
siRNA-Mediated Timp1 Silencing Inhibited the Inflammatory Phenotype during Acute Lung Injury

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results
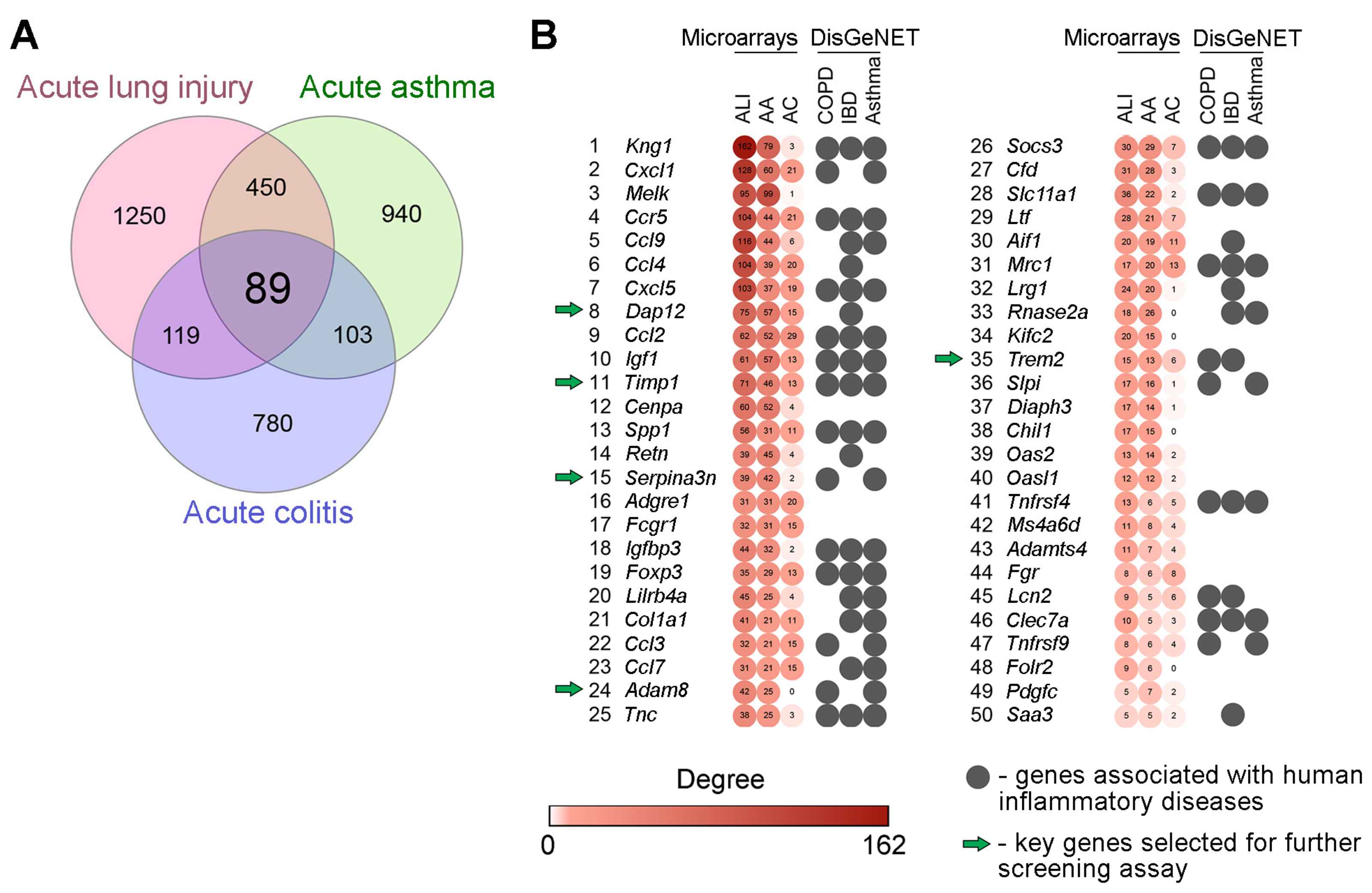
2.1. Selection of Inflammation-Associated Hub Genes as Potential Targets
2.2. Screening for the Expression of Selected Genes in Cell Lines of Different Origin Stimulated by LPS
2.3. Functional Analysis of Timp1 and Its First Neighbors in Gene Networks
2.4. Silencing of Timp1 in Macrophages RAW264.7 with siRNA
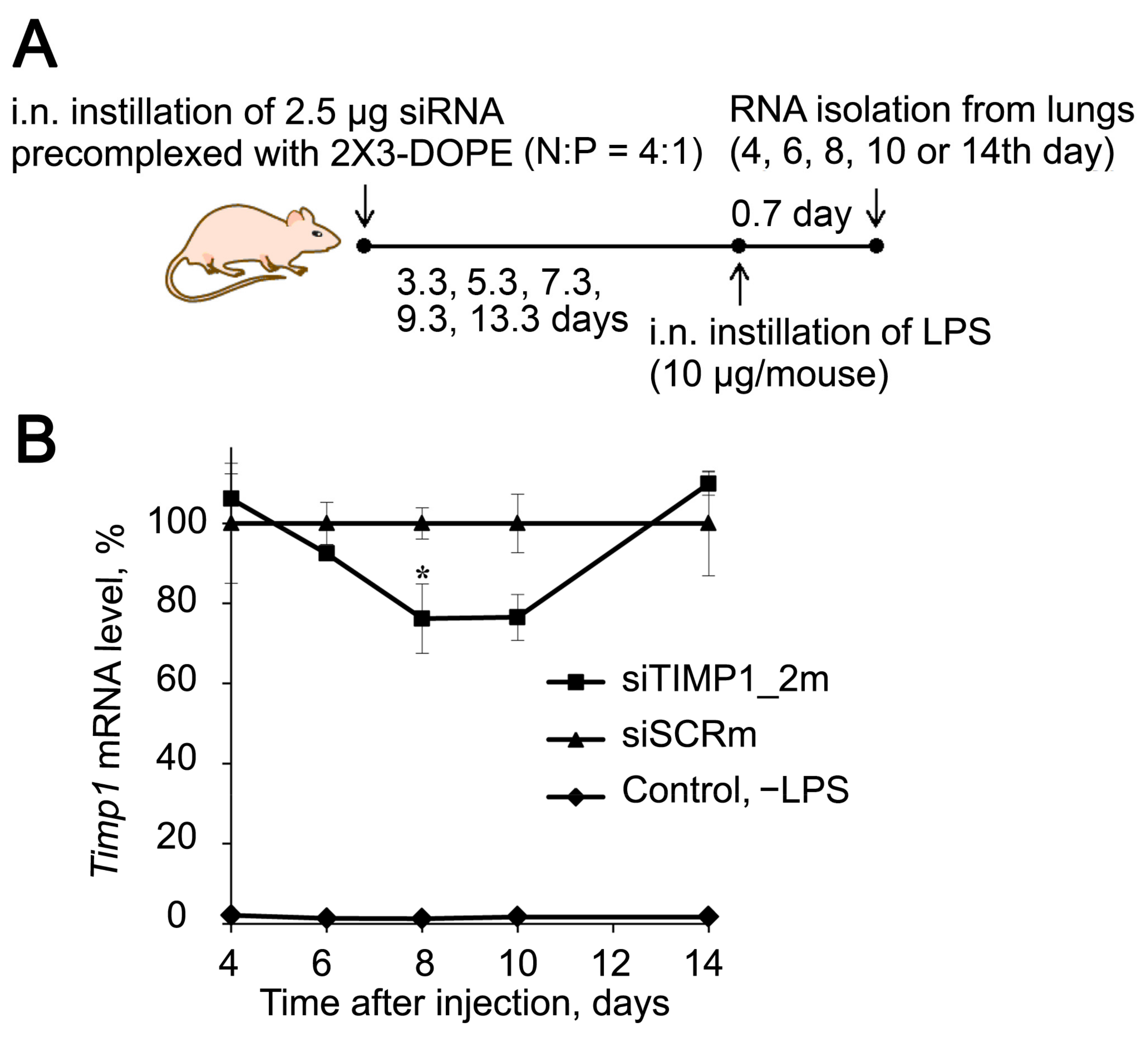
2.5. Dynamics of Timp1 Expression and Silencing in the Lung Tissue of Mice during ALI Development
2.6. Anti-Timp1 siRNA Effectively Suppress LPS-induced Lung Inflammation In Vivo
3. Discussion
4. Materials and Methods
4.1. Identification of Hub Genes, Their Functional Analysis and PPI Network Reconstruction
4.2. Cell Culture
4.3. LPS Treatment
4.4. Mice
4.5. Real-Time Quantitative PCR (RT-qPCR) Assay
4.6. ELISA
4.7. Synthesis of siRNAs and Duplex Annealing
4.8. Transfection of siRNA
4.9. Western Blotting
4.10. In Vivo Model of Acute Lung Injury (ALI)
4.11. Silencing of Timp1 in Mice with siRNA
4.12. Bronchoalveolar Lavage (BAL) Fluid Analysis
4.13. Histology
4.14. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, E.R.; Matthay, M.A. Acute Lung Injury: Epidemiology, Pathogenesis, and Treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243. [Google Scholar] [CrossRef] [PubMed]
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute Respiratory Distress in Adults. Lancet 1967, 290, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Avecillas, J.F.; Freire, A.X.; Arroliga, A.C. Clinical Epidemiology of Acute Lung Injury and Acute Respiratory Distress Syndrome: Incidence, Diagnosis, and Outcomes. Clin. Chest Med. 2006, 27, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Gorman, E.A.; O’Kane, C.M.; McAuley, D.F. Acute respiratory distress syndrome in adults: Diagnosis, outcomes, long-term sequelae, and management. Lancet 2022, 400, 1157–1170. [Google Scholar] [CrossRef]
- Ajibowo, A.O.; Kolawole, O.A.; Sadia, H.; Amedu, O.S.; Chaudhry, H.A.; Hussaini, H.; Hambolu, E.; Khan, T.; Kauser, H.; Khan, A. A Comprehensive Review of the Management of Acute Respiratory Distress Syndrome. Cureus 2022, 14, e30669. [Google Scholar] [CrossRef]
- Barile, M. Pulmonary Edema: A Pictorial Review of Imaging Manifestations and Current Understanding of Mechanisms of Disease. Eur. J. Radiol. Open 2020, 7, 100274. [Google Scholar] [CrossRef]
- Silva, P.L.; Pelosi, P.; Rocco, P.R.M. Personalized pharmacological therapy for ARDS: A light at the end of the tunnel. Expert Opin. Investig. Drugs 2020, 29, 49–61. [Google Scholar] [CrossRef]
- Kaku, S.; Nguyen, C.D.; Htet, N.N.; Tutera, D.; Barr, J.; Paintal, H.S.; Kuschner, W.G. Acute Respiratory Distress Syndrome: Etiology, Pathogenesis, and Summary on Management. J. Intensive Care Med. 2020, 35, 723–737. [Google Scholar] [CrossRef]
- Keskinidou, C.; Vassiliou, A.G.; Dimopoulou, I.; Kotanidou, A.; Orfanos, S.E. Mechanistic Understanding of Lung Inflammation: Recent Advances and Emerging Techniques. J. Inflamm. Res. 2022, 15, 3501–3546. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Griffiths, M.J.D.; McAuley, D.F.; Perkins, G.D.; Barrett, N.; Blackwood, B.; Boyle, A.; Chee, N.; Connolly, B.; Dark, P.; Finney, S.; et al. Guidelines on the management of acute respiratory distress syndrome. BMJ Open Respir. Res. 2019, 6, e000420. [Google Scholar] [CrossRef]
- Ragaller, M.; Richter, T. Acute lung injury and acute respiratory distress syndrome. J. Emerg. Trauma Shock 2010, 3, 43. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zimmerman, G.A. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am. J. Respir. Cell Mol. Biol. 2005, 33, 319–327. [Google Scholar] [CrossRef]
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 31–39. [Google Scholar] [CrossRef]
- Bos, L.D.J.; Ware, L.B. Acute respiratory distress syndrome: Causes, pathophysiology, and phenotypes. Lancet 2022, 400, 1145–1156. [Google Scholar] [CrossRef]
- Gouda, M.M.; Shaikh, S.B.; Bhandary, Y.P. Inflammatory and Fibrinolytic System in Acute Respiratory Distress Syndrome. Lung 2018, 196, 609–616. [Google Scholar] [CrossRef]
