
Prophage-Derived Regions in Curtobacterium Genomes: Good Things, Small Packages

 , ,
, ,  , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Results
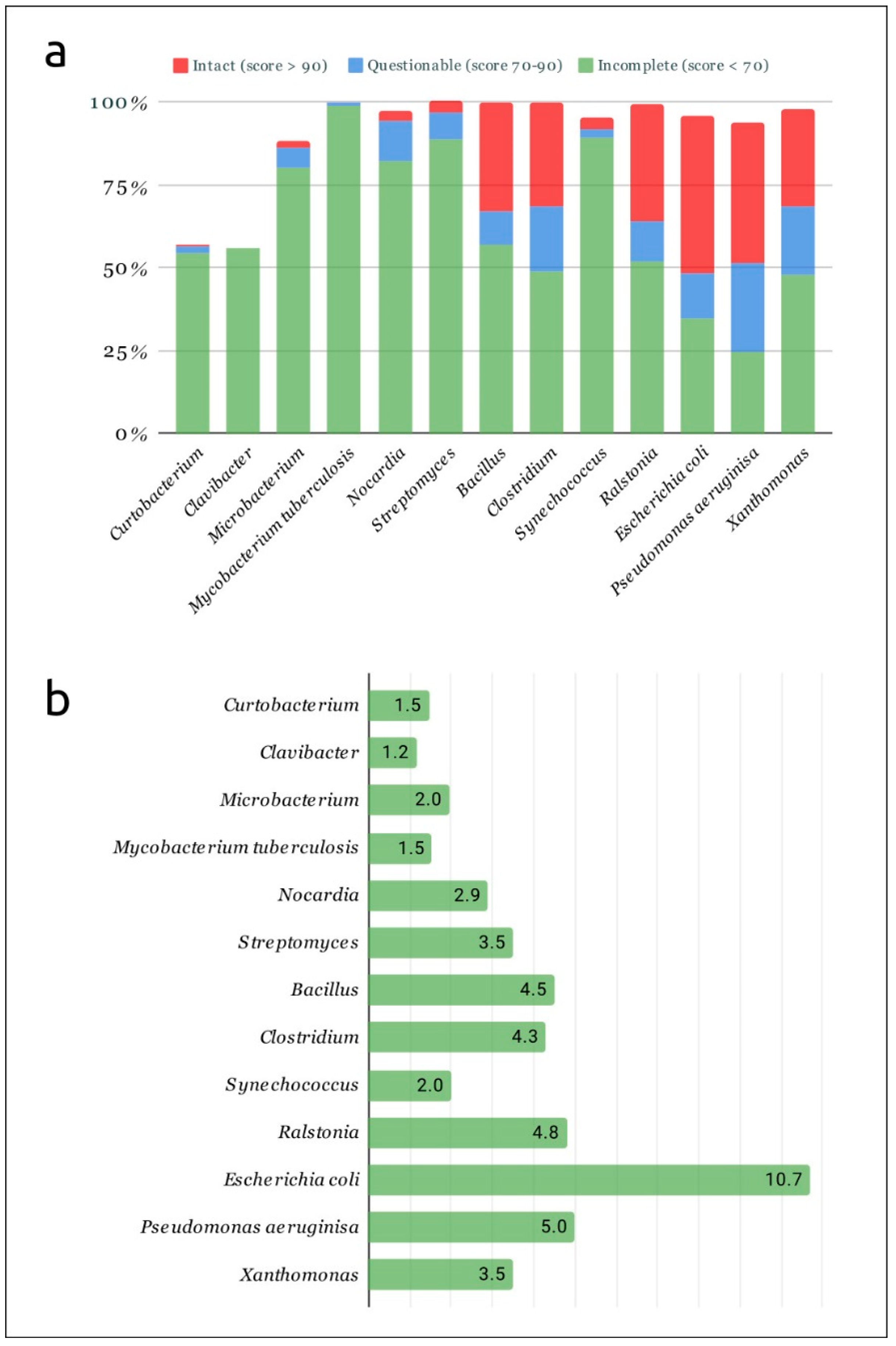
2.1. Predicted Prophages in Curtobacterial and Other Bacterial Genomes
2.2. Post-Processing of Phaster and PhiSpy Results
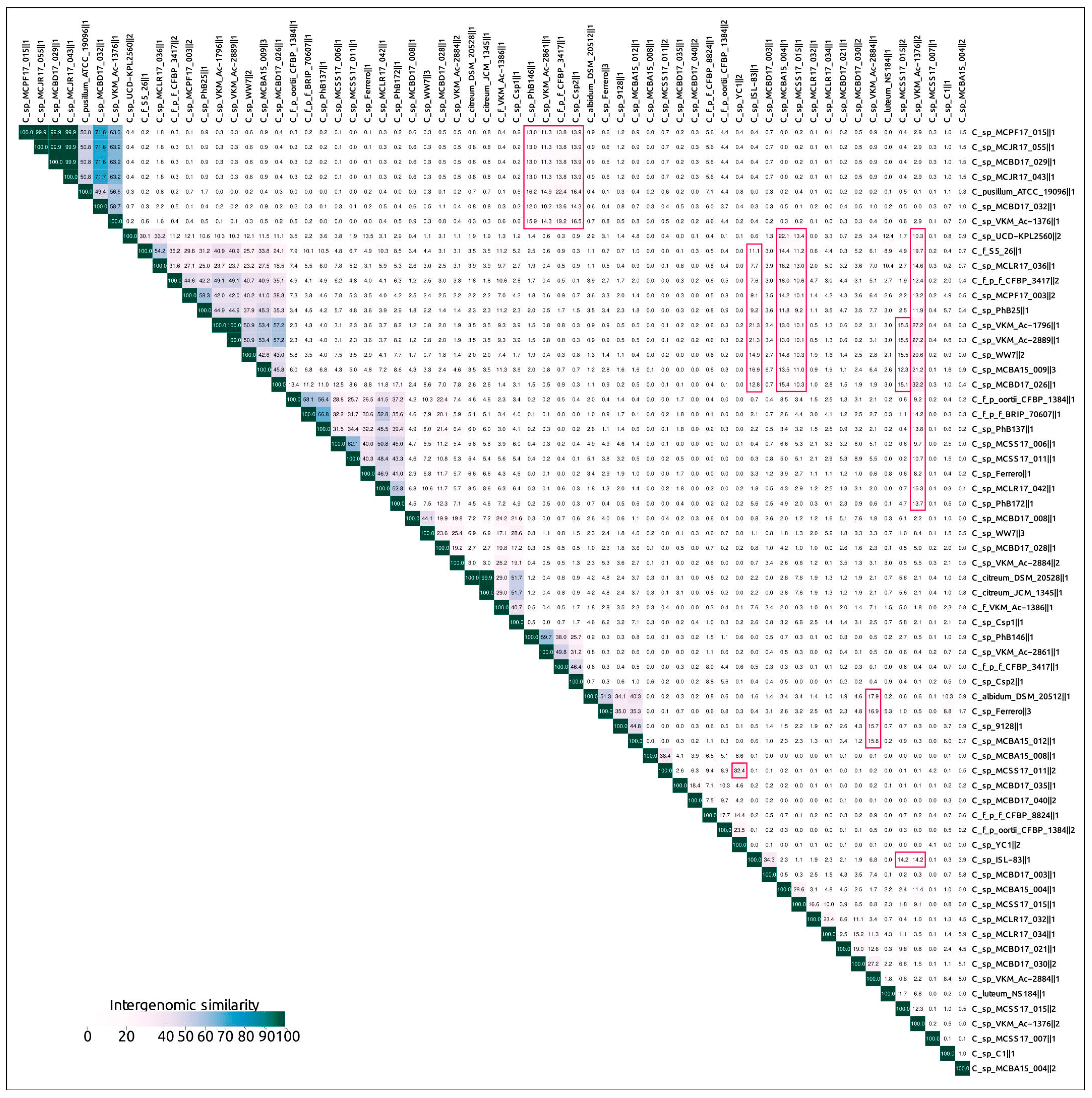
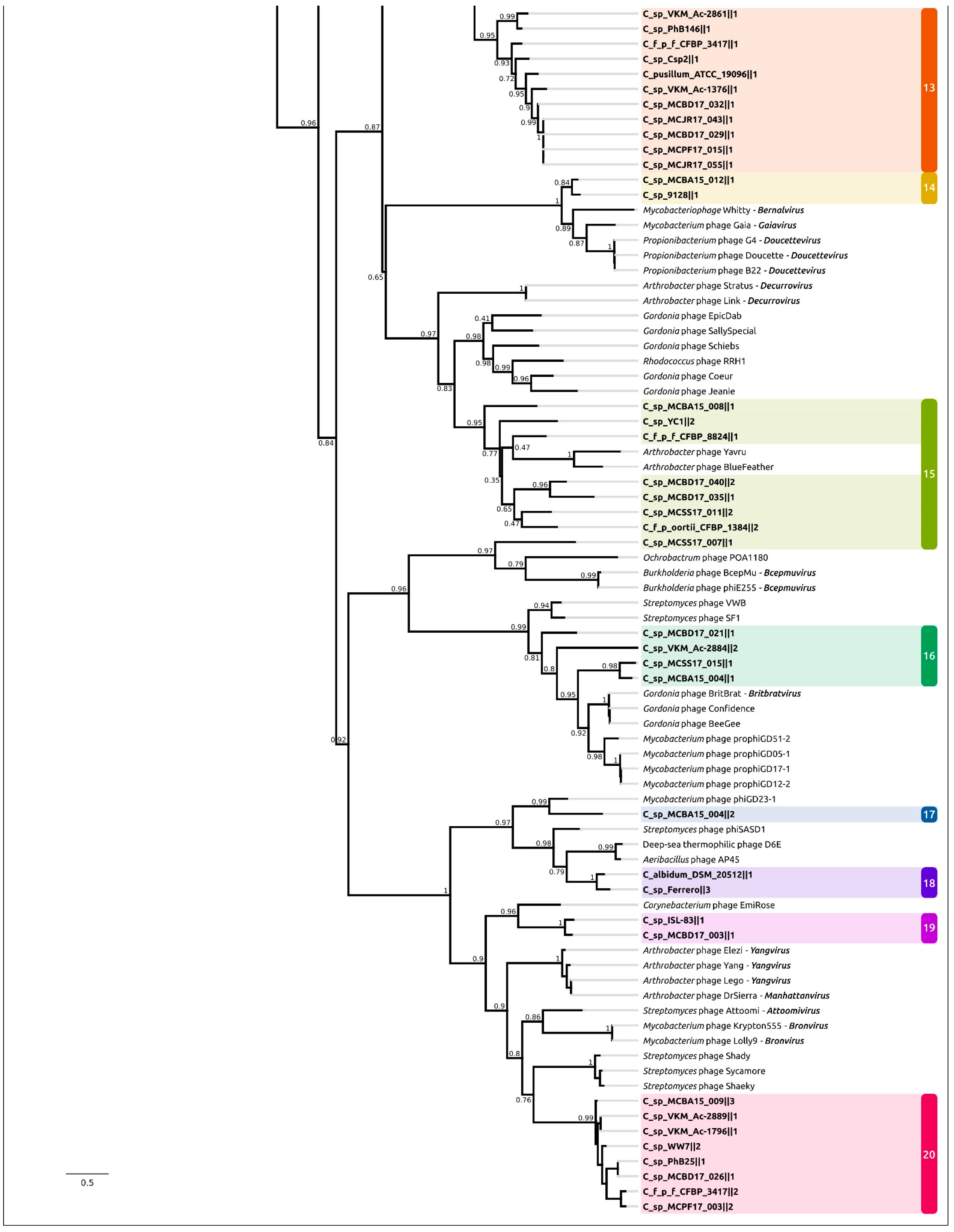
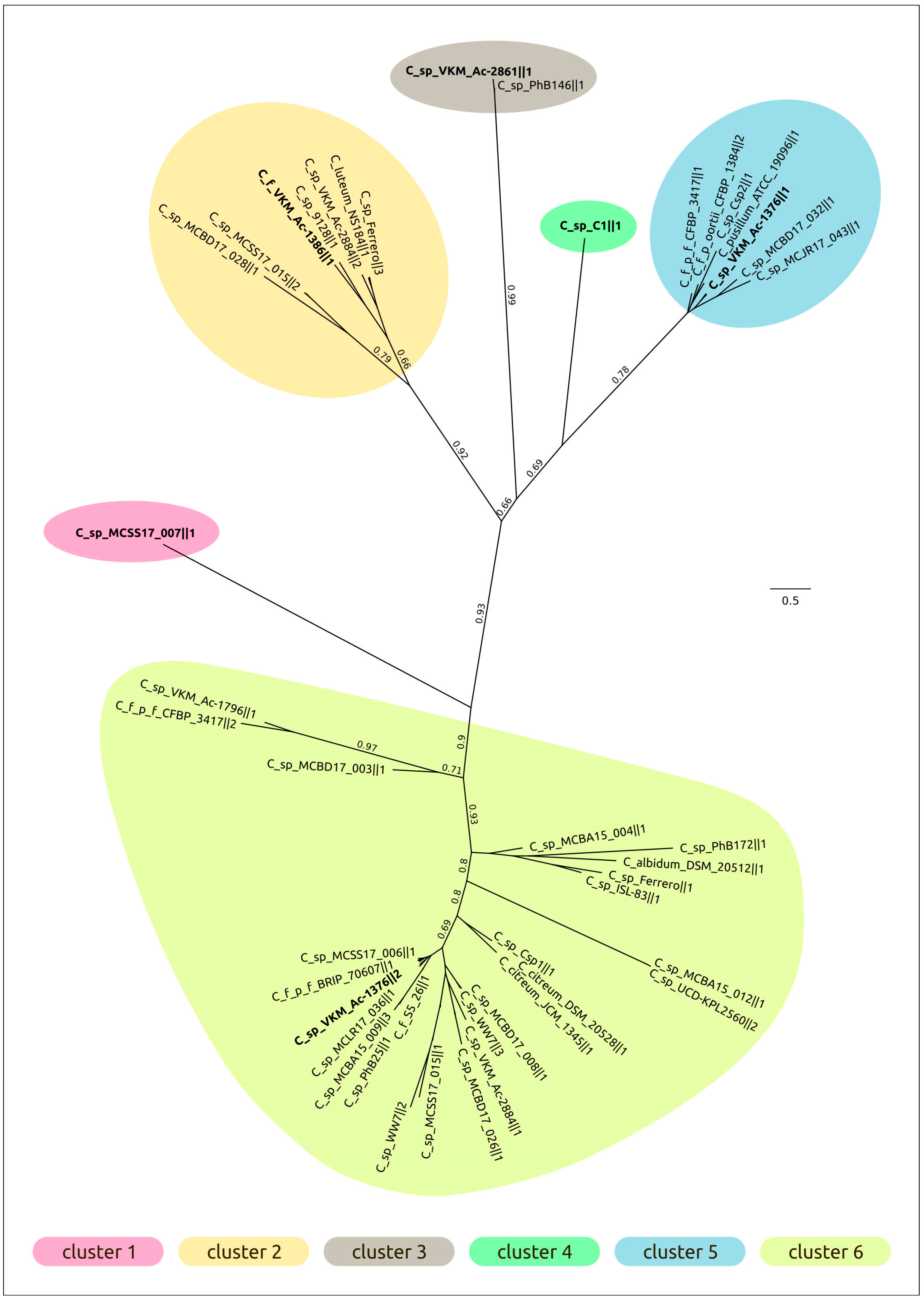
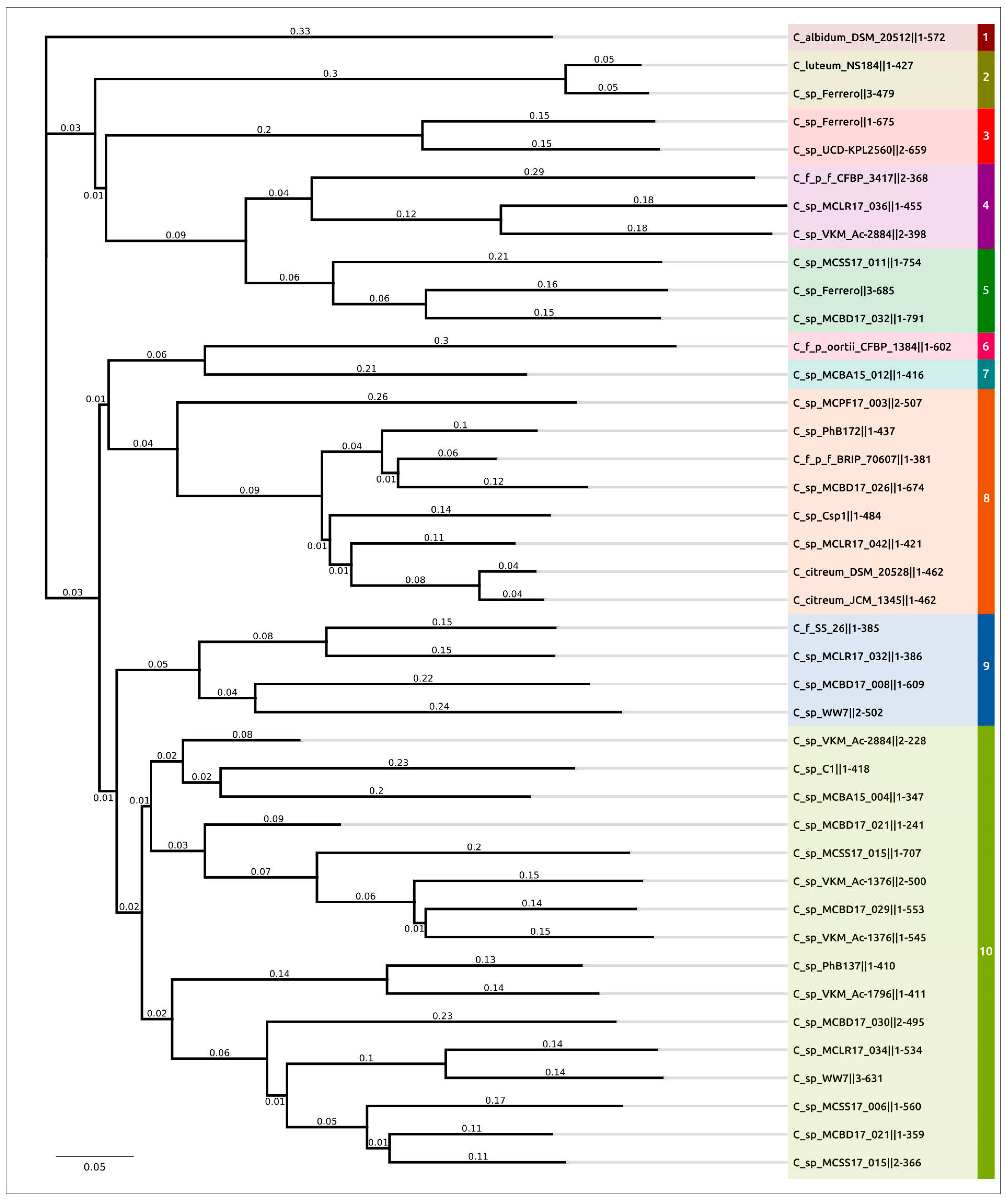
2.3. Intergenomic Comparison, Phylogenetic Analysis, Annotations and General Genomic Features
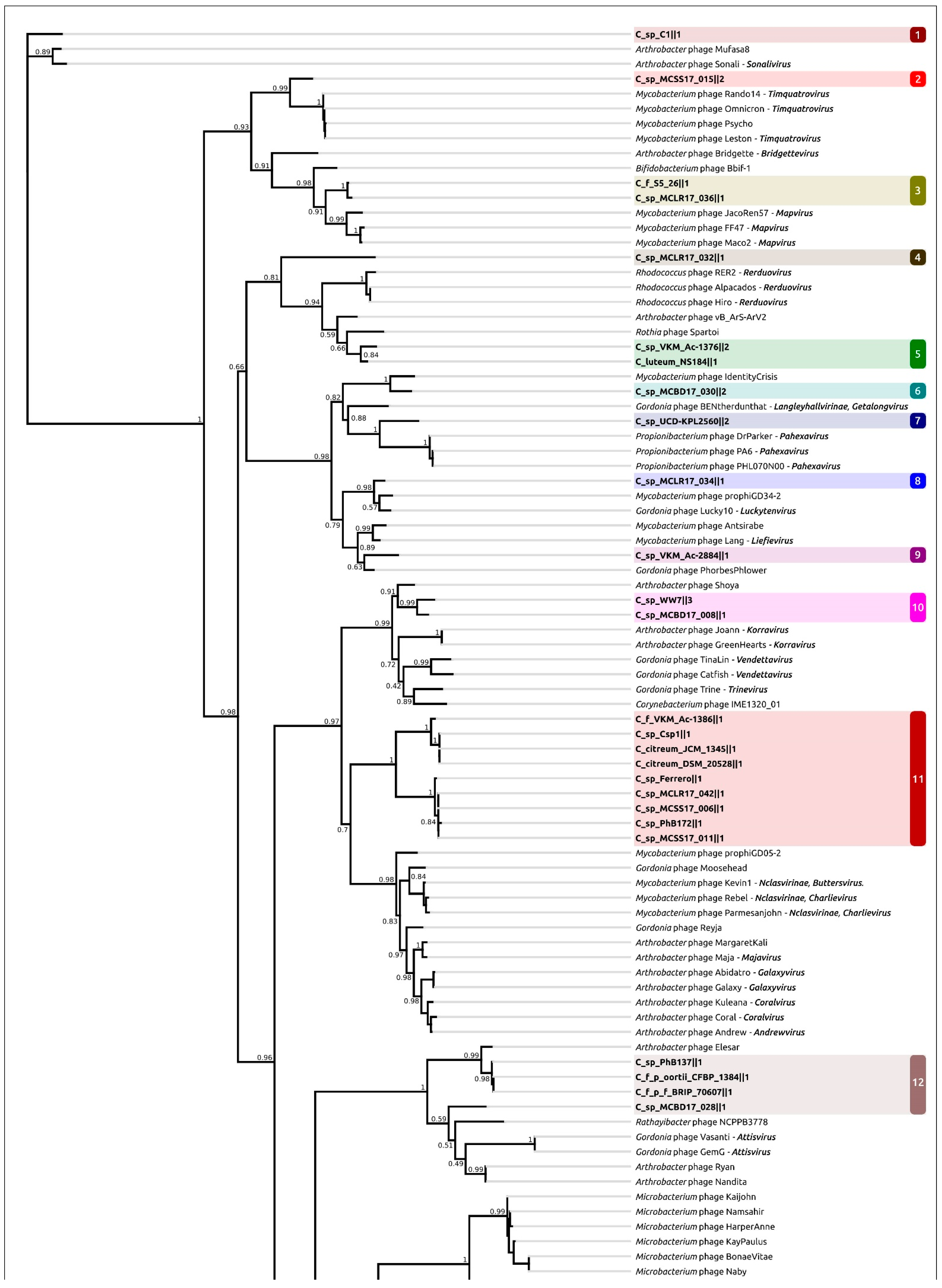
2.4. Taxonomy of Related Phages
2.5. Prophage Induction
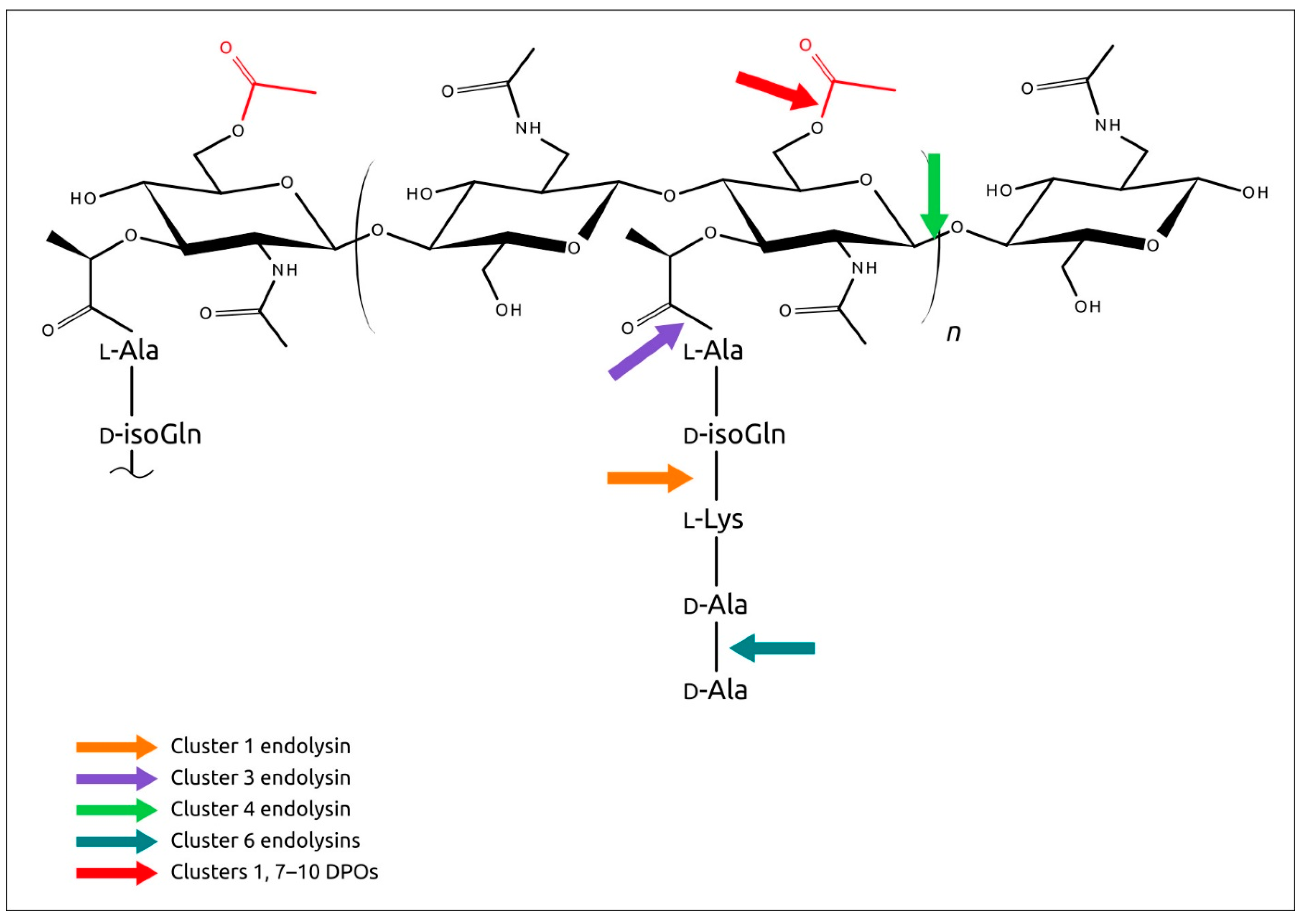
2.6. Analysis of Phage Endolysins Encoded in PDRs
2.7. Analysis of Other Glycopolymer-Degrading Enzymes Encoded in PDRs
3. Discussion
4. Materials and Methods
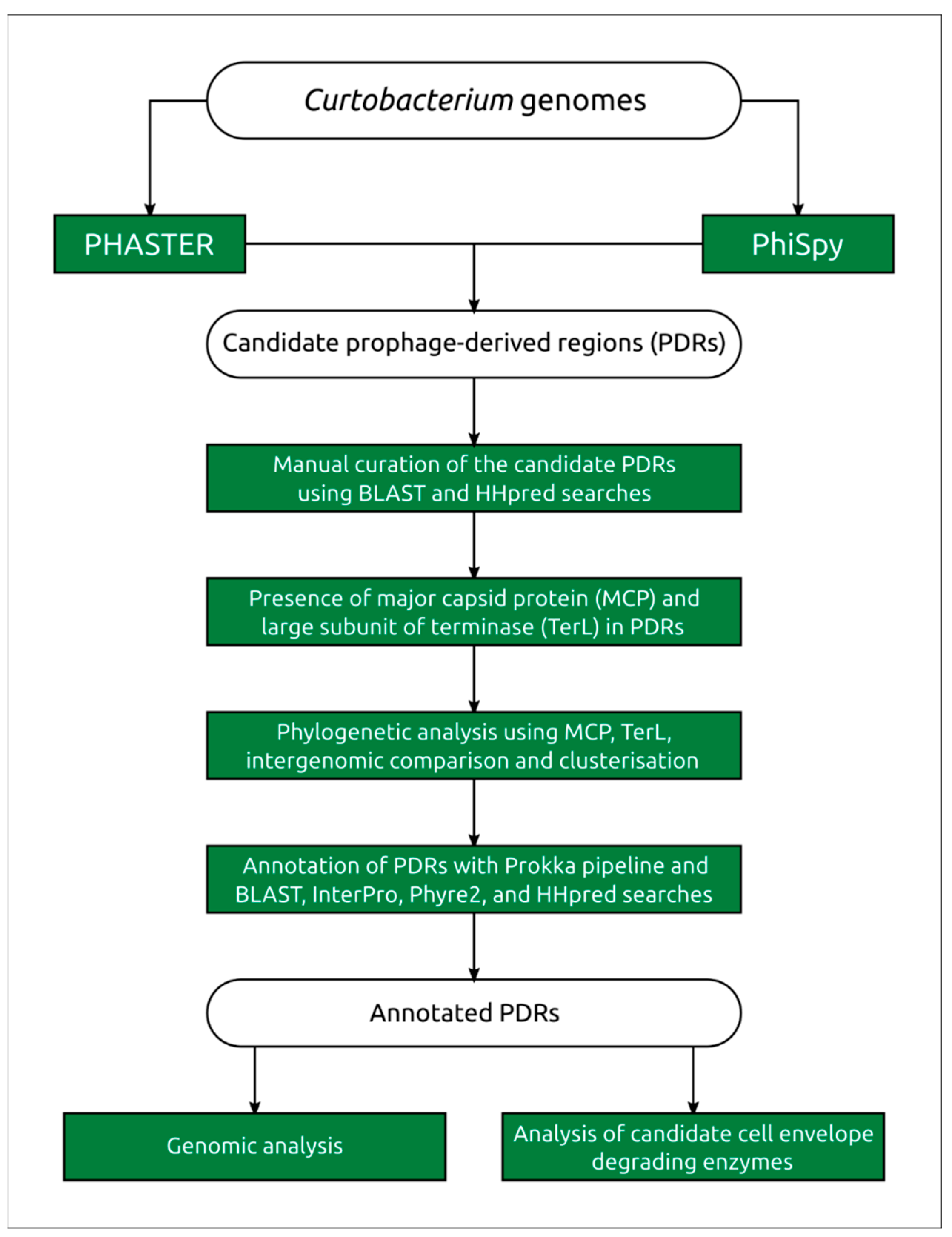
4.1. Search for Prophage-Derived Sequences
4.2. Prophage Annotation
4.3. Genomic and Phylogenetic Analysis
4.4. Prophage Induction Assay with Mitomycin C
4.5. Electron Microscopy
4.6. PCR Analysis
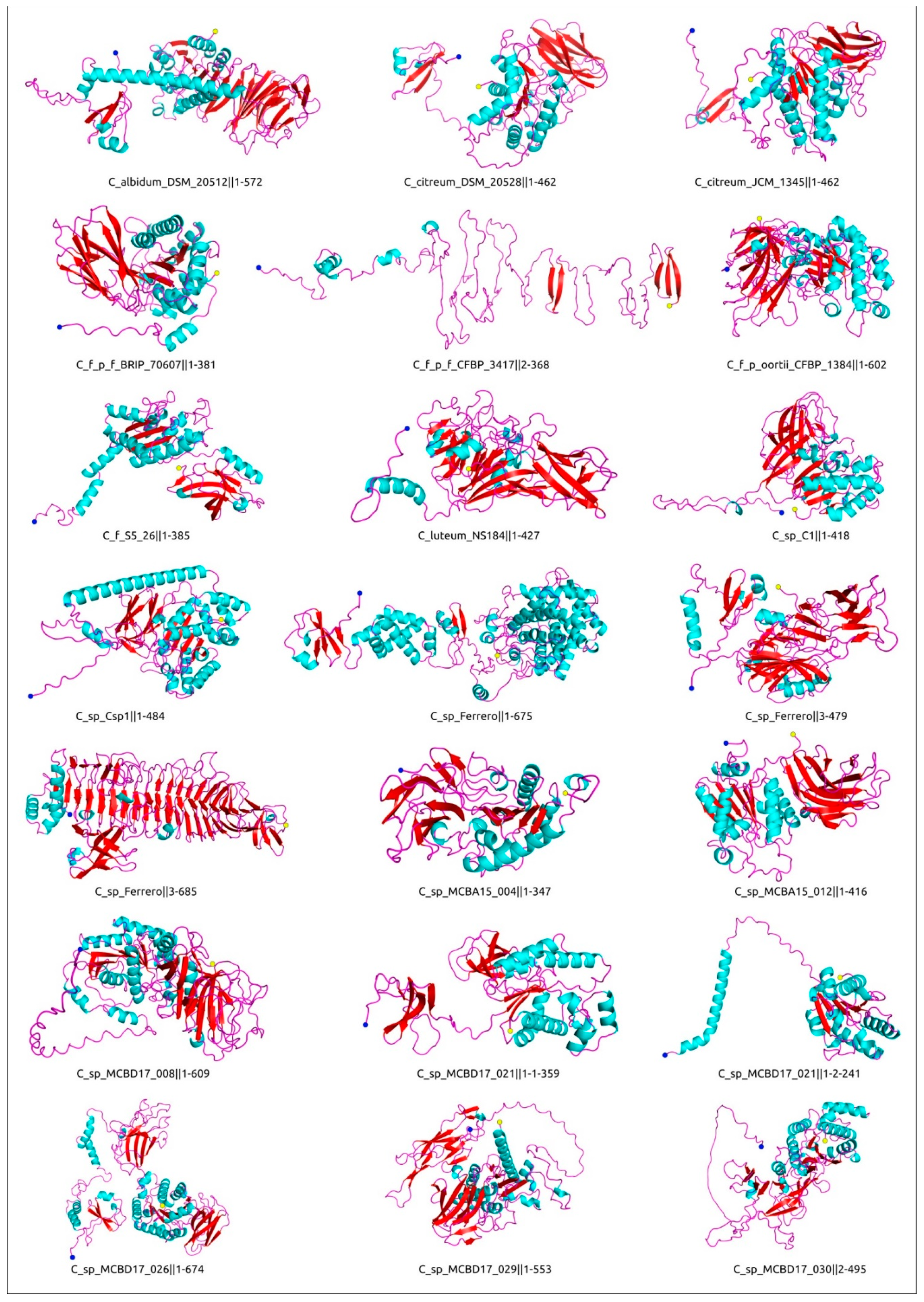
4.7. Computational Modelling and Analysis of Protein Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chase, A.B.; Arevalo, P.; Polz, M.F.; Berlemont, R.; Martiny, J.B.H. Evidence for Ecological Flexibility in the Cosmopolitan Genus Curtobacterium. Front. Microbiol. 2016, 7, 1874. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.B.; Gomez-Lunar, Z.; Lopez, A.E.; Li, J.; Allison, S.D.; Martiny, A.C.; Martiny, J.B.H. Emergence of Soil Bacterial Ecotypes along a Climate Gradient. Environ. Microbiol. 2018, 20, 4112–4126. [Google Scholar] [CrossRef]
- Undabarrena, A.; Beltrametti, F.; Claverías, F.P.; González, M.; Moore, E.R.B.; Seeger, M.; Cámara, B. Exploring the Diversity and Antimicrobial Potential of Marine Actinobacteria from the Comau Fjord in Northern Patagonia, Chile. Front. Microbiol. 2016, 7, 1135. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.J.; Bryan, N.C.; Christner, B.C.; Reisch, C.R. Draft Genome Sequences of Actinobacterial and Betaproteobacterial Strains Isolated from the Stratosphere. Microbiol. Resour. Announc. 2021, 10, e01009-21. [Google Scholar] [CrossRef] [PubMed]
- Scales, N.C.; Chase, A.B.; Finks, S.S.; Malik, A.A.; Weihe, C.; Allison, S.D.; Martiny, A.C.; Martiny, J.B.H. Differential Response of Bacterial Microdiversity to Simulated Global Change. Appl. Environ. Microbiol. 2022, 88, e0242921. [Google Scholar] [CrossRef]
- Liu, J.; Song, M.; Wei, X.; Zhang, H.; Bai, Z.; Zhuang, X. Responses of Phyllosphere Microbiome to Ozone Stress: Abundance, Community Compositions and Functions. Microorganisms 2022, 10, 680. [Google Scholar] [CrossRef]
- Hone, H.; Mann, R.; Yang, G.; Kaur, J.; Tannenbaum, I.; Li, T.; Spangenberg, G.; Sawbridge, T. Profiling, Isolation and Characterisation of Beneficial Microbes from the Seed Microbiomes of Drought Tolerant Wheat. Sci. Rep. 2021, 11, 11916. [Google Scholar] [CrossRef]
