRRK2 Deletion Reduces COX-2 Expression
2.6. LRRK2 Influences Glycolysis following LPS Stimulation
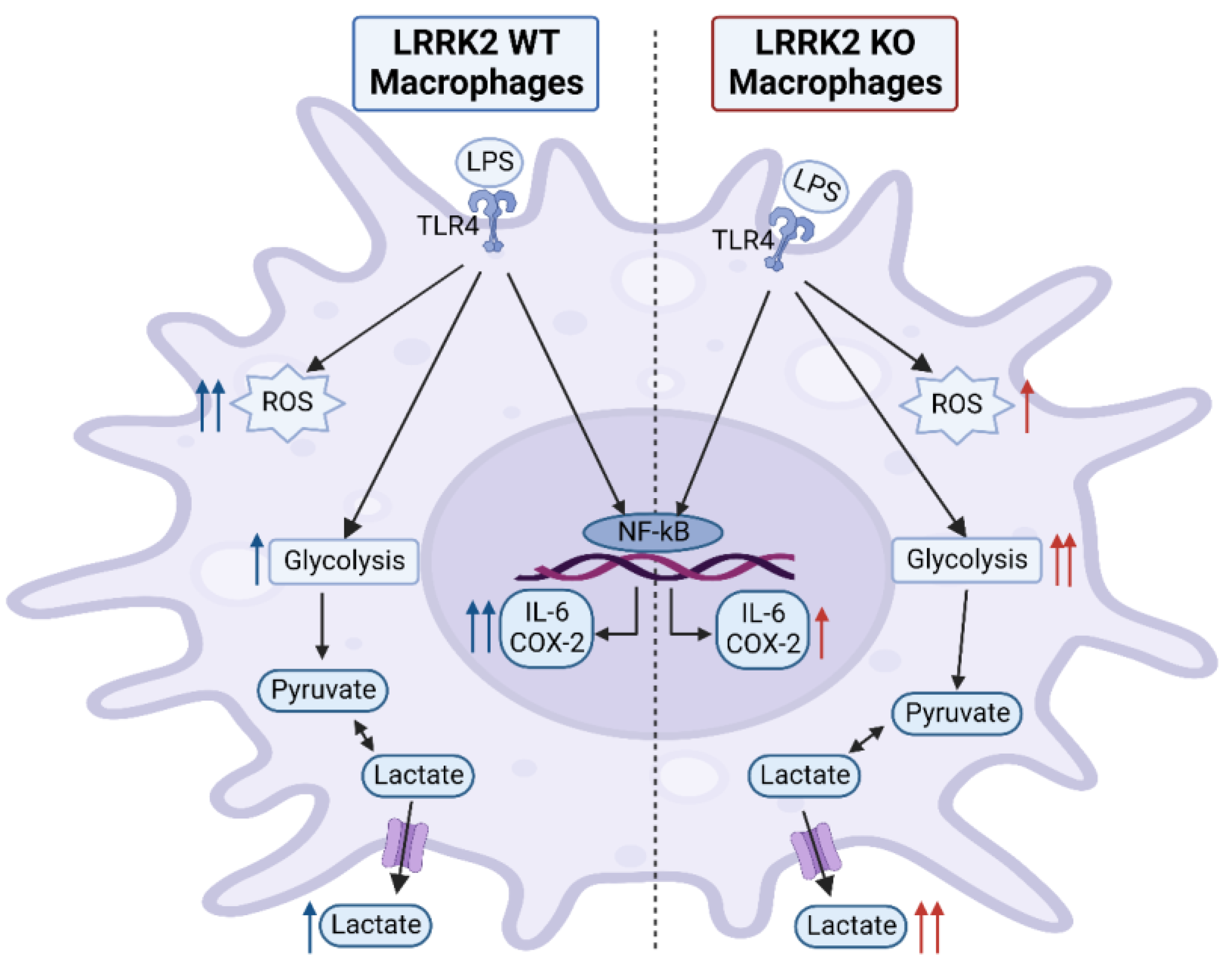
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Treatment
4.3. Western Blotting
4.4. Real-Time Cell Impedance Measurements
4.5. Flow Cytometry Measurement
4.5.1. Reactive Oxygen Species (ROS) Levels
4.5.2. Cyclooxygenase-2 (COX-2) Detection
4.6. RNA Extraction and Reverse Transcription Quantitative Real-Time PCR (RT-qPCR)
4.7. Enzyme-Linked Immunosorbent Assay (ELISA) for IL-6 and TNF-α
4.8. Lactate Measurement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxynonenal |
| ANOVA | analysis of variance |
| AUC | area under curve |
| BMDMs | bone-marrow-derived macrophage |
| COX-2 | cyclooxygenase-2 |
| DCF | dichlorodihydrofluorescein |
| DCFDA | 2′,7′-dichlorodihydrofluorescein diacetate |
| ECAR | extracellular acidification rate |
| ELISA | enzyme-linked immunosorbent assay |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| IL-1β | interleukin-1 beta |
| IL-6 | interleukin 6 |
| INF-γ | interferon-gamma |
| iPSC | induced pluripotent stem cell |
| LLOMe | L-leucyl-L-leucine methyl ester |
| LRRK2 | leucine rich-repeat kinase 2 |
| MAPK | mitogen-activated protein kinase |
| MEFs | mouse embryonic fibroblasts |
| NF-κB | nuclear factor-kappa B |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| PD | Parkinson’s disease |
| PI3Kγ | phosphatidylinositol 3-kinase γ |
| qPCR | reverse transcription quantitative real-time polymerase chain reaction |
| ROS | reactive oxygen species |
| RPL13A | ribosomal protein L13A |
| SD | standard deviation |
| SEM | standard error of the mean |
| SNpc | substantia nigra pars compacta |
| TLR4 | toll-like receptor 4 |
| TNF-α | tumor necrosis factor alpha. |
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Greffard, S.; Verny, M.; Bonnet, A.M.; Beinis, J.Y.; Gallinari, C.; Meaume, S.; Piette, F.; Hauw, J.J.; Duyckaerts, C. Motor Score of the Unified Parkinson Disease Rating Scale as a Good Predictor of Lewy Body-Associated Neuronal Loss in the Substantia Nigra. Arch. Neurol. 2006, 63, 584–588. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Reed, X.; Bandrés-Ciga, S.; Blauwendraat, C.; Cookson, M.R. The Role of Monogenic Genes in Idiopathic Parkinson’s Disease. Neurobiol. Dis. 2019, 124, 230–239. [Google Scholar] [CrossRef]
- Guadagnolo, D.; Piane, M.; Torrisi, M.R.; Pizzuti, A.; Petrucci, S. Genotype-Phenotype Correlations in Monogenic Parkinson Disease: A Review on Clinical and Molecular Findings. Front. Neurol. 2021, 12, 648588. [Google Scholar] [CrossRef]
- Marín, I. The Parkinson Disease Gene LRRK2: Evolutionary and Structural Insights. Mol. Biol. Evol. 2006, 23, 2423–2433. [Google Scholar] [CrossRef]
