

Global Impairment of Immediate-Early Genes Expression in Rett Syndrome Models and Patients Linked to Myelination Defects
 , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
2. Results
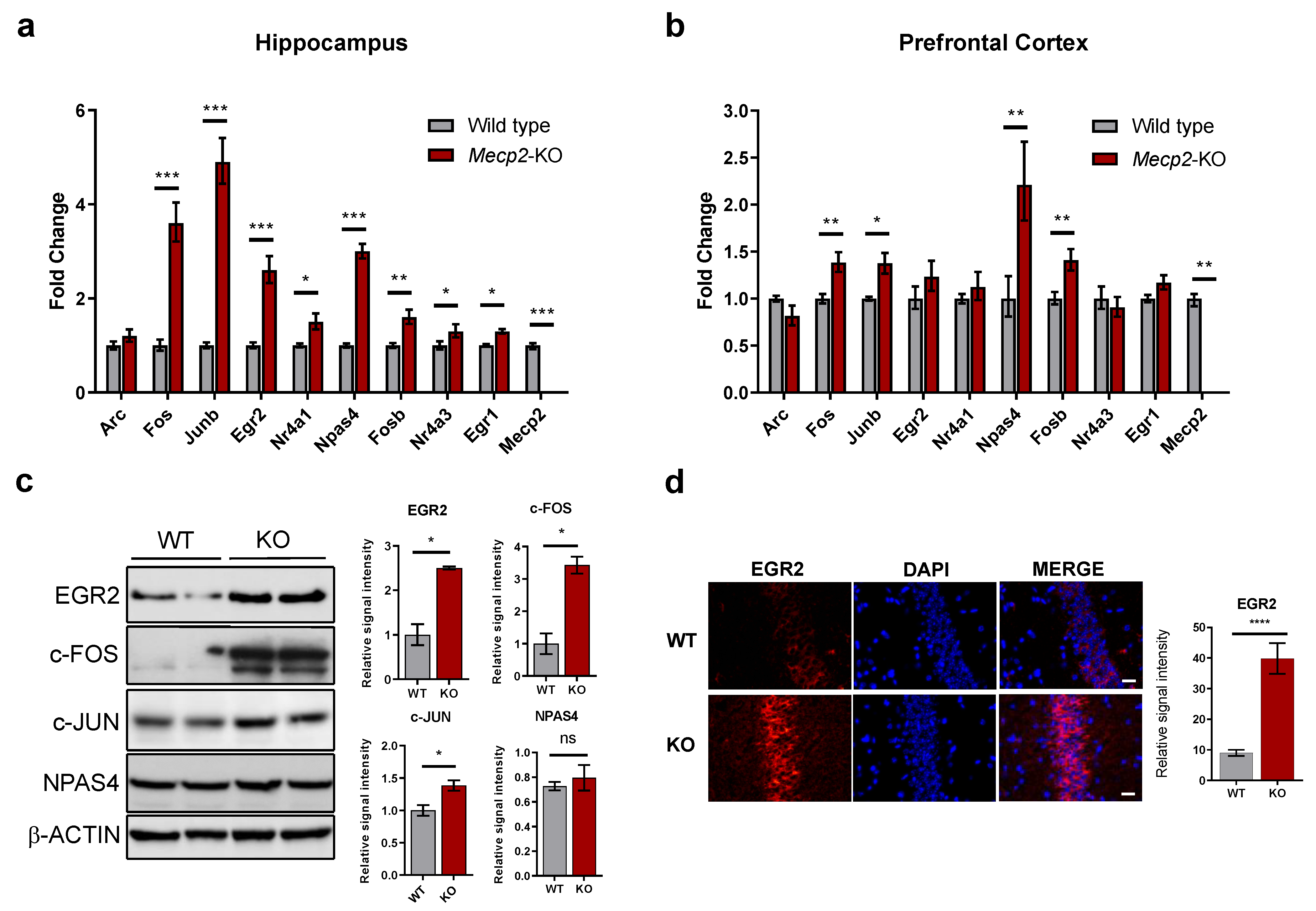
2.1. RNA-Seq Analysis Reveals Dysregulation of IEGs in a Mouse Model of RTT
2.2. MeCP2 Has a Potential Repressive Role on IEGs
2.3. IEGs Response to Activation Is Misregulated in Mecp2-KO Neurons
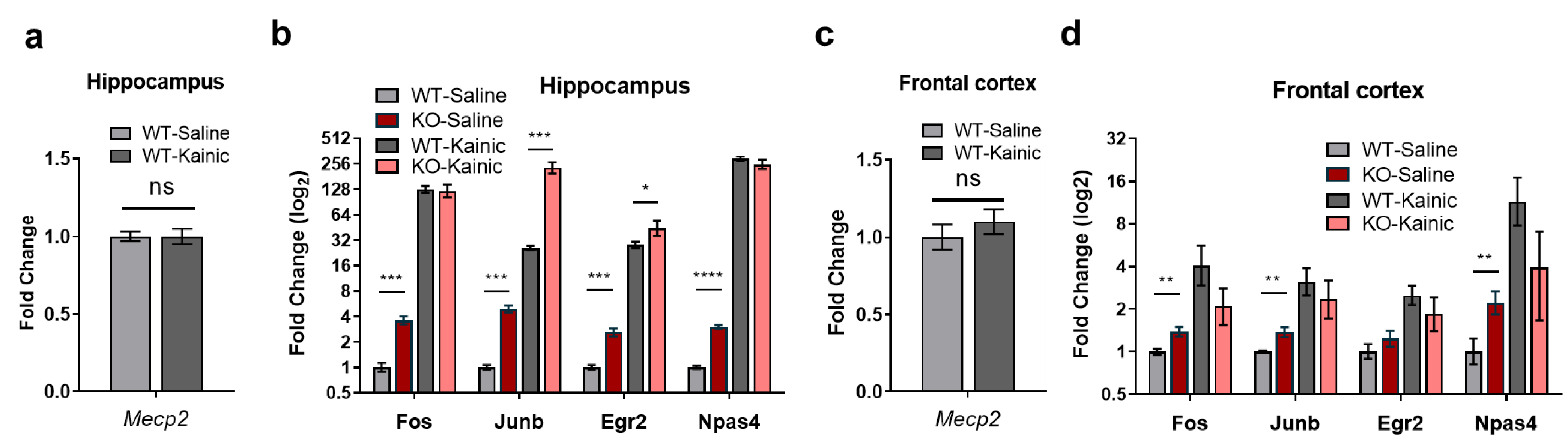
2.4. IEGs Are Over-Activated in Kainate-Stimulated Hippocampus In Vivo
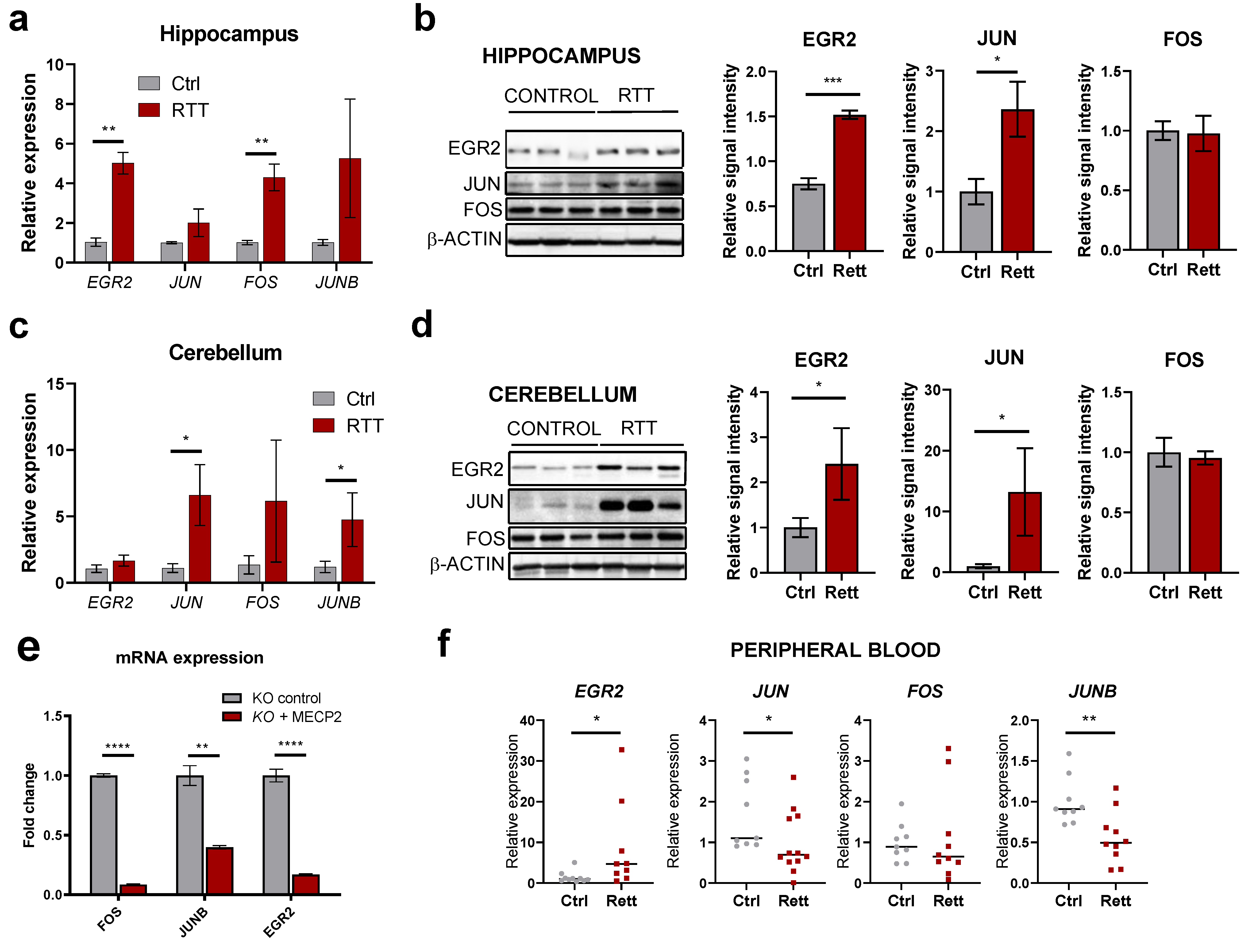
2.5. IEGs Are Dysregulated in Different Regions from Human RTT Post-Mortem Brains and Patient’s Peripheral Blood
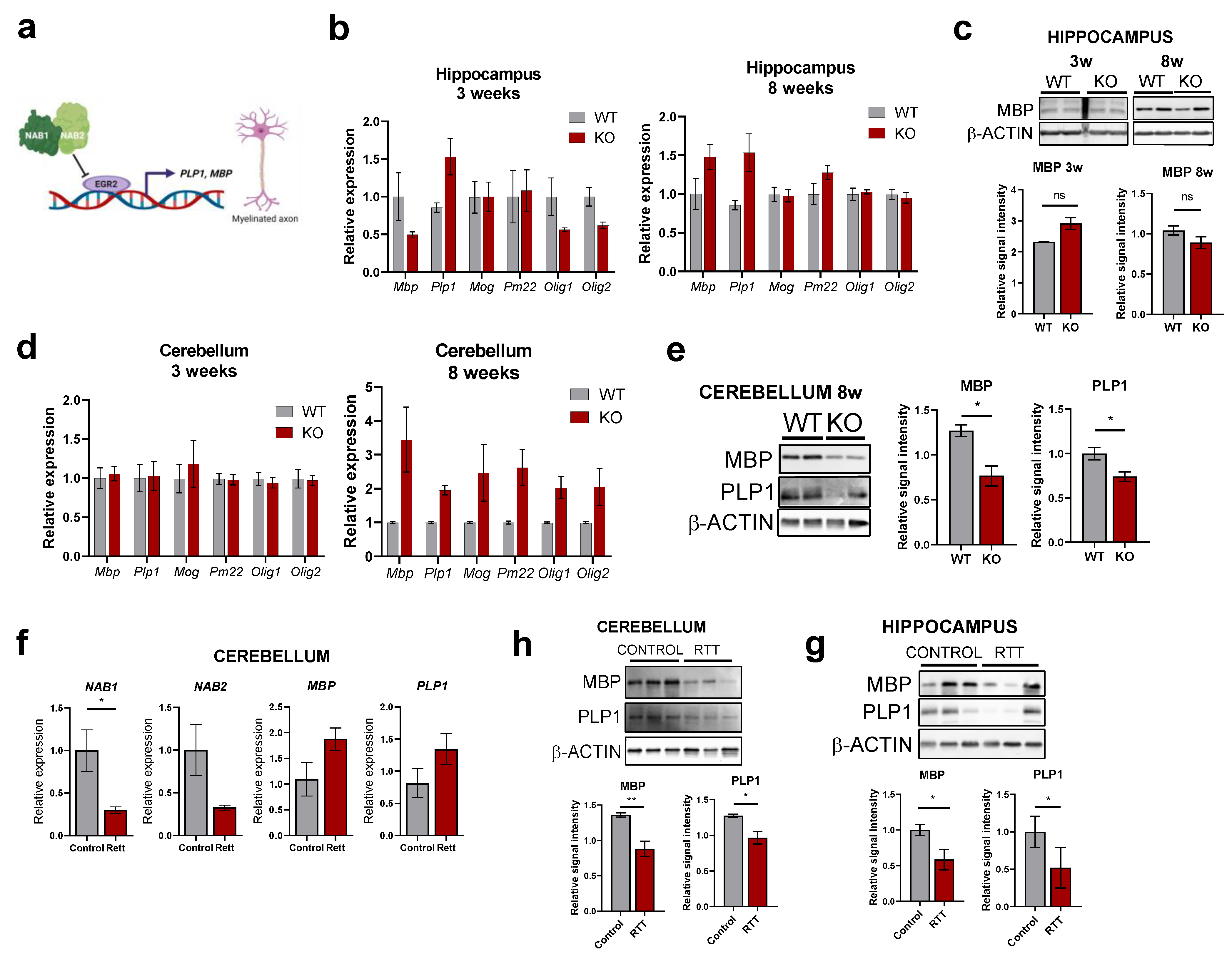
2.6. Aberrant EGR2 Expression Correlates with Altered Myelination Pathways in RTT Patients
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Human Tissues
4.3. Total RNA Isolation
4.4. RNA-Seq Analysis
4.5. Quantitative Real-Time PCR
4.6. Chromatin Immunoprecipitation
4.7. Chromatin Accessibility Assay