- Gerna, D.; Clara, D.; Allwardt, D.; Mitter, B.; Roach, T. Tailored Media Are Key to Unlocking the Diversity of Endophytic Bacteria in Distinct Compartments of Germinating Seeds. Microbiol. Spectr. 2022, 10, e0017222. [Google Scholar] [CrossRef]
- Bziuk, N.; Maccario, L.; Straube, B.; Wehner, G.; Sørensen, S.J.; Schikora, A.; Smalla, K. The Treasure inside Barley Seeds: Microbial Diversity and Plant Beneficial Bacteria. Environ. Microbiome 2021, 16, 20. [Google Scholar] [CrossRef]
- Schillaci, M.; Raio, A.; Sillo, F.; Zampieri, E.; Mahmood, S.; Anjum, M.; Khalid, A.; Centritto, M. Pseudomonas and Curtobacterium Strains from Olive Rhizosphere Characterized and Evaluated for Plant Growth Promoting Traits. Plants 2022, 11, 2245. [Google Scholar] [CrossRef]
- Díez-Méndez, A.; Rivas, R. Improvement of Saffron Production Using Curtobacterium Herbarum as a Bioinoculant under Greenhouse Conditions. AIMS Microbiol. 2017, 3, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A.; Salanoubat, M.; Partensky, F.; Artiguenave, F.; Axmann, I.M.; Barbe, V.; Duprat, S.; Galperin, M.Y.; Koonin, E.V.; Le Gall, F.; et al. Genome Sequence of the Cyanobacterium Prochlorococcus Marinus SS120, a Nearly Minimal Oxyphototrophic Genome. Proc. Natl. Acad. Sci. USA 2003, 100, 10020–10025. [Google Scholar] [CrossRef]
- Janisiewicz, W.J.; Buyer, J.S. Culturable Bacterial Microflora Associated with Nectarine Fruit and Their Potential for Control of Brown Rot. Can. J. Microbiol. 2010, 56, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Osdaghi, E.; Young, A.J.; Harveson, R.M. Bacterial Wilt of Dry Beans Caused by Curtobacterium Flaccumfaciens Pv. Flaccumfaciens: A New Threat from an Old Enemy. Mol. Plant Pathol. 2020, 21, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Mallick, P. A Rare Case of Curtobacterium Flaccumfaciens Infection in the Eye: A Case Report. Trop. Med. Health 2022, 50, 64. [Google Scholar] [CrossRef]
- Francis, M.J.; Doherty, R.R.; Patel, M.; Hamblin, J.F.; Ojaimi, S.; Korman, T.M. Curtobacterium Flaccumfaciens Septic Arthritis Following Puncture with a Coxspur Hawthorn Thorn. J. Clin. Microbiol. 2011, 49, 2759–2760. [Google Scholar] [CrossRef]
- Buonaurio, R.; Moretti, C.; da Silva, D.P.; Cortese, C.; Ramos, C.; Venturi, V. The Olive Knot Disease as a Model to Study the Role of Interspecies Bacterial Communities in Plant Disease. Front. Plant Sci. 2015, 6, 434. [Google Scholar] [CrossRef]
- Kamei, I.; Yoshida, T.; Enami, D.; Meguro, S. Coexisting Curtobacterium Bacterium Promotes Growth of White-Rot Fungus Stereum sp. Curr. Microbiol. 2012, 64, 173–178. [Google Scholar] [CrossRef]