- Myasnikov, A.; Zhu, H.; Hixson, P.; Xie, B.; Yu, K.; Pitre, A.; Peng, J.; Sun, J. Structural Analysis of the Full-Length Human LRRK2. Cell 2021, 184, 3519–3527.e10. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Z.; Ye, T.; Mabrouk, O.S.; Maltbie, T.; Aasly, J.; West, A.B. Elevated LRRK2 Autophosphorylation in Brain-Derived and Peripheral Exosomes in LRRK2 Mutation Carriers. Acta Neuropathol. Commun. 2017, 5, 86. [Google Scholar] [CrossRef]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 Activation in Idiopathic Parkinson’s Disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef]
- Wallings, R.; Manzoni, C.; Bandopadhyay, R. Cellular Processes Associated with LRRK2 Function and Dysfunction. FEBS J. 2015, 282, 2806–2826. [Google Scholar] [CrossRef]
- Martin, I.; Kim, J.W.; Dawson, V.L.; Dawson, T.M. LRRK2 Pathobiology in Parkinson’s Disease. J. Neurochem. 2014, 131, 554–565. [Google Scholar] [CrossRef]
- Tsafaras, G.; Baekelandt, V. The Role of LRRK2 in the Periphery: Link with Parkinson’s Disease and Inflammatory Diseases. Neurobiol. Dis. 2022, 172, 105806. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s Disease and Its Potential as Therapeutic Target. Transl. Neurodegener. 2015, 4, 1–9. [Google Scholar] [CrossRef]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 Is Involved in the IFN-γ Response and Host Response to Pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef]
- Hakimi, M.; Selvanantham, T.; Swinton, E.; Padmore, R.F.; Tong, Y.; Kabbach, G.; Venderova, K.; Girardin, S.E.; Bulman, D.E.; Scherzer, C.R.; et al. Parkinson’s Disease-Linked LRRK2 Is Expressed in Circulating and Tissue Immune Cells and Upregulated Following Recognition of Microbial Structures. J. Neural Transm. 2011, 118, 795–808. [Google Scholar] [CrossRef]
- Miklossy, J.; Arai, T.; Guo, J.P.; Klegeris, A.; Yu, S.; McGeer, E.G.; McGeer, P.L. LRRK2 Expression in Normal and Pathologic Human Brain and in Human Cell Lines. J. Neuropathol. Exp. Neurol. 2006, 65, 953–963. [Google Scholar] [CrossRef]
- Atashrazm, F.; Hammond, D.; Perera, G.; Bolliger, M.F.; Matar, E.; Halliday, G.M.; Schüle, B.; Lewis, S.J.G.; Nichols, R.J.; Dzamko, N. LRRK2-Mediated Rab10 Phosphorylation in Immune Cells from Parkinson’s Disease Patients. Mov. Disord. 2019, 34, 406–415. [Google Scholar] [CrossRef]
- Cook, D.A.; Kannarkat, G.T.; Cintron, A.F.; Butkovich, L.M.; Fraser, K.B.; Chang, J.; Grigoryan, N.; Factor, S.A.; West, A.B.; Boss, J.M.; et al. LRRK2 Levels in Immune Cells Are Increased in Parkinson’s Disease. NPJ Park. Dis. 2017, 3, 1–12. [Google Scholar] [CrossRef]
- Zhang, F.-R.; Huang, W.; Chen, S.-M.; Sun, L.-D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.-X.; Yang, H.-T.; Yang, R.-D.; et al. Genomewide Association Study of Leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional Variants in the LRRK2 Gene Confer Shared Effects on Risk for Crohn’s Disease and Parkinson’s Disease. Sci. Transl. Med. 2018, 10, eaai7795. [Google Scholar] [CrossRef]
- Deng, I.; Corrigan, F.; Zhai, G.; Zhou, X.-F.; Bobrovskaya, L. Lipopolysaccharide Animal Models of Parkinson’s Disease: Recent Progress and Relevance to Clinical Disease. Brain Behav. Immun.—Health 2020, 4, 100060. [Google Scholar] [CrossRef] [PubMed]