- Dang, W.; Tao, Y.; Xu, X.; Zhao, H.; Zou, L.; Li, Y. The role of lung macrophages in acute respiratory distress syndrome. Inflamm. Res. 2022, 71, 1417–1432. [Google Scholar] [CrossRef]
- Vichare, R.; Janjic, J.M. Macrophage-Targeted Nanomedicines for ARDS/ALI: Promise and Potential. Inflammation 2022, 45, 2124–2141. [Google Scholar] [CrossRef]
- Butt, Y.; Kurdowska, A.; Allen, T.C. Acute Lung Injury: A Clinical and Molecular Review. Arch. Pathol. Lab. Med. 2016, 140, 345–350. [Google Scholar] [CrossRef]
- Metwaly, S.M.; Winston, B.W. Systems Biology ARDS Research with a Focus on Metabolomics. Metabolites 2020, 10, 207. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xiang, D.; Zhang, H.; Yao, H.; Wang, Y. Hypoxia-Inducible Factor-1: A Potential Target to Treat Acute Lung Injury. Oxid. Med. Cell. Longev. 2020, 2020, 8871476. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Ferrando, C.; Tusman, G.; Berra, L.; Rodríguez-Suárez, P.; Suárez-Sipmann, F. Unsuccessful and Successful Clinical Trials in Acute Respiratory Distress Syndrome: Addressing Physiology-Based Gaps. Front. Physiol. 2021, 12, 2147. [Google Scholar] [CrossRef] [PubMed]
- Banavasi, H.; Nguyen, P.; Osman, H.; Soubani, A.O. Management of ARDS—What Works and What Does Not. Am. J. Med. Sci. 2021, 362, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dean, D.A. Gene Therapy for Acute Respiratory Distress Syndrome. Front. Physiol. 2021, 12, 2277. [Google Scholar] [CrossRef]
- Zhang, X.-P.; Zhang, W.-T.; Qiu, Y.; Ju, M.-J.; Tu, G.-W.; Luo, Z. Understanding Gene Therapy in Acute Respiratory Distress Syndrome. Curr. Gene Ther. 2019, 19, 93–99. [Google Scholar] [CrossRef]
- McAuliffe, P.F.; Murday, M.E.; Efron, P.A.; Scumpia, P.O.; Ungaro, R.; Abouhamze, A.; Tannahill, C.L.; Hutchins, B.; LaFace, D.; Moldawer, L.L. Dose-dependent improvements in outcome with adenoviral expression of interleukin-10 in a murine model of multisystem organ failure. Gene Ther. 2005, 13, 276–282. [Google Scholar] [CrossRef]
- Greenberger, M.J.; Kunkel, S.L.; Strieter, R.M.; Lukacs, N.W.; Bramson, J.; Gauldie, J.; Graham, F.L.; Hitt, M.; Danforth, J.M.; Standiford, T.J. IL-12 gene therapy protects mice in lethal Klebsiella pneumonia. J. Immunol. 1996, 157, 3006–3012. [Google Scholar] [CrossRef]
- Bowler, R.P.; Nicks, M.; Tran, K.; Tanner, G.; Chang, L.Y.; Young, S.K.; Worthen, G.S. Extracellular Superoxide Dismutase Attenuates Lipopolysaccharide-Induced Neutrophilic Inflammation. Am. J. Respir. Cell Mol. Biol. 2012, 31, 432–439. [Google Scholar] [CrossRef]
- Gong, Q.; Yin, H.; Fang, M.; Xiang, Y.; Yuan, C.; Zheng, G.Y.; Yang, H.; Xiong, P.; Chen, G.; Gong, F.-l.; et al. Heme oxygenase-1 upregulation significantly inhibits TNF-α and Hmgb1 releasing and attenuates lipopolysaccharide-induced acute lung injury in mice. Int. Immunopharmacol. 2008, 8, 792–798. [Google Scholar] [CrossRef]
- Ando, M.; Murakami, Y.; Kojima, F.; Endo, H.; Kitasato, H.; Hashimoto, A.; Kobayashi, H.; Majima, M.; Inoue, M.; Kondo, H.; et al. Retrovirally Introduced Prostaglandin D2 Synthase Suppresses Lung Injury Induced by Bleomycin. Am. J. Respir. Cell Mol. Biol. 2012, 28, 582–591. [Google Scholar] [CrossRef]
- Murata, T.; Aritake, K.; Tsubosaka, Y.; Maruyama, T.; Nakagawa, T.; Hori, M.; Hirai, H.; Nakamura, M.; Narumiya, S.; Urade, Y.; et al. Anti-inflammatory role of PGD2 in acute lung inflammation and therapeutic application of its signal enhancement. Proc. Natl. Acad. Sci. USA 2013, 110, 5205–5210. [Google Scholar] [CrossRef]
- Ballarín-González, B.; Thomsen, T.B.; Howard, K.A. Clinical translation of RNAi-based treatments for respiratory diseases. Drug Deliv. Transl. Res. 2012, 3, 84–99. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Liang, W.; Chan, H.K. Pulmonary delivery of therapeutic siRNA. Adv. Drug Deliv. Rev. 2012, 64, 1–15. [Google Scholar] [CrossRef]
- Dua, K.; Wadhwa, R.; Singhvi, G.; Rapalli, V.; Shukla, S.D.; Shastri, M.D.; Gupta, G.; Satija, S.; Mehta, M.; Khurana, N.; et al. The potential of siRNA based drug delivery in respiratory disorders: Recent advances and progress. Drug Dev. Res. 2019, 80, 714–730. [Google Scholar] [CrossRef]