- Evseev, P.; Lukianova, A.; Tarakanov, R.; Tokmakova, A.; Shneider, M.; Ignatov, A.; Miroshnikov, K. Curtobacterium spp. and Curtobacterium flaccumfaciens: Phylogeny, Genomics-Based Taxonomy, Pathogenicity, and Diagnostics. Curr. Issues Mol. Biol. 2022, 44, 889–927. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and Bacterial Genomics: What Have We Learned so Far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Canchaya, C.; Fournous, G.; Brüssow, H. The Impact of Prophages on Bacterial Chromosomes. Mol. Microbiol. 2004, 53, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Argov, T.; Azulay, G.; Pasechnek, A.; Stadnyuk, O.; Ran-Sapir, S.; Borovok, I.; Sigal, N.; Herskovits, A.A. Temperate Bacteriophages as Regulators of Host Behavior. Curr. Opin. Microbiol. 2017, 38, 81–87. [Google Scholar] [CrossRef]
- Davies, E.V.; Winstanley, C.; Fothergill, J.L.; James, C.E. The Role of Temperate Bacteriophages in Bacterial Infection. FEMS Microbiol. Lett. 2016, 363, fnw015. [Google Scholar] [CrossRef]
- Schroven, K.; Aertsen, A.; Lavigne, R. Bacteriophages as Drivers of Bacterial Virulence and Their Potential for Biotechnological Exploitation. FEMS Microbiol. Rev. 2021, 45, fuaa041. [Google Scholar] [CrossRef]
- Home-Genome-NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome (accessed on 11 November 2021).
- Arndt, D.; Marcu, A.; Liang, Y.; Wishart, D.S. PHAST, PHASTER and PHASTEST: Tools for Finding Prophage in Bacterial Genomes. Brief. Bioinform. 2017, 20, 1560–1567. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A Novel Algorithm for Finding Prophages in Bacterial Genomes That Combines Similarity- and Composition-Based Strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef] [PubMed]
- Sirén, K.; Millard, A.; Petersen, B.; Gilbert, M.T.P.; Clokie, M.R.J.; Sicheritz-Pontén, T. Rapid Discovery of Novel Prophages Using Biological Feature Engineering and Machine Learning. NAR Genom. Bioinform. 2021, 3, lqaa109. [Google Scholar] [CrossRef]
- Roach, M.J.; McNair, K.; Michalczyk, M.; Giles, S.K.; Inglis, L.K.; Pargin, E.; Barylski, J.; Roux, S.; Decewicz, P.; Edwards, R.A. Philympics 2021: Prophage Predictions Perplex Programs. F1000 Res. 2022, 10, 758. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.-Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef] [PubMed]
- Gokey, T.; Halavaty, A.S.; Minasov, G.; Anderson, W.F.; Kuhn, M.L. Structure of the Bacillus Anthracis DTDP-L-Rhamnose Biosynthetic Pathway Enzyme: DTDP-α-D-Glucose 4,6-Dehydratase, RfbB. J. Struct. Biol. 2018, 202, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, I.; Courtin, P.; Palussière, S.; Kulakauskas, S.; Bidnenko, E.; Péchoux, C.; Fenaille, F.; Penno, C.; Mahony, J.; van Sinderen, D.; et al. A Dual-Chain Assembly Pathway Generates the High Structural Diversity of Cell-Wall Polysaccharides in Lactococcus Lactis. J. Biol. Chem. 2019, 294, 17612–17625. [Google Scholar] [CrossRef] [PubMed]
- Balzaretti, S.; Taverniti, V.; Guglielmetti, S.; Fiore, W.; Minuzzo, M.; Ngo, H.N.; Ngere, J.B.; Sadiq, S.; Humphreys, P.N.; Laws, A.P. A Novel Rhamnose-Rich Hetero-Exopolysaccharide Isolated from Lactobacillus Paracasei DG Activates THP-1 Human Monocytic Cells. Appl. Environ. Microbiol. 2017, 83, e02702-16. [Google Scholar] [CrossRef] [PubMed]