- Beier, E.E.; Neal, M.; Alam, G.; Edler, M.; Wu, L.J.; Richardson, J.R. Alternative Microglial Activation Is Associated with Cessation of Progressive Dopamine Neuron Loss in Mice Systemically Administered Lipopolysaccharide. Neurobiol. Dis. 2017, 108, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Bodea, L.G.; Wang, Y.; Linnartz-Gerlach, B.; Kopatz, J.; Sinkkonen, L.; Musgrove, R.; Kaoma, T.; Muller, A.; Vallar, L.; Di Monte, D.A.; et al. Neurodegeneration by Activation of the Microglial Complement–Phagosome Pathway. J. Neurosci. 2014, 34, 8546–8556. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, C.; Wu, W.Y.; Chen, F.; Wu, Y.Y.; Li, W.Z.; Chen, H.Q.; Yin, Y.Y. Biochanin A Protects Dopaminergic Neurons against Lipopolysaccharide-Induced Damage and Oxidative Stress in a Rat Model of Parkinson’s Disease. Pharmacol. Biochem. Behav. 2015, 138, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Castaño, A.; Herrera, A.J.; Cano, J.; Machado, A. Lipopolysaccharide Intranigral Injection Induces Inflammatory Reaction and Damage in Nigrostriatal Dopaminergic System. J. Neurochem. 1998, 70, 1584–1592. [Google Scholar] [CrossRef]
- Machado, A.; Herrera, A.J.; Venero, J.L.; Santiago, M.; de Pablos, R.M.; Villarán, R.F.; Espinosa-Oliva, A.M.; Argüelles, S.; Sarmiento, M.; Delgado-Cortés, M.J.; et al. Inflammatory Animal Model for Parkinson’s Disease: The Intranigral Injection of LPS Induced the Inflammatory Process along with the Selective Degeneration of Nigrostriatal Dopaminergic Neurons. ISRN Neurol. 2011, 2011, 1–16. [Google Scholar] [CrossRef]
- Kuss, M.; Adamopoulou, E.; Kahle, P.J. Interferon-γ Induces Leucine-Rich Repeat Kinase LRRK2 via Extracellular Signal-Regulated Kinase ERK5 in Macrophages. J. Neurochem. 2014, 129, 980–987. [Google Scholar] [CrossRef]
- Gillardon, F.; Schmid, R.; Draheim, H. Parkinson’s Disease-Linked Leucine-Rich Repeat Kinase 2(R1441G) Mutation Increases Proinflammatory Cytokine Release from Activated Primary Microglial Cells and Resultant Neurotoxicity. Neuroscience 2012, 208, 41–48. [Google Scholar] [CrossRef]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; Desilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 Inhibition Attenuates Microglial Inflammatory Responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef]
- Daher, J.P.L.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of α-Synuclein -Mediated Dopaminergic Neurodegeneration in LRRK2-Deficient Rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef]
- Dzamko, N.; Inesta-Vaquera, F.; Zhang, J.; Xie, C.; Cai, H.; Arthur, S.; Tan, L.; Choi, H.; Gray, N.; Cohen, P.; et al. The IkappaB Kinase Family Phosphorylates the Parkinson’s Disease Kinase LRRK2 at Ser935 and Ser910 during Toll-Like Receptor Signaling. PLoS ONE 2012, 7, e39132. [Google Scholar] [CrossRef]
- Schapansky, J.; Nardozzi, J.D.; Felizia, F.; LaVoie, M.J. Membrane Recruitment of Endogenous LRRK2 Precedes Its Potent Regulation of Autophagy. Hum. Mol. Genet. 2014, 23, 4201–4214. [Google Scholar] [CrossRef]