- Zoulikha, M.; Xiao, Q.; Boafo, G.F.; Sallam, M.A.; Chen, Z.; He, W. Pulmonary delivery of siRNA against acute lung injury/acute respiratory distress syndrome. Acta Pharm. Sin. B 2022, 12, 600–620. [Google Scholar] [CrossRef]
- Meng, L.; Liao, X.; Wang, Y.; Chen, L.; Gao, W.; Wang, M.; Dai, H.; Yan, N.; Gao, Y.; Wu, X.; et al. Pharmacologic therapies of ARDS: From natural herb to nanomedicine. Front. Pharmacol. 2022, 13, 930593. [Google Scholar] [CrossRef]
- Sen’kova, A.V.; Savin, I.A.; Brenner, E.V.; Zenkova, M.A.; Markov, A.V. Core genes involved in the regulation of acute lung injury and their association with COVID-19 and tumor progression: A bioinformatics and experimental study. PLoS ONE 2021, 16, e0260450. [Google Scholar] [CrossRef]
- Lomas-Neira, J.L.; Chung, C.-S.; Wesche, D.E.; Perl, M.; Ayala, A. In vivo gene silencing (with siRNA) of pulmonary expression of MIP-2 versus KC results in divergent effects on hemorrhage-induced, neutrophil-mediated septic acute lung injury. J. Leukoc. Biol. 2005, 77, 846–853. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Jha, P.; Das, H. KLF2 in regulation of NF-κB-mediated immune cell function and inflammation. Int. J. Mol. Sci. 2017, 18, 2383. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Hattori, Y.; Jesmin, S.; Gando, S. Nuclear Factor-κB Decoy Oligodeoxynucleotides Prevent Acute Lung Injury in Mice with Cecal Ligation and Puncture-Induced Sepsis. Mol. Pharmacol. 2005, 67, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Song, Y.; Zhao, W.; Han, T.; Lin, S.; Ramirez, O.; Liang, L. Small interfering RNA targeting NF-κB attenuates lipopolysaccharide-induced acute lung injury in rats. BMC Physiol. 2016, 16, 7. [Google Scholar] [CrossRef]
- Chawla, M.; Roy, P.; Basak, S. Role of the NF-κB system in context-specific tuning of the inflammatory gene response. Curr. Opin. Immunol. 2021, 68, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Lanier, L.L.; Bakker, A.B.H. The ITAM-bearing transmembrane adaptor DAP12 in lymphoid and myeloid cell function. Immunol. Today 2000, 21, 611–614. [Google Scholar] [CrossRef]
- Zhong, L.; Zhang, Z.L.; Li, X.; Liao, C.; Mou, P.; Wang, T.; Wang, Z.; Wang, Z.; Wei, M.; Xu, H.; et al. TREM2/DAP12 complex regulates inflammatory responses in Microglia via the JNK signaling pathway. Front. Aging Neurosci. 2017, 9, 204. [Google Scholar] [CrossRef]
- Aoki, N.; Zganiacz, A.; Margetts, P.; Xing, Z. Differential Regulation of DAP12 and Molecules Associated with DAP12 during Host Responses to Mycobacterial Infection. Infect. Immun. 2004, 72, 2477–2483. [Google Scholar] [CrossRef]
- Jeyanathan, M.; Mccormick, S.; Lai, R.; Afkhami, S.; Shaler, C.R.; Horvath, C.N.; Damjanovic, D.; Zganiacz, A.; Barra, N.; Ashkar, A.; et al. Pulmonary M. tuberculosis infection delays Th1 immunity via immunoadaptor DAP12-regulated IRAK-M and IL-10 expression in antigen-presenting cells. Mucosal Immunol. 2014, 7, 670–683. [Google Scholar] [CrossRef]
- Heung, L.J.; Hohl, T.M. DAP12 inhibits pulmonary immune responses to Cryptococcus neoformans. Infect. Immun. 2016, 84, 1879–1886. [Google Scholar] [CrossRef]
- Turnbull, I.R.; Colonna, M. Activating and inhibitory functions of DAP12. Nat. Rev. Immunol. 2007, 7, 155–161. [Google Scholar] [CrossRef]
- Kim, K.-H.; Burkhart, K.; Chen, P.; Frevert, C.W.; Randolph-Habecker, J.; Hackman, R.C.; Soloway, P.D.; Madtes, D.K. Tissue Inhibitor of Metalloproteinase-1 Deficiency Amplifies Acute Lung Injury in Bleomycin-Exposed Mice. Am. J. Respir. Cell Mol. Biol. 2005, 33, 271. [Google Scholar] [CrossRef]
- Ries, C. Cytokine functions of TIMP-1. Cell. Mol. Life Sci. 2014, 71, 659–672. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Costa, S.; Ragusa, M.A.; Lo Buglio, G.; Scilabra, S.D.; Nicosia, A. The Repertoire of Tissue Inhibitors of Metalloproteases: Evolution, Regulation of Extracellular Matrix Proteolysis, Engineering and Therapeutic Challenges. Life 2022, 12, 1145. [Google Scholar] [CrossRef]
- Schoeps, B.; Frädrich, J.; Krüger, A. Cut loose TIMP-1: An emerging cytokine in inflammation. Trends Cell Biol. 2022, in press. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta—Protein Struct. Mol. Enzymol. 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef]