- Zatyka, M.; Thomas, C.M. Control of Genes for Conjugative Transfer of Plasmids and Other Mobile Elements. FEMS Microbiol. Rev. 1998, 21, 291–319. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Czajkowski, R. May the Phage Be With You? Prophage-Like Elements in the Genomes of Soft Rot Pectobacteriaceae: Pectobacterium spp. and Dickeya spp. Front. Microbiol. 2019, 10, 138. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Juhala, R.J.; Ford, M.E.; Duda, R.L.; Youlton, A.; Hatfull, G.F.; Hendrix, R.W. Genomic Sequences of Bacteriophages HK97 and HK022: Pervasive Genetic Mosaicism in the Lambdoid Bacteriophages. J. Mol. Biol. 2000, 299, 27–51. [Google Scholar] [CrossRef]
- Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J.A.; Jacobs-Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N.R.; et al. Origins of Highly Mosaic Mycobacteriophage Genomes. Cell 2003, 113, 171–182. [Google Scholar] [CrossRef]
- Evseev, P.; Sykilinda, N.; Gorshkova, A.; Kurochkina, L.; Ziganshin, R.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage PaBG—A Jumbo Member of an Old Parasite Family. Viruses 2020, 12, 721. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Rossmann, M.G. Molecular Architecture of Tailed Double-Stranded DNA Phages. Bacteriophage 2014, 4, e28281. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Shneider, M.; Miroshnikov, K. Evolution of Phage Tail Sheath Protein. Viruses 2022, 14, 1148. [Google Scholar] [CrossRef]
- Shao, Q.; Trinh, J.T.; Zeng, L. High-Resolution Studies of Lysis–Lysogeny Decision-Making in Bacteriophage Lambda. J. Biol. Chem. 2019, 294, 3343–3349. [Google Scholar] [CrossRef] [PubMed]
- Bose, B.; Auchtung, J.M.; Lee, C.A.; Grossman, A.D. A Conserved Anti-Repressor Controls Horizontal Gene Transfer by Proteolysis. Mol. Microbiol. 2008, 70, 570–582. [Google Scholar] [CrossRef] [PubMed]
- LeBowitz, J.H.; McMacken, R. The Bacteriophage Lambda O and P Protein Initiators Promote the Replication of Single-Stranded DNA. Nucleic Acids Res. 1984, 12, 3069–3088. [Google Scholar] [CrossRef]
- Ravin, V.; Ravin, N.; Casjens, S.; Ford, M.E.; Hatfull, G.F.; Hendrix, R.W. Genomic Sequence and Analysis of the Atypical Temperate Bacteriophage N15. J. Mol. Biol. 2000, 299, 53–73. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Multiple Origins of Viral Capsid Proteins from Cellular Ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]
- Taylor, A.L. BACTERIOPHAGE-INDUCED MUTATION IN ESCHERICHIA COLI*. Proc. Natl. Acad. Sci. USA 1963, 50, 1043–1051. [Google Scholar] [CrossRef]
- Harshey, R.M. The Mu Story: How a Maverick Phage Moved the Field Forward. Mob. DNA 2012, 3, 21. [Google Scholar] [CrossRef]