- Li, T.; Ning, B.; Kong, L.; Dai, B.; He, X.; Thomas, J.M.; Sawa, A.; Ross, C.A.; Smith, W.W. A Lrrk2 Gtp Binding Inhibitor, 68, Reduces Lps-induced Signaling Events and Tnf-α Release in Human Lymphoblasts. Cells 2021, 10, 480. [Google Scholar] [CrossRef]
- Ho, D.H.; Je, A.R.; Lee, H.; Son, I.; Kweon, H.S.; Kim, H.G.; Seol, W. LRRK2 Kinase Activity Induces Mitochondrial Fission in Microglia via Drp1 and Modulates Neuroinflammation. Exp. Neurobiol. 2018, 27, 171–180. [Google Scholar] [CrossRef]
- Russo, I.; Berti, G.; Plotegher, N.; Bernardo, G.; Filograna, R.; Bubacco, L.; Greggio, E. Leucine-Rich Repeat Kinase 2 Positively Regulates Inflammation and down-Regulates NF-ΚB P50 Signaling in Cultured Microglia Cells. J. Neuroinflamm. 2015, 12, 230. [Google Scholar] [CrossRef]
- Kim, B.; Yang, M.S.; Choi, D.; Kim, J.H.; Kim, H.S.; Seol, W.; Choi, S.; Jou, I.; Kim, E.Y.; Joe, E. hye Impaired Inflammatory Responses in Murine Lrrk2-Knockdown Brain Microglia. PLoS ONE 2012, 7, e34693. [Google Scholar] [CrossRef]
- Fan, C.; Wu, L.H.; Zhang, G.F.; Xu, F.; Zhang, S.; Zhang, X.; Sun, L.; Yu, Y.; Zhang, Y.; Ye, R.D. 4’-Hydroxywogonin Suppresses Lipopolysaccharide-Induced Inflammatory Responses in RAW 264.7 Macrophages and Acute Lung Injury Mice. PLoS ONE 2017, 12, e0181191. [Google Scholar] [CrossRef]
- Yauger, Y.J.; Bermudez, S.; Moritz, K.E.; Glaser, E.; Stoica, B.; Byrnes, K.R. Iron Accentuated Reactive Oxygen Species Release by NADPH Oxidase in Activated Microglia Contributes to Oxidative Stress in Vitro. J. Neuroinflamm. 2019, 16, 41. [Google Scholar] [CrossRef]
- More, G.K.; Makola, R.T. In-Vitro Analysis of Free Radical Scavenging Activities and Suppression of LPS-Induced ROS Production in Macrophage Cells by Solanum Sisymbriifolium Extracts. Sci. Rep. 2020, 10, 6493. [Google Scholar] [CrossRef]
- Jakubowski, W.; Bartosz, G. 2,7-Dichlorofluorescin Oxidation and Reactive Oxygen Species: What Does It Measure? Cell Biol. Int. 2000, 24, 757–760. [Google Scholar] [CrossRef]
- Sakai, J.; Cammarota, E.; Wright, J.A.; Cicuta, P.; Gottschalk, R.A.; Li, N.; Fraser, I.D.C.; Bryant, C.E. Lipopolysaccharide-Induced NF-ΚB Nuclear Translocation Is Primarily Dependent on MyD88, but TNFα Expression Requires TRIF and MyD88. Sci. Rep. 2017, 7, 1428. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-ΚB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Teismann, P.; Tieu, K.; Choi, D.K.; Wu, D.C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 Is Instrumental in Parkinson’s Disease Neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory Regulators in Parkinson’s Disease: INOS, Lipocortin-1, and Cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef]
- Sánchez-Pernaute, R.; Ferree, A.; Cooper, O.; Yu, M.; Brownell, A.L.; Isacson, O. Selective COX-2 Inhibition Prevents Progressive Dopamine Neuron Degeneration in a Rat Model of Parkinson’s Disease. J. Neuroinflamm. 2004, 1, 6. [Google Scholar] [CrossRef]
- Vijitruth, R.; Liu, M.; Choi, D.Y.; Nguyen, X.V.; Hunter, R.L.; Bing, G. Cyclooxygenase-2 Mediates Microglial Activation and Secondary Dopaminergic Cell Death in the Mouse MPTP Model of Parkinson’s Disease. J. Neuroinflamm. 2006, 3, 6. [Google Scholar] [CrossRef]