- Guay, C.; Laviolette, M.; Tremblay, G. Targeting serine proteases in asthma. Curr. Top. Med. Chem. 2006, 6, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Kelly-robinson, G.A.; Reihill, J.A.; Lundy, F.T.; McGarvey, L.P.; Lockhart, J.C.; Litherland, G.J.; Thornbury, K.D.; Martin, S.L. The Serpin Superfamily and Their Role in the Regulation and Dysfunction of Serine Protease Activity in COPD and Other Chronic Lung Diseases. Int. J. Mol. Sci. 2021, 22, 6351. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.C.; Macgregor, I.; Zamani, A.; Gordon, M.W.G.; Robertson, C.E.; Steedman, D.J.; Little, K.; Haslett, C. Plasma elastase levels and the development of the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2012, 151, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Askew, D.J.; Silverman, G.A. Intracellular and extracellular serpins modulate lung disease. J. Perinatol. 2008, 28 (Suppl. 3), S127–S135. [Google Scholar] [CrossRef] [PubMed]
- Kalsheker, N.; Morley, S.; Morgan, K. Gene regulation of the serine proteinase inhibitors α1-antitrypsin and α1-antichymotrypsin. Biochem. Soc. Trans. 2002, 30, 93–98. [Google Scholar] [CrossRef]
- Heit, C.; Jackson, B.C.; McAndrews, M.; Wright, M.W.; Thompson, D.C.; Silverman, G.A.; Nebert, D.W.; Vasiliou, V. Update of the human and mouse SERPIN gene superfamily. Hum. Genom. 2013, 7, 22. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, X.; Duan, Y.; Long, J.; Bartsch, J.W.; Deng, L. ADAM8 in asthma. Friend or foe to airway inflammation? Am. J. Respir. Cell Mol. Biol. 2013, 49, 875–884. [Google Scholar] [CrossRef]
- Dreymueller, D.; Pruessmeyer, J.; Schumacher, J.; Fellendorf, S.; Hess, F.M.; Seifert, A.; Babendreyer, A.; Bartsch, J.W.; Ludwig, A. The metalloproteinase ADAM8 promotes leukocyte recruitment in vitro and in acute lung inflammation. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2017, 313, L602–L614. [Google Scholar] [CrossRef]
- Conrad, C.; Yildiz, D.; Cleary, S.J.; Margraf, A.; Cook, L.; Schlomann, U.; Panaretou, B.; Bowser, J.L.; Karmouty-Quintana, H.; Li, J.; et al. ADAM8 signaling drives neutrophil migration and ARDS severity. JCI Insight 2022, 7, e149870. [Google Scholar] [CrossRef]
- Schlomann, U.; Wildeboer, D.; Webster, A.; Antropova, O.; Zeuschner, D.; Knight, C.G.; Docherty, A.J.P.; Lambert, M.; Skelton, L.; Jockusch, H.; et al. The metalloprotease disintegrin ADAM8. Processing by autocatalysis is required for proteolytic activity and cell adhesion. J. Biol. Chem. 2002, 277, 48210–48219. [Google Scholar] [CrossRef]
- Amour, A.; English, W.R.; Knäuper, V.; Murphy, G.; Webster, A.; Slocombe, P.M.; Docherty, A.J.P.; Becherer, J.D. The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett. 2002, 524, 154–158. [Google Scholar] [CrossRef]
- Chen, G.; Ge, D.; Zhu, B.; Shi, H.; Ma, Q. Upregulation of matrix metalloproteinase 9 (MMP9)/tissue inhibitor of metalloproteinase 1 (TIMP1) and MMP2/TIMP2 ratios may be involved in lipopolysaccharide-induced acute lung injury. J. Int. Med. Res. 2020, 48, 0300060520919592. [Google Scholar] [CrossRef]
- Ricklin, D.; Reis, E.S.; Mastellos, D.C.; Gros, P.; Lambris, J.D. Complement component C3—The “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 2016, 274, 33. [Google Scholar] [CrossRef]
- Somashekar, A.R.; Ramakrishnan, K.G.; Gowda, V. Complement Factor (C3) Level as Marker of Inflammation in Paediatric Asthma. J. Lung Health Dis. 2019, 3, 36–38. [Google Scholar] [CrossRef]
- Marc, M.M.; Korosec, P.; Kosnik, M.; Kern, I.; Flezar, M.; Suskovic, S.; Sorli, J. Complement factors c3a, c4a, and c5a in chronic obstructive pulmonary disease and asthma. Am. J. Respir. Cell Mol. Biol. 2004, 31, 216–219. [Google Scholar] [CrossRef]
- Kerr, A.R.; Paterson, G.K.; Riboldi-Tunnicliffe, A.; Mitchell, T.J. Innate Immune Defense against Pneumococcal Pneumonia Requires Pulmonary Complement Component C3. Infect. Immun. 2005, 73, 4245. [Google Scholar] [CrossRef]
- Okamoto, T.; Mathai, S.K.; Hennessy, C.E.; Hancock, L.A.; Walts, A.D.; Stefanski, A.L.; Brown, K.K.; Lynch, D.A.; Cosgrove, G.P.; Groshong, S.D.; et al. The relationship between complement C3 expression and the MUC5B genotype in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L1–L10. [Google Scholar] [CrossRef]