- Young, R. Phage Lysis: Three Steps, Three Choices, One Outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Ramanculov, E.; Young, R. An Ancient Player Unmasked: T4 RI Encodes a t-Specific Antiholin. Mol. Microbiol. 2001, 41, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Xin, Y.; Zhang, C.; Kong, J. A Cytoplasmic Antiholin Is Embedded In Frame with the Holin in a Lactobacillus Fermentum Bacteriophage. Appl. Environ. Microbiol. 2018, 84, e02518-17. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-Encoded Virion-Associated Enzymes to Overcome the Carbohydrate Barriers during the Infection Process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ouk Kim, Y.; Park, S.-C.; Chun, J. OrthoANI: An Improved Algorithm and Software for Calculating Average Nucleotide Identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Tse, V.Y.; Liu, H.; Kapinos, A.; Torres, C.; Cayabyab, B.C.S.; Fett, S.N.; Nakashima, L.G.; Rahman, M.; Vargas, A.S.; Reddi, K.; et al. Exploring Genomic Conservation in Actinobacteriophages with Small Genomes. IEEE Access 2022, 7, 87931–87942. [Google Scholar]
- Taxonomy. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 27 April 2022).
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Dion, M.B.; Plante, P.-L.; Zufferey, E.; Shah, S.A.; Corbeil, J.; Moineau, S. Streamlining CRISPR Spacer-Based Bacterial Host Predictions to Decipher the Viral Dark Matter. Nucleic Acids Res. 2021, 49, 3127–3138. [Google Scholar] [CrossRef]
- Stern, A.; Mick, E.; Tirosh, I.; Sagy, O.; Sorek, R. CRISPR Targeting Reveals a Reservoir of Common Phages Associated with the Human Gut Microbiome. Genome Res. 2012, 22, 1985–1994. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Levy-Assaraf, M.; Voronov-Goldman, M.; Rozman Grinberg, I.; Weiserman, G.; Shimon, L.J.W.; Jindou, S.; Borovok, I.; White, B.A.; Bayer, E.A.; Lamed, R.; et al. Crystal Structure of an Uncommon Cellulosome-Related Protein Module from Ruminococcus Flavefaciens That Resembles Papain-Like Cysteine Peptidases. PLoS ONE 2013, 8, e56138. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Kerff, F.; Petrella, S.; Mercier, F.; Sauvage, E.; Herman, R.; Pennartz, A.; Zervosen, A.; Luxen, A.; Frère, J.-M.; Joris, B.; et al. Specific Structural Features of the N-Acetylmuramoyl-L-Alanine Amidase AmiD from Escherichia Coli and Mechanistic Implications for Enzymes of This Family. J. Mol. Biol. 2010, 397, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, D.; Zhou, B.; Zhen, X.; Ouyang, S. Structural and Biochemical Analyses of the Tetrameric Cell Binding Domain of Lys170 from Enterococcal Phage F170/08. Eur. Biophys. J. 2021, 50, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Lemak, S.; Tchigvintsev, A.; Petit, P.; Flick, R.; Singer, A.U.; Brown, G.; Evdokimova, E.; Egorova, O.; Gonzalez, C.F.; Chernikova, T.N.; et al. Structure and Activity of the Cold-Active and Anion-Activated Carboxyl Esterase OLEI01171 from the Oil-Degrading Marine Bacterium Oleispira Antarctica. Biochem. J. 2012, 445, 193–203. [Google Scholar] [CrossRef]
- Gorelik, A.; Illes, K.; Nagar, B. Crystal Structure of the Mannose-6-Phosphate Uncovering Enzyme. Structure 2020, 28, 426–436.e3. [Google Scholar] [CrossRef]