- Lopez de Maturana, R.; Aguila, J.C.; Sousa, A.; Vazquez, N.; del Rio, P.; Aiastui, A.; Gorostidi, A.; Lopez de Munain, A.; Sanchez-Pernaute, R. Leucine-Rich Repeat Kinase 2 Modulates Cyclooxygenase 2 and the Inflammatory Response in Idiopathic and Genetic Parkinson’s Disease. Neurobiol. Aging 2014, 35, 1116–1124. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic Reprogramming in Macrophage Responses. Biomark. Res. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Wallings, R.L.; Herrick, M.K.; Tansey, M.G. LRRK2 at the Interface Between Peripheral and Central Immune Function in Parkinson’s. Front. Neurosci. 2020, 14, 443. [Google Scholar] [CrossRef]
- Su, X.; Federoff, H.J. Immune Responses in Parkinson’s Disease: Interplay between Central and Peripheral Immune Systems. Biomed. Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Oun, A.; Sabogal-Guaqueta, A.M.; Galuh, S.; Alexander, A.; Kortholt, A.; Dolga, A. The Multifaceted Role of LRRK2 in Parkinson’s Disease: From Human IPSC to Organoids. Neurobiol. Dis. 2022, 173, 105837. [Google Scholar] [CrossRef]
- Nazish, I.; Arber, C.; Piers, T.M.; Warner, T.T.; Hardy, J.A.; Lewis, P.A.; Pocock, J.M.; Bandopadhyay, R. Abrogation of LRRK2 Dependent Rab10 Phosphorylation with TLR4 Activation and Alterations in Evoked Cytokine Release in Immune Cells. Neurochem. Int. 2021, 147, 105070. [Google Scholar] [CrossRef]
- Herbst, S.; Campbell, P.; Harvey, J.; Bernard, E.M.; Papayannopoulos, V.; Wood, N.W.; Morris, H.R.; Gutierrez, M.G. LRRK 2 Activation Controls the Repair of Damaged Endomembranes in Macrophages. EMBO J. 2020, 39, e104494. [Google Scholar] [CrossRef]
- Wall, A.A.; Luo, L.; Hung, Y.; Tong, S.J.; Condon, N.D.; Blumenthal, A.; Sweet, M.J.; Stow, J.L. Small GTPase Rab8a-Recruited Phosphatidylinositol 3-Kinase γ Regulates Signaling and Cytokine Outputs from Endosomal Toll-like Receptors. J. Biol. Chem. 2017, 292, 4411–4422. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Kim, J.; Pajarillo, E.; Rizor, A.; Son, D.S.; Lee, J.; Aschner, M.; Lee, E. LRRK2 Kinase Plays a Critical Role in Manganese-Induced Inflammation and Apoptosis in Microglia. PLoS ONE 2019, 14, e0210248. [Google Scholar] [CrossRef]
- Helton, L.G.; Soliman, A.; Von Zweydorf, F.; Kentros, M.; Manschwetus, J.T.; Hall, S.; Gilsbach, B.; Ho, F.Y.; Athanasopoulos, P.S.; Singh, R.K.; et al. Allosteric Inhibition of Parkinson’s-Linked LRRK2 by Constrained Peptides. ACS Chem. Biol. 2021, 16, 2326–2338. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Bonet-Ponce, L.; Blesa, J.R.J.; Romero, F.J.F.J.; Murphy, M.P.M.; Jordan, J.; Galindo, M.F.M. The LRRK2 Inhibitor GSK2578215A Induces Protective Autophagy in SH-SY5Y Cells: Involvement of Drp-1-Mediated Mitochondrial Fission and Mitochondrial-Derived ROS Signaling. Cell Death Dis. 2014, 5, e1368-10. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral Cytokines Profile in Parkinson’s Disease. Brain. Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. Inflammatory Process in Parkinsons Disease: Role for Cytokines. Curr. Pharm. Des. 2005, 11, 999–1016. [Google Scholar] [CrossRef]