- Hambleton, J.; Weinstein, S.L.; Lem, L.; Defranco, A.L. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. USA 1996, 93, 2774–2778. [Google Scholar] [CrossRef]
- Fujihara, M.; Muroi, M.; Tanamoto, K.I.; Suzuki, T.; Azuma, H.; Ikeda, H. Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: Roles of the receptor complex. Pharmacol. Ther. 2003, 100, 171–194. [Google Scholar] [CrossRef]
- Deakin, A.M.; Payne, A.N.; Whittle, B.J.R.; Moncada, S. The modulation of IL-6 and TNF-α release by nitric oxide following stimulation of J774 cells with LPS and IFN-γ. Cytokine 1995, 7, 408–416. [Google Scholar] [CrossRef]
- Kossakowska, A.E.; Edwards, D.R.; Prusinkiewicz, C.; Zhang, M.C.; Guo, D.; Urbanski, S.J.; Grogan, T.; Marquez, L.A.; Janowska-Wieczorek, A. Interleukin-6 regulation of matrix metalloproteinase (MMP-2 and MMP-9) and tissue inhibitor of metalloproteinase (TIMP-1) expression in malignant non-Hodgkin’s lymphomas. Blood 1999, 94, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, S.B.; Christensen, S.L.T.; Würtz, S.T.; Lundberg, M.; Nielsen, B.S.; Vinther, L.; Knowles, M.; Gee, N.; Fredriksson, S.; Møller, S.; et al. Plasma levels of the MMP-9: TIMP-1 complex as prognostic biomarker in breast cancer: A retrospective study. BMC Cancer 2013, 13, 598. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, J.Z.; Marusich, L.R. Repeated measures correlation. Front. Psychol. 2017, 8, 456. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, M.; Feng, T.; Zhan, L.; Zhou, L.; Yu, G. Use ggbreak to Effectively Utilize Plotting Space to Deal With Large Datasets and Outliers. Front. Genet. 2021, 12, 2122. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.; Guerne, P.A. Interleukin-6 induces the synthesis of tissue inhibitor of metalloproteinases-1/erythroid potentiating activity (TIMP-1/EPA). J. Biol. Chem. 1991, 266, 2017–2020. [Google Scholar] [CrossRef]
- Zheng, X.; Xu, M.; Yao, B.; Wang, C.; Jia, Y.; Liu, Q. IL-6/STAT3 axis initiated CAFs via up-regulating TIMP-1 which was attenuated by acetylation of STAT3 induced by PCAF in HCC microenvironment. Cell. Signal. 2016, 28, 1314–1324. [Google Scholar] [CrossRef]
- Ding, X.; Cao, Y.; Xing, Y.; Ge, S.; Lin, M.; Li, J. TIMP-1 Mediates Inflammatory and Immune Response to IL-6 in Adult Orbital Xanthogranulomatous Disease. Ocul. Immunol. Inflamm. 2020, 28, 288–297. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, L.; Howard, J.; Kolhe, R.; Rojiani, A.M.; Rojiani, M.V. TIMP-1-Mediated Chemoresistance via Induction of IL-6 in NSCLC. Cancers 2019, 11, 1184. [Google Scholar] [CrossRef]
- Hassler, M.R.; Turanov, A.A.; Alterman, J.F.; Haraszti, R.A.; Coles, A.H.; Osborn, M.F.; Echeverria, D.; Nikan, M.; Salomon, W.E.; Roux, L.; et al. Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018, 46, 2185–2196. [Google Scholar] [CrossRef]
- Semizarov, D.; Frost, L.; Sarthy, A.; Kroeger, P.; Halbert, D.N.; Fesik, S.W. Specificity of short interfering RNA determined through gene expression signatures. Proc. Natl. Acad. Sci. USA 2003, 100, 6347. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, X.; Lu, Z.; Perry, D.M.; Li, Y.; Russo, S.B.; Cowart, L.A.; Hannun, Y.A.; Huang, Y. Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E853. [Google Scholar] [CrossRef]
- Jia, J.; Sun, Y.; Hu, Z.; Li, Y.; Ruan, X. Propofol inhibits the release of interleukin-6, 8 and tumor necrosis factor-α correlating with high-mobility group box 1 expression in lipopolysaccharides-stimulated RAW 264.7 cells. BMC Anesthesiol. 2017, 17, 148. [Google Scholar] [CrossRef]
- Dong, J.; Li, J.; Cui, L.; Wang, Y.; Lin, J.; Qu, Y.; Wang, H. Cortisol modulates inflammatory responses in LPS-stimulated RAW264.7 cells via the NF-κB and MAPK pathways. BMC Vet. Res. 2018, 14, 30. [Google Scholar] [CrossRef]
- Kabilova, T.O.; Shmendel, E.V.; Gladkikh, D.V.; Chernolovskaya, E.L.; Markov, O.V.; Morozova, N.G.; Maslov, M.A.; Zenkova, M.A. Targeted delivery of nucleic acids into xenograft tumors mediated by novel folate-equipped liposomes. Eur. J. Pharm. Biopharm. 2018, 123, 59–70. [Google Scholar] [CrossRef]