- Ji, S.; Dix, S.R.; Aziz, A.A.; Sedelnikova, S.E.; Baker, P.J.; Rafferty, J.B.; Bullough, P.A.; Tzokov, S.B.; Agirre, J.; Li, F.-L.; et al. The Molecular Basis of Endolytic Activity of a Multidomain Alginate Lyase from Defluviitalea Phaphyphila, a Representative of a New Lyase Family, PL39. J. Biol. Chem. 2019, 294, 18077–18091. [Google Scholar] [CrossRef]
- Leiman, P.G.; Kanamaru, S.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure and Morphogenesis of Bacteriophage T4. Cell Mol. Life Sci. 2003, 60, 2356–2370. [Google Scholar] [CrossRef]
- Garcia-Doval, C.; van Raaij, M.J. Structure of the Receptor-Binding Carboxy-Terminal Domain of Bacteriophage T7 Tail Fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Drobiazko, A.Y.; Kasimova, A.A.; Evseev, P.V.; Shneider, M.M.; Klimuk, E.I.; Shashkov, A.S.; Dmitrenok, A.S.; Chizhov, A.O.; Slukin, P.V.; Skryabin, Y.P.; et al. Capsule-Targeting Depolymerases Derived from Acinetobacter Baumannii Prophage Regions. Int. J. Mol. Sci. 2022, 23, 4971. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.L.; Taylor, E.J.; Lindsay, A.-M.; Charnock, S.J.; Turkenburg, J.P.; Dodson, E.J.; Davies, G.J.; Black, G.W. Structure of a Group A Streptococcal Phage-Encoded Virulence Factor Reveals a Catalytically Active Triple-Stranded β-Helix. Proc. Natl. Acad. Sci. USA 2005, 102, 17652–17657. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, J.; Shang, X.; Luo, H.; Zhou, Y.; Heselpoth, R.D.; Nelson, D.C.; Herzberg, O. Structure and Tailspike Glycosidase Machinery of ORF212 from E. Coli O157:H7 Phage CBA120 (TSP3). Sci. Rep. 2019, 9, 7349. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Kauppinen, S.; Larsen, S. The Crystal Structure of Rhamnogalacturonase A from Aspergillus Aculeatus: A Right-Handed Parallel Beta Helix. Structure 1997, 5, 533–544. [Google Scholar] [CrossRef]
- Kovalová, T.; Koval, T.; Benešová, E.; Vodicková, P.; Spiwok, V.; Lipovová, P.; Dohnálek, J. Active Site Complementation and Hexameric Arrangement in the GH Family 29; a Structure-Function Study of α-l-Fucosidase Isoenzyme 1 from Paenibacillus Thiaminolyticus. Glycobiology 2019, 29, 59–73. [Google Scholar] [CrossRef]
- Zhou, S.; Ko, T.-P.; Huang, J.-W.; Liu, W.; Zheng, Y.; Wu, S.; Wang, Q.; Xie, Z.; Liu, Z.; Chen, C.-C.; et al. Structure of a Gut Microbial Diltiazem-Metabolizing Enzyme Suggests Possible Substrate Binding Mode. Biochem. Biophys. Res. Commun. 2020, 527, 799–804. [Google Scholar] [CrossRef]
- Tarakanov, R.I.; Lukianova, A.A.; Evseev, P.V.; Pilik, R.I.; Tokmakova, A.D.; Kulikov, E.E.; Toshchakov, S.V.; Ignatov, A.N.; Dzhalilov, F.S.-U.; Miroshnikov, K.A. Ayka, a Novel Curtobacterium Bacteriophage, Provides Protection against Soybean Bacterial Wilt and Tan Spot. Int. J. Mol. Sci. 2022, 23, 10913. [Google Scholar] [CrossRef]
- Four Novel Curtobacterium Phages Isolated from Environmental Samples. Available online: https://www.researchsquare.com (accessed on 7 November 2022).