- Tansey, M.G.; Frank-Cannon, T.C.; McCoy, M.K.; Jae, K.L.; Martinez, T.N.; McAlpine, F.E.; Ruhn, K.A.; Tran, T.A. Neuroinflammation in Parkinson’s Disease: Is There Sufficient Evidence for Mechanism-Based Interventional Therapy? Front. Biosci. 2008, 13, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Apel, A.; Schulte, C.; Schneiderhan-Marra, N.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.C.; Cormier, F.; et al. Inflammatory Profile in LRRK2-Associated Prodromal and Clinical PD. J. Neuroinflamm. 2016, 13, 122. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Schulte, C.; Schneiderhan-Marra, N.; Apel, A.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.-C.; Cormier, F.; et al. Inflammatory Profile Discriminates Clinical Subtypes in LRRK2-Associated Parkinson’s Disease. Eur. J. Neurol. 2017, 24, 427.e6. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakopoulou, V.; Ivanyuk, D.; De Cicco, S.; Haq, W.; Arsić, A.; Yu, C.; Messelodi, D.; Oldrati, M.; Schöndorf, D.C.; Perez, M.J.; et al. Interferon-γ Signaling Synergizes with LRRK2 in Neurons and Microglia Derived from Human Induced Pluripotent Stem Cells. Nat. Commun. 2020, 11, 5163. [Google Scholar] [CrossRef]
- Ahmadi Rastegar, D.; Hughes, L.P.; Perera, G.; Keshiya, S.; Zhong, S.; Gao, J.; Halliday, G.M.; Schüle, B.; Dzamko, N. Effect of LRRK2 Protein and Activity on Stimulated Cytokines in Human Monocytes and Macrophages. NPJ Park. Dis. 2022, 8, 34. [Google Scholar] [CrossRef]
- Dwyer, Z.; Rudyk, C.; Thompson, A.; Farmer, K.; Fenner, B.; Fortin, T.; Derksen, A.; Sun, H.; Hayley, S. Leucine-Rich Repeat Kinase-2 (LRRK2) Modulates Microglial Phenotype and Dopaminergic Neurodegeneration. Neurobiol. Aging 2020, 91, 45–55. [Google Scholar] [CrossRef]
- Kubo, M.; Nagashima, R.; Kurihara, M.; Kawakami, F.; Maekawa, T.; Eshima, K.; Ohta, E.; Kato, H.; Obata, F. Leucine-Rich Repeat Kinase 2 Controls Inflammatory Cytokines Production through NF-ΚB Phosphorylation and Antigen Presentation in Bone Marrow-Derived Dendritic Cells. Int. J. Mol. Sci. 2020, 21, 1890. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The Pro- and Anti-Inflammatory Properties of the Cytokine Interleukin-6. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef]
- San Luciano, M.; Tanner, C.M.; Meng, C.; Marras, C.; Goldman, S.M.; Lang, A.E.; Tolosa, E.; Schüle, B.; Langston, J.W.; Brice, A.; et al. Nonsteroidal Anti-Inflammatory Use and LRRK2 Parkinson’s Disease Penetrance. Mov. Disord. 2020, 35, 1755–1764. [Google Scholar] [CrossRef]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Fernandez-Carasa, I.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing Glycolysis Attenuates Parkinson’s Disease Progression in Models and Clinical Databases. J. Clin. Investig. 2019, 129, 4539–4549. [Google Scholar] [CrossRef]