- Kabilova, T.; Shmendel, E.; Gladkikh, D.; Morozova, N.; Maslov, M.; Chernolovskaya, E.; Vlassov, V.; Zenkova, M. Novel PEGylated liposomes enhance immunostimulating activity of isRNA. Molecules 2018, 23, 3101. [Google Scholar] [CrossRef]
- Gvozdeva, O.V.; Gladkih, D.V.; Chernikov, I.V.; Meschaninova, M.I.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Nuclease-resistant 63-bp trimeric siRNAs simultaneously silence three different genes in tumor cells. FEBS Lett. 2018, 592, 122–129. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Hsu, A.T.; Barrett, C.D.; DeBusk, G.M.; Ellson, C.D.; Gautam, S.; Talmor, D.S.; Gallagher, D.C.; Yaffe, M.B. Kinetics and role of plasma matrix metalloproteinase-9 expression in acute lung injury and the acute respiratory distress syndrome. Shock 2015, 44, 128–136. [Google Scholar] [CrossRef]
- Setten, R.L.; Rossi, J.J.; Han, S. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Z.; Zhang, G. FBN1 promotes DLBCL cell migration by activating the Wnt/β-catenin signaling pathway and regulating TIMP1. Am. J. Transl. Res. 2020, 12, 7340. [Google Scholar]
- Tan, Y.; Li, X.; Tian, Z.; Chen, S.; Zou, J.; Lian, G.; Chen, S.; Huang, K.; Chen, Y. TIMP1 down-regulation enhances gemcitabine sensitivity and reverses chemoresistance in pancreatic cancer. Biochem. Pharmacol. 2021, 189, 114085. [Google Scholar] [CrossRef] [PubMed]
- Toricelli, M.; Melo, F.H.M.; Hunger, A.; Zanatta, D.; Strauss, B.E.; Jasiulionis, M.G. Timp1 Promotes Cell Survival by Activating the PDK1 Signaling Pathway in Melanoma. Cancers 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, K.; Gao, L.; Gao, W. TIMP1 Is A Potential Key Gene Associated With The Pathogenesis And Prognosis Of Ulcerative Colitis-Associated Colorectal Cancer. OncoTargets Ther. 2019, 12, 8895–8904. [Google Scholar] [CrossRef]
- Wang, K.; Lin, B.; Brems, J.J.; Gamelli, R.L. Hepatic apoptosis can modulate liver fibrosis through TIMP1 pathway. Apoptosis 2013, 18, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Fowell, A.J.; Collins, J.E.; Duncombe, D.R.; Pickering, J.A.; Rosenberg, W.M.C.; Benyon, R.C. Silencing tissue inhibitors of metalloproteinases (TIMPs) with short interfering RNA reveals a role for TIMP-1 in hepatic stellate cell proliferation. Biochem. Biophys. Res. Commun. 2011, 407, 277–282. [Google Scholar] [CrossRef]
- Egea, V.; Zahler, S.; Rieth, N.; Neth, P.; Popp, T.; Kehe, K.; Jochum, M.; Ries, C. Tissue inhibitor of metalloproteinase-1 (TIMP-1) regulates mesenchymal stem cells through let-7f microRNA and Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E309–E316. [Google Scholar] [CrossRef]
- Aoki, M.; Matsumoto, N.M.; Dohi, T.; Kuwahawa, H.; Akaishi, S.; Okubo, Y.; Ogawa, R.; Yamamoto, H.; Takabe, K. Direct Delivery of Apatite Nanoparticle-Encapsulated siRNA Targeting TIMP-1 for Intractable Abnormal Scars. Mol. Ther. Nucleic Acids 2020, 22, 50–61. [Google Scholar] [CrossRef]
- Ding, L.; Tang, S.; Wyatt, T.A.; Knoell, D.L.; Oupický, D. Pulmonary siRNA delivery for lung disease: Review of recent progress and challenges. J. Control. Release 2021, 330, 977–991. [Google Scholar] [CrossRef]
- Ge, C.; Yang, J.; Duan, S.; Liu, Y.; Meng, F.; Yin, L. Fluorinated α-Helical Polypeptides Synchronize Mucus Permeation and Cell Penetration toward Highly Efficient Pulmonary siRNA Delivery against Acute Lung Injury. Nano Lett. 2020, 20, 1738–1746. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Q.; Bai, J.; Tao, T.; Tang, L.; Chen, Y.; Chung, C.S.; Fallon, E.A.; Ayala, A. Blockade of endothelial, but not epithelial, cell expression of PD-L1 following severe shock attenuates the development of indirect acute lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L801–L812. [Google Scholar] [CrossRef]
- Dong, J.; Liao, W.; Tan, L.H.; Yong, A.; Peh, W.Y.; Wong, W.S.F. Gene silencing of receptor-interacting protein 2 protects against cigarette smoke-induced acute lung injury. Pharmacol. Res. 2019, 139, 560–568. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Wang, X.; Rai, A.; Groot, M.; Jin, Y. Exosome-Mediated Small RNA Delivery: A Novel Therapeutic Approach for Inflammatory Lung Responses. Mol. Ther. 2018, 26, 2119–2130. [Google Scholar] [CrossRef]