- Carstens, A.B.; Djurhuus, A.M.; Kot, W.; Jacobs-Sera, D.; Hatfull, G.F.; Hansen, L.H. Unlocking the Potential of 46 New Bacteriophages for Biocontrol of Dickeya Solani. Viruses 2018, 10, 621. [Google Scholar] [CrossRef]
- Miroshnikov, K.A.; Evseev, P.V.; Lukianova, A.A.; Ignatov, A.N. Tailed Lytic Bacteriophages of Soft Rot Pectobacteriaceae. Microorganisms 2021, 9, 1819. [Google Scholar] [CrossRef]
- Runtuvuori-Salmela, A.; Kunttu, H.M.T.; Laanto, E.; Almeida, G.M.F.; Mäkelä, K.; Middelboe, M.; Sundberg, L.-R. Prevalence of Genetically Similar Flavobacterium Columnare Phages across Aquaculture Environments Reveals a Strong Potential for Pathogen Control. Environ. Microbiol. 2022, 24, 2404–2420. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.C.; Burnett, S.H.; Carson, S.; Caruso, S.M.; Clase, K.; DeJong, R.J.; Dennehy, J.J.; Denver, D.R.; Dunbar, D.; Elgin, S.C.R.; et al. A Broadly Implementable Research Course in Phage Discovery and Genomics for First-Year Undergraduate Students. mBio 2014, 5, e01051-13. [Google Scholar] [CrossRef]
- Hatfull, G.F. Mycobacteriophages. Microbiol. Spectr. 2018, 6, 1029–1055. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Pires, D.P.; Costa, A.R.; Azeredo, J. Phage Therapy: Going Temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef]
- Cobb, L.H.; Park, J.; Swanson, E.A.; Beard, M.C.; McCabe, E.M.; Rourke, A.S.; Seo, K.S.; Olivier, A.K.; Priddy, L.B. CRISPR-Cas9 Modified Bacteriophage for Treatment of Staphylococcus Aureus Induced Osteomyelitis and Soft Tissue Infection. PLoS ONE 2019, 14, e0220421. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; São-José, C.; Azeredo, J. Phage-Derived Peptidoglycan Degrading Enzymes: Challenges and Future Prospects for In Vivo Therapy. Viruses 2018, 10, 292. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B. Bacteriophages and Phage-Derived Proteins--Application Approaches. Curr. Med. Chem. 2015, 22, 1757–1773. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb Database and Tools to Display CRISPRs and to Generate Dictionaries of Spacers and Repeats. BMC Bioinform. 2007, 8, 172. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An Updated Evolutionary Classification of CRISPR–Cas Systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Zaychikov, V.A.; Potekhina, N.V.; Dmitrenok, A.S.; Fan, D.; Tul’skaya, E.M.; Dorofeeva, L.V.; Evtushenko, L.I. Cell Wall Rhamnan in Actinobacteria of the Genus Curtobacterium. Microbiology 2021, 90, 343–348. [Google Scholar] [CrossRef]
- Rivera Starr, C.; Engleberg, N.C. Role of Hyaluronidase in Subcutaneous Spread and Growth of Group A Streptococcus. Infect. Immun. 2006, 74, 40–48. [Google Scholar] [CrossRef]
- Di Lallo, G.; Falconi, M.; Iacovelli, F.; Frezza, D.; D’Addabbo, P. Analysis of Four New Enterococcus Faecalis Phages and Modeling of a Hyaluronidase Catalytic Domain from Saphexavirus. PHAGE 2021, 2, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Hynes, W.L.; Walton, S.L. Hyaluronidases of Gram-Positive Bacteria. FEMS Microbiol. Lett. 2000, 183, 201–207. [Google Scholar] [CrossRef]
- Dowah, A.S.A.; Clokie, M.R.J. Review of the Nature, Diversity and Structure of Bacteriophage Receptor Binding Proteins That Target Gram-Positive Bacteria. Biophys. Rev. 2018, 10, 535–542. [Google Scholar] [CrossRef]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host Receptors for Bacteriophage Adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef]
- Rostøl, J.T.; Marraffini, L. (Ph)Ighting Phages–How Bacteria Resist Their Parasites. Cell Host Microbe 2019, 25, 184–194. [Google Scholar] [CrossRef]
- Brockhurst, M.A.; Koskella, B.; Zhang, Q.-G. Bacteria-Phage Antagonistic Coevolution and the Implications for Phage Therapy. In Bacteriophages: Biology, Technology, Therapy; Harper, D., Abedon, S., Burrowes, B., McConville, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–21. ISBN 978-3-319-40598-8. [Google Scholar]
- Sychantha, D.; Brott, A.S.; Jones, C.S.; Clarke, A.J. Mechanistic Pathways for Peptidoglycan O-Acetylation and De-O-Acetylation. Front. Microbiol. 2018, 9, 2332. [Google Scholar] [CrossRef] [PubMed]
- Russo, T.A.; Luke, N.R.; Beanan, J.M.; Olson, R.; Sauberan, S.L.; MacDonald, U.; Schultz, L.W.; Umland, T.C.; Campagnari, A.A. The K1 Capsular Polysaccharide of Acinetobacter Baumannii Strain 307-0294 Is a Major Virulence Factor. Infect. Immun. 2010, 78, 3993–4000. [Google Scholar] [CrossRef] [PubMed]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2020, 10, 2949. [Google Scholar] [CrossRef]
- Hitri, K.; Kuttel, M.M.; De Benedetto, G.; Lockyer, K.; Gao, F.; Hansal, P.; Rudd, T.R.; Beamish, E.; Rijpkema, S.; Ravenscroft, N.; et al. O-Acetylation of Typhoid Capsular Polysaccharide Confers Polysaccharide Rigidity and Immunodominance by Masking Additional Epitopes. Vaccine 2019, 37, 3866–3875. [Google Scholar] [CrossRef]
- Fiebig, T.; Cramer, J.T.; Bethe, A.; Baruch, P.; Curth, U.; Führing, J.I.; Buettner, F.F.R.; Vogel, U.; Schubert, M.; Fedorov, R.; et al. Structural and Mechanistic Basis of Capsule O-Acetylation in Neisseria Meningitidis Serogroup A. Nat. Commun. 2020, 11, 4723. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Peng, Q.; Gao, M. Characteristics of a Broad Lytic Spectrum Endolysin from Phage BtCS33 of Bacillus Thuringiensis. BMC Microbiol. 2012, 12, 297. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F.; Menzi, C.; Moreillon, P.; Resch, G. The Multidomain Architecture of a Bacteriophage Endolysin Enables Intramolecular Synergism and Regulation of Bacterial Lysis. J. Biol. Chem. 2021, 296, 100639. [Google Scholar] [CrossRef]
- Santos, S.B.; Oliveira, A.; Melo, L.D.R.; Azeredo, J. Identification of the First Endolysin Cell Binding Domain (CBD) Targeting Paenibacillus Larvae. Sci. Rep. 2019, 9, 2568. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Geneious Bioinformatics Software for Sequence Data Analysis. Available online: https://www.geneious.com/ (accessed on 11 November 2022).
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- MinCED-Mining CRISPRs in Environmental Datasets 2022. Available online: https://github.com/ctSkennerton/minced (accessed on 11 November 2022).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Edler, D.; Klein, J.; Antonelli, A.; Silvestro, D. RaxmlGUI 2.0: A Graphical Interface and Toolkit for Phylogenetic Analyses Using RAxML. Methods Ecol. Evol. 2021, 12, 373–377. [Google Scholar] [CrossRef]
- Veerassamy, S.; Smith, A.; Tillier, E.R.M. A Transition Probability Model for Amino Acid Substitutions from Blocks. J. Comput. Biol. 2003, 10, 997–1010. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- Ackermann, H.-W. Basic Phage Electron Microscopy. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions; Clokie, M.R.J., Kropinski, A.M., Eds.; Methods in Molecular BiologyTM; Humana Press: Totowa, NJ, USA, 2009; pp. 113–126. ISBN 978-1-60327-164-6. [Google Scholar]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- PyMOL Pymol.Org. Available online: https://pymol.org/2/ (accessed on 11 November 2021).