- Nikonova, E.V.; Xiong, Y.; Tanis, K.Q.; Dawson, V.L.; Vogel, R.L.; Finney, E.M.; Stone, D.J.; Reynolds, I.J.; Kern, J.T.; Dawson, T.M. Transcriptional Responses to Loss or Gain of Function of the Leucine-Rich Repeat Kinase 2 (LRRK2) Gene Uncover Biological Processes Modulated by LRRK2 Activity. Hum. Mol. Genet. 2012, 21, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Nabar, N.R.; Heijjer, C.N.; Shi, C.S.; Hwang, I.Y.; Ganesan, S.; Karlsson, M.C.I.; Kehrl, J.H. LRRK2 Is Required for CD38-Mediated NAADP-Ca2+ Signaling and the Downstream Activation of TFEB (Transcription Factor EB) in Immune Cells. Autophagy 2022, 18, 204–222. [Google Scholar] [CrossRef] [PubMed]
- Teslaa, T.; Teitell, M.A. Techniques to Monitor Glycolysis. Methods Enzymol. 2014, 542, 91–114. [Google Scholar] [CrossRef]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK2 Regulates Endoplasmic Reticulum–Mitochondrial Tethering through the PERK -mediated Ubiquitination Pathway. EMBO J. 2020, 39, e100875. [Google Scholar] [CrossRef]
- Weindel, C.G.; Bell, S.L.; Vail, K.J.; West, K.O.; Patrick, K.L.; Watson, R.O. LRRK2 Maintains Mitochondrial Homeostasis and Regulates Innate Immune Responses to Mycobacterium Tuberculosis. Elife 2020, 9, e51071. [Google Scholar] [CrossRef]
- Diemert, S.; Dolga, A.M.M.; Tobaben, S.; Grohm, J.; Pfeifer, S.; Oexler, E.; Culmsee, C. Impedance Measurement for Real Time Detection of Neuronal Cell Death. J. Neurosci. Methods 2012, 203, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Krabbendam, I.E.; Honrath, B.; Dilberger, B.; Iannetti, E.F.; Branicky, R.S.; Meyer, T.; Evers, B.; Dekker, F.J.; Koopman, W.J.H.; Beyrath, J.; et al. SK Channel-Mediated Metabolic Escape to Glycolysis Inhibits Ferroptosis and Supports Stress Resistance in C. Elegans. Cell Death Dis. 2020, 11, 263. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oun, A.; Hoeksema, E.; Soliman, A.; Brouwer, F.; García-Reyes, F.; Pots, H.; Trombetta-Lima, M.; Kortholt, A.; Dolga, A.M. Characterization of Lipopolysaccharide Effects on LRRK2 Signaling in RAW Macrophages. Int. J. Mol. Sci. 2023, 24, 1644. https://doi.org/10.3390/ijms24021644
Oun A, Hoeksema E, Soliman A, Brouwer F, García-Reyes F, Pots H, Trombetta-Lima M, Kortholt A, Dolga AM. Characterization of Lipopolysaccharide Effects on LRRK2 Signaling in RAW Macrophages. International Journal of Molecular Sciences. 2023; 24(2):1644. https://doi.org/10.3390/ijms24021644
Chicago/Turabian StyleOun, Asmaa, Emmy Hoeksema, Ahmed Soliman, Famke Brouwer, Fabiola García-Reyes, Henderikus Pots, Marina Trombetta-Lima, Arjan Kortholt, and Amalia M. Dolga. 2023. "Characterization of Lipopolysaccharide Effects on LRRK2 Signaling in RAW Macrophages" International Journal of Molecular Sciences 24, no. 2: 1644. https://doi.org/10.3390/ijms24021644
APA StyleOun, A., Hoeksema, E., Soliman, A., Brouwer, F., García-Reyes, F., Pots, H., Trombetta-Lima, M., Kortholt, A., & Dolga, A. M. (2023). Characterization of Lipopolysaccharide Effects on LRRK2 Signaling in RAW Macrophages. International Journal of Molecular Sciences, 24(2), 1644. https://doi.org/10.3390/ijms24021644