- Fu, P.; Usatyuk, P.V.; Lele, A.; Harijith, A.; Gregorio, C.C.; Garcia, J.G.N.; Salgia, R.; Natarajan, V. c-abl mediated tyrosine phosphorylation of paxillin regulates LPS-induced endothelial dysfunction and lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1025–L1038. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Roux, L.; Coles, A.H.; Turanov, A.A.; Alterman, J.F.; Echeverria, D.; Godinho, B.M.D.C.; Aronin, N.; Khvorova, A. 5′-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res. 2017, 45, 7581–7592. [Google Scholar] [CrossRef]
- Fujimoto, M.; Takagi, Y.; Aoki, T.; Hayase, M.; Marumo, T.; Gomi, M.; Nishimura, M.; Kataoka, H.; Hashimoto, N.; Nozaki, K. Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 1674–1685. [Google Scholar] [CrossRef]
- Mandel, E.R.; Uchida, C.; Nwadozi, E.; Makki, A.; Haas, T.L. Tissue Inhibitor of Metalloproteinase 1 Influences Vascular Adaptations to Chronic Alterations in Blood Flow. J. Cell. Physiol. 2017, 232, 831–841. [Google Scholar] [CrossRef]
- Takawale, A.; Zhang, P.; Patel, V.B.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63-Integrin β1 Interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef]
- Cong, M.; Liu, T.; Wang, P.; Fan, X.; Yang, A.; Bai, Y.; Peng, Z.; Wu, P.; Tong, X.; Chen, J.; et al. Antifibrotic Effects of a Recombinant Adeno-Associated Virus Carrying Small Interfering RNA Targeting TIMP-1 in Rat Liver Fibrosis. Am. J. Pathol. 2013, 182, 1607–1616. [Google Scholar] [CrossRef]
- Aoki, M.; Miyake, K.; Ogawa, R.; Dohi, T.; Akaishi, S.; Hyakusoku, H.; Shimada, T. siRNA knockdown of tissue inhibitor of metalloproteinase-1 in keloid fibroblasts leads to degradation of collagen type I. J. Investig. Dermatol. 2014, 134, 818–826. [Google Scholar] [CrossRef]
- Ma, B.; Ueda, H.; Okamoto, K.; Bando, M.; Fujimoto, S.; Okada, Y.; Kawaguchi, T.; Wada, H.; Miyamoto, H.; Shimada, M.; et al. TIMP1 promotes cell proliferation and invasion capability of right-sided colon cancers via the FAK/Akt signaling pathway. Cancer Sci. 2022, 113, 4244–4257. [Google Scholar] [CrossRef]
- Luo, C.H.; Shi, Y.; Liu, Y.Q.; Liu, Q.; Mao, M.; Luo, M.; Yang, K.D.; Wang, W.Y.; Chen, C.; Niu, Q.; et al. High levels of TIMP1 are associated with increased extracellular matrix stiffness in isocitrate dehydrogenase 1-wild type gliomas. Lab. Investig. 2022, 102, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, B.; Harant, V.; Schaten, S.; Frühschütz, M.; Spallek, R.; Höchst, B.; Stutzer, K.; Berchtold, S.; Erkan, M.; Prokopchuk, O.; et al. Pancreatic Premalignant Lesions Secrete Tissue Inhibitor of Metalloproteinases-1, Which Activates Hepatic Stellate Cells via CD63 Signaling to Create a Premetastatic Niche in the Liver. Gastroenterology 2016, 151, 1011–1024.e7. [Google Scholar] [CrossRef] [PubMed]
- Are, E.B.; Song, Y.; Stockdale, J.E.; Tupper, P.; Colijn, C. COVID-19 endgame: From pandemic to endemic? Vaccination, reopening and evolution in low- and high-vaccinated populations. J. Theor. Biol. 2023, 559, 111368. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.; Subbian, S. Critical Determinants of Cytokine Storm and Type I Interferon Response in COVID-19 Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e00299-20. [Google Scholar] [CrossRef] [PubMed]
- Domscheit, H.; Hegeman, M.A.; Carvalho, N.; Spieth, P.M. Molecular Dynamics of Lipopolysaccharide-Induced Lung Injury in Rodents. Front. Physiol. 2020, 11, 36. [Google Scholar] [CrossRef]
- Videira, M.A.; Llop, J.; Sousa, C.; Kreutzer, B.; Cossío, U.; Forbes, B.; Vieira, I.; Gil, N.; Silva-Lima, B. Pulmonary Administration: Strengthening the Value of Therapeutic Proximity. Front. Med. 2020, 7, 50. [Google Scholar] [CrossRef]
- Su, Y.; Sun, B.; Gao, X.; Dong, X.; Fu, L.; Zhang, Y.; Li, Z.; Wang, Y.; Jiang, H.; Han, B. Intranasal Delivery of Targeted Nanoparticles Loaded With miR-132 to Brain for the Treatment of Neurodegenerative Diseases. Front. Pharmacol. 2020, 11, 1165. [Google Scholar] [CrossRef]
- Fan, Y.; Yang, Z. Inhaled siRNA Formulations for Respiratory Diseases: From Basic Research to Clinical Application. Pharmaceutics 2022, 14, 1193. [Google Scholar] [CrossRef]