- Dong, R.; Peng, Z.; Zhang, Y.; Yang, J. MTM-Align: An Algorithm for Fast and Accurate Multiple Protein Structure Alignment. Bioinformatics 2018, 34, 1719–1725. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | PDR | Size, kb | GC, % | Bacterial Strain |
|---|---|---|---|---|
| 1 | C_sp_C1‖1 | 47.1 | 69.5 | Curtobacterium sp. C1 |
| 2 | C_sp_MCSS17_015‖2 | 31.5 | 67.4 | Curtobacterium sp. MCSS17_015 |
| 3 | C_f_S5_26‖1 | 27.2 | 68.3 | Curtobacterium flaccumfaciens S5_26 |
| C_sp_MCLR17_036‖1 | 26.6 | 68.6 | Curtobacterium sp. MCLR17_036 | |
| 4 | C_sp_MCLR17_032‖1 | 38.3 | 62.5 | Curtobacterium sp. MCLR17_032 |
| 5 | C_luteum_NS184‖1 | 22.5 | 62.3 | Curtobacterium luteum NS184 |
| C_sp_VKM_Ac-1376‖2 | 39.4 | 64.1 | Curtobacterium sp. VKM Ac-1376 | |
| 6 | C_sp_MCBD17_030‖2 | 39.4 | 65.6 | Curtobacterium sp. MCBD17_030 |
| 7 | C_sp_UCD-KPL2560‖2 | 23.2 | 66.6 | Curtobacterium sp. UCD-KPL2560 |
| 8 | C_sp_MCLR17_034‖1 | 40 | 61.3 | Curtobacterium sp. MCLR17_034 |
| 9 | C_sp_VKM_Ac-2884‖1 | 43 | 69 | Curtobacterium sp. VKM_Ac-2884 |
| 10 | C_sp_MCBD17_008‖1 | 35.7 | 69.7 | Curtobacterium sp. MCBD17_008 |
| C_sp_WW7‖3 | 34.8 | 69.7 | Curtobacterium sp. WW7 | |
| 11 | C_citreum_DSM_20528‖1 | 36 | 69.6 | Curtobacterium citreum DSM 20528 |
| C_citreum_JCM_1345‖1 | 35.9 | 69.6 | Curtobacterium citreum JCM 1345 | |
| C_f_VKM_Ac-1386‖1 | 38.3 | 69.3 | Curtobacterium flaccumfaciens VKM Ac-1386 | |
| C_sp_Csp1‖1 | 37.5 | 70.1 | Curtobacterium sp. Csp1 | |
| C_sp_Ferrero‖1 | 34.9 | 65.4 | Curtobacterium sp. Ferrero | |
| C_sp_MCLR17_042‖1 | 35.5 | 65.8 | Curtobacterium sp. MCLR17_042 | |
| C_sp_MCSS17_006‖1 | 35.6 | 65.7 | Curtobacterium sp. MCSS17_006 | |
| C_sp_MCSS17_011‖1 | 36.7 | 65.4 | Curtobacterium sp. MCSS17_011 | |
| C_sp_PhB172‖1 | 35.9 | 65.3 | Curtobacterium sp. PhB172 | |
| 12 | C_f_p_f_BRIP_70607‖1 | 34.6 | 65.8 | Curtobacterium flaccumfaciens pv. flaccumfaciens BRIP_70607 |
| C_f_p_oortii_CFBP_1384‖1 | 36.3 | 65.6 | Curtobacterium flaccumfaciens pv. oortii CFBP 1384 | |
| C_sp_MCBD17_028‖1 | 39.3 | 69.4 | Curtobacterium sp. MCBD17_028 | |
| C_sp_PhB137‖1 | 34.1 | 65.2 | Curtobacterium sp. PhB137 | |
| 13 | C_f_p_f_CFBP_3417‖1 | 17.5 | 67.8 | Curtobacterium flaccumfaciens pv. flaccumfaciens CFBP 3417 |
| C_pusillum_ATCC_19096‖1 | 17 | 68.5 | Curtobacterium pusillum ATCC 19096 | |
| C_sp_Csp2‖1 | 16.8 | 68.9 | Curtobacterium sp. Csp2‖1 | |
| C_sp_MCBD17_029‖1 | 17.2 | 69.1 | Curtobacterium sp. MCBD17_029‖1 | |
| C_sp_MCBD17_032‖1 | 17.7 | 69.3 | Curtobacterium sp. MCBD17_032‖1 | |
| C_sp_MCJR17_043‖1 | 17.2 | 69.2 | Curtobacterium sp. MCJR17_043‖1 | |
| C_sp_MCJR17_055‖1 | 17.2 | 69.2 | Curtobacterium sp. MCJR17_055‖1 | |
| C_sp_MCPF17_015‖1 | 17.2 | 69.2 | Curtobacterium sp. MCPF17_015‖1 | |
| C_sp_PhB146‖1 | 17.8 | 69.1 | Curtobacterium sp. PhB146‖1 | |
| C_sp_VKM_Ac-1376‖1 | 17.2 | 69.5 | Curtobacterium sp. VKM Ac-1376 | |
| C_sp_VKM_Ac-2861‖1 | 17.5 | 69.2 | Curtobacterium sp. VKM Ac-2861 | |
| 14 | C_sp_9128‖1 | 38.4 | 69.2 | Curtobacterium sp. 9128 |
| C_sp_MCBA15_012‖1 | 41.2 | 70.8 | Curtobacterium sp. MCBA15_012 | |
| 15 | C_f_p_f_CFBP_8824‖1 | 16.5 | 69.5 | Curtobacterium flaccumfaciens pv. flaccumfaciens CFBP 8824 |
| C_f_p_oortii_CFBP_1384‖2 | 16.5 | 68.3 | Curtobacterium flaccumfaciens pv. oortii_CFBP 1384 | |
| C_sp_MCBA15_008‖1 | 15.2 | 64.6 | Curtobacterium sp. MCBA15_008 | |
| C_sp_MCBD17_035‖1 | 16.5 | 66 | Curtobacterium sp. MCBD17_035 | |
| C_sp_MCBD17_040‖2 | 16.2 | 66.5 | Curtobacterium sp. MCBD17_040 | |
| C_sp_MCSS17_011‖2 | 16.5 | 65.3 | Curtobacterium sp. MCSS17_011 | |
| C_sp_YC1‖2 | 17.2 | 65.2 | Curtobacterium sp. YC1 | |
| 16 | C_sp_MCSS17_007‖1 | 37.9 | 62.1 | Curtobacterium sp. MCSS17_007 |
| 17 | C_sp_MCBA15_004‖1 | 46.3 | 65.4 | Curtobacterium sp. MCBA15_004 |
| C_sp_MCBD17_021‖1 | 47.1 | 67.8 | Curtobacterium sp. MCBD17_021 | |
| C_sp_MCSS17_015‖1 | 45 | 63.8 | Curtobacterium sp. MCSS17_015 | |
| C_sp_VKM_Ac-2884‖2 | 43.9 | 69.2 | Curtobacterium sp. VKM Ac-2884 | |
| 18 | C_albidum_DSM_20512‖1 | 38.2 | 70.2 | Curtobacterium albidum DSM 20512 |
| C_sp_Ferrero‖3 | 41 | 70 | Curtobacterium sp. Ferrero | |
| C_sp_MCBA15_004‖2 | 14 | 69.8 | Curtobacterium sp. MCBA15_004 | |
| 19 | C_sp_ISL-83‖1 | 36.7 | 66.9 | Curtobacterium sp. ISL-83 |
| C_sp_MCBD17_003‖1 | 24.7 | 68.5 | Curtobacterium sp. MCBD17_003 | |
| 20 | C_f_p_f_CFBP_3417‖2 | 37 | 67.3 | Curtobacterium flaccumfaciens pv. flaccumfaciens CFBP 3417 |
| C_sp_MCBA15_009‖3 | 36.8 | 67.8 | Curtobacterium sp. MCBA15_009 | |
| C_sp_MCBD17_026‖1 | 36.8 | 67 | Curtobacterium sp. MCBD17_026 | |
| C_sp_MCPF17_003‖2 | 37.3 | 68.5 | Curtobacterium sp. MCPF17_003 | |
| C_sp_PhB25‖1 | 36.6 | 69 | Curtobacterium sp. PhB25 | |
| C_sp_VKM_Ac-1796‖1 | 37.2 | 67.3 | Curtobacterium sp. VKM Ac-1796 | |
| C_sp_VKM_Ac-2889‖1 | 37.2 | 67.3 | Curtobacterium sp. VKM Ac-2889 | |
| C_sp_WW7‖2 | 36.4 | 66.6 | Curtobacterium sp. WW7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evseev, P.; Lukianova, A.; Tarakanov, R.; Tokmakova, A.; Popova, A.; Kulikov, E.; Shneider, M.; Ignatov, A.; Miroshnikov, K. Prophage-Derived Regions in Curtobacterium Genomes: Good Things, Small Packages. Int. J. Mol. Sci. 2023, 24, 1586. https://doi.org/10.3390/ijms24021586
Evseev P, Lukianova A, Tarakanov R, Tokmakova A, Popova A, Kulikov E, Shneider M, Ignatov A, Miroshnikov K. Prophage-Derived Regions in Curtobacterium Genomes: Good Things, Small Packages. International Journal of Molecular Sciences. 2023; 24(2):1586. https://doi.org/10.3390/ijms24021586
Chicago/Turabian StyleEvseev, Peter, Anna Lukianova, Rashit Tarakanov, Anna Tokmakova, Anastasia Popova, Eugene Kulikov, Mikhail Shneider, Alexander Ignatov, and Konstantin Miroshnikov. 2023. "Prophage-Derived Regions in Curtobacterium Genomes: Good Things, Small Packages" International Journal of Molecular Sciences 24, no. 2: 1586. https://doi.org/10.3390/ijms24021586
APA StyleEvseev, P., Lukianova, A., Tarakanov, R., Tokmakova, A., Popova, A., Kulikov, E., Shneider, M., Ignatov, A., & Miroshnikov, K. (2023). Prophage-Derived Regions in Curtobacterium Genomes: Good Things, Small Packages. International Journal of Molecular Sciences, 24(2), 1586. https://doi.org/10.3390/ijms24021586