- Youngren-Ortiz, S.R.; Gandhi, N.S.; España-Serrano, L.; Chougule, M.B. Aerosol Delivery of siRNA to the Lungs. Part 1: Rationale for Gene Delivery Systems. KONA Powder Part. J. 2016, 33, 63–85. [Google Scholar] [CrossRef]
- Cortez-Jugo, C.; Masoumi, S.; Chan, P.P.Y.; Friend, J.; Yeo, L. Nebulization of siRNA for inhalation therapy based on a microfluidic surface acoustic wave platform. Ultrason. Sonochem. 2022, 88, 106088. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Maslov, M.A.; Kabilova, T.O.; Petukhov, I.A.; Morozova, N.G.; Serebrennikova, G.A.; Vlassov, V.V.; Zenkova, M.A. Novel cholesterol spermine conjugates provide efficient cellular delivery of plasmid DNA and small interfering RNA. J. Control. Release 2012, 160, 182–193. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Designation 1 | Sequence 2 | |
|---|---|---|
| siTIMP1_1 | S | CUUmGUUmGCUmAUCmACUmGAUmAGC |
| AS | UmAUCmAGUmGAUmAGCmAACmAAGAG | |
| siTIMP1_2 | S | GCCUmAAGGAACGGAAAUUUCG |
| AS | AAAUUUCCGUUCCUUmAGGCGG | |
| siTIMP1_2m | S | GmCmCmUmAmAmGfGmAfAfCfGmGmAmAmAmUmUmUmCmGm |
| AS | AmAfAmUmUmUfCmCmGmUmUmCmCmUfUmAfGmGmCmGmGm | |
| siSCRm | S | CmCmAmCmUmAmCfAmUfAfCfGmAmGmAmCmUmUmGmUmUm |
| AS | CmAfAmGmUmCfUmCmGmUmAmUmGmUfAmGfUmGmGmUmUm | |
| Gene | Primer/Probe | Sequence 5′–3′ |
|---|---|---|
| Tnf-α | probe | ((5,6)-FAM)-CACAGAAAGCATGATCCGCGACG–BHQ1 |
| forward | CCCTCCAGAAAAGACACCATG | |
| reverse | GCCACAAGCAGGAATGAGAAG | |
| Il6 | probe | ((5,6)-FAM)-TTGTCACCAGCATCAGTCCCAAGAA-BHQ1 |
| forward | AAACCGCTATGAAGTTCCTCTC | |
| reverse | GTGGTATCCTCTGTGAAGTCTC | |
| C3 | probe | ((5,6)-FAM)-ACACCCTGATTGGAGCTAGTGGC-BHQ1 |
| forward | GTTTATTCCTTCATTTCGCCTGG | |
| reverse | GATGGTTATCTCTTGGGTCACC | |
| Serpina3a | probe | ((5,6)-FAM)-ACTGTGGATGGTCTGTGTCAGGC-BHQ1 |
| forward | GGCTTCTATCCTCTGATTGGC | |
| reverse | CCCAGGAATATGTGCTAGTGATG | |
| Dap12 | probe | ((5,6)-FAM)-CCTTCCGCTGTCCCTTGACCTC-BHQ1 |
| forward | GGTGACTTGGTGTTGACTCTG | |
| reverse | GACCCTGAAGCTCCTGATAAG | |
| Adam8 | probe | ((5,6)-FAM)-TCATCTGATACATCTGCCAGCCGC-BHQ1 |
| forward | TATGCAACCACAAGAGGGAG | |
| reverse | ACCAAGACCACAACCACAC | |
| Timp1 | probe | ((5,6)-FAM)-ACTCACTGTTTGTGGACGGATCAGG-BHQ1 |
| forward | CTCAAAGACCTATAGTGCTGGC | |
| reverse | CAAAGTGACGGCTCTGGTAG | |
| Trem2 | probe | ((5,6)-FAM)-ATCTTGCACAAGGTCCCCTCCG-BHQ1 |
| forward | GCTTGGTCATCTCTTTTCTGC | |
| reverse | GTTGAGGGCTTGGGACAG | |
| Mmp9 | probe | ((5,6)-FAM)-TAGCGGTACAAGTATGCCTCTGCC-BHQ1 |
| forward | ACCTGAAAACCTCCAACCTC | |
| reverse | TCGAATGGCCTTTAGTGTCTG | |
| Hprt | probe | ((5,6)-ROX)-CTTGCTGGTGAAAAGGACCTCTCGAA-BHQ2 |
| forward | CCCCAAAATGGTTAAGGTTGC | |
| reverse | AACAAAGTCTGGCCTGTATCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernikov, I.V.; Staroseletz, Y.Y.; Tatarnikova, I.S.; Sen’kova, A.V.; Savin, I.A.; Markov, A.V.; Logashenko, E.B.; Chernolovskaya, E.L.; Zenkova, M.A.; Vlassov, V.V. siRNA-Mediated Timp1 Silencing Inhibited the Inflammatory Phenotype during Acute Lung Injury. Int. J. Mol. Sci. 2023, 24, 1641. https://doi.org/10.3390/ijms24021641
Chernikov IV, Staroseletz YY, Tatarnikova IS, Sen’kova AV, Savin IA, Markov AV, Logashenko EB, Chernolovskaya EL, Zenkova MA, Vlassov VV. siRNA-Mediated Timp1 Silencing Inhibited the Inflammatory Phenotype during Acute Lung Injury. International Journal of Molecular Sciences. 2023; 24(2):1641. https://doi.org/10.3390/ijms24021641
Chicago/Turabian StyleChernikov, Ivan V., Yaroslav Yu. Staroseletz, Irina S. Tatarnikova, Aleksandra V. Sen’kova, Innokenty A. Savin, Andrey V. Markov, Evgeniya B. Logashenko, Elena L. Chernolovskaya, Marina A. Zenkova, and Valentin V. Vlassov. 2023. "siRNA-Mediated Timp1 Silencing Inhibited the Inflammatory Phenotype during Acute Lung Injury" International Journal of Molecular Sciences 24, no. 2: 1641. https://doi.org/10.3390/ijms24021641
APA StyleChernikov, I. V., Staroseletz, Y. Y., Tatarnikova, I. S., Sen’kova, A. V., Savin, I. A., Markov, A. V., Logashenko, E. B., Chernolovskaya, E. L., Zenkova, M. A., & Vlassov, V. V. (2023). siRNA-Mediated Timp1 Silencing Inhibited the Inflammatory Phenotype during Acute Lung Injury. International Journal of Molecular Sciences, 24(2), 1641. https://doi.org/10.3390/ijms24021641