4.8. Primary Neuronal Hippocampal and Cortical Cultures
4.9. In Vivo Kainic Acid Administration
4.10. Western Blot
4.11. Brain Tissue Immunofluorescence
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lyst, M.; Bird, A. Rett syndrome: A complex disorder with simple roots. Nat. Rev. Genet. 2015, 16, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Flavell, S.W.; Greenberg, M.E. Signaling Mechanisms Linking Neuronal Activity to Gene Expression and Plasticity of the Nervous System. Annu. Rev. Neurosci. 2008, 31, 563–590. [Google Scholar] [CrossRef]
- Ebert, D.H.; Greenberg, M.E. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013, 493, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Smrt, R.D.; Eaves-Egenes, J.; Barkho, B.Z.; Santistevan, N.J.; Zhao, C.; Aimone, J.B.; Gage, F.H.; Zhao, X. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol. Dis. 2007, 27, 77–89. [Google Scholar] [CrossRef]
- Banerjee, A.; Castro, J.; Sur, M. Rett Syndrome: Genes, Synapses, Circuits, and Therapeutics. Front. Psychiatry 2012, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.D. Neuropathology of Rett Syndrome. J. Child Neurol. 2005, 20, 747–753. [Google Scholar] [CrossRef]
- Tropea, D.; Giacometti, E.; Wilson, N.R.; Beard, C.; McCurry, C.; Fu, D.D.; Flannery, R.; Jaenisch, R.; Sur, M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2029–2034. [Google Scholar] [CrossRef]
- Jorge-Torres, O.C.; Szczesna, K.; Roa, L.; Casal, C.; Gonzalez-Somermeyer, L.; Soler, M.; Velasco, C.D.; Segundo, P.M.-S.; Petazzi, P.; Sáez, M.A.; et al. Inhibition of Gsk3b Reduces Nfkb1 Signaling and Rescues Synaptic Activity to Improve the Rett Syndrome Phenotype in Mecp2-Knockout Mice. Cell Rep. 2018, 23, 1665–1677. [Google Scholar] [CrossRef]
- Chao, H.-T.; Zoghbi, H.Y.; Rosenmund, C. MeCP2 Controls Excitatory Synaptic Strength by Regulating Glutamatergic Synapse Number. Neuron 2007, 56, 58–65. [Google Scholar] [CrossRef]
- Zhang, Z.-W.; Zak, J.D.; Liu, H. MeCP2 Is Required for Normal Development of GABAergic Circuits in the Thalamus. J. Neurophysiol. 2010, 103, 2470–2481. [Google Scholar] [CrossRef]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to Methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Campoy, F.J.; Bird, A. MeCP2 Is a Transcriptional Repressor with Abundant Binding Sites in Genomic Chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, F.; Rossi, R.L.; Galli, F.; Gigli, C.C.; Gandaglia, A.; Kilstrup-Nielsen, C.; Landsberger, N. Rett syndrome and the urge of novel approaches to study MeCP2 functions and mechanisms of action. Neurosci. Biobehav. Rev. 2014, 46, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Ausió, J.; de Paz, A.M.; Esteller, M. MeCP2: The long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 2014, 20, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Mellén, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 Binds to 5hmC Enriched within Active Genes and Accessible Chromatin in the Nervous System. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Papin, C.; Mohideen-Abdul, K.; Le Gras, S.; Stoll, I.; Bronner, C.; Dimitrov, S.; Klaholz, B.P.; Hamiche, A. MeCP2 is a microsatellite binding protein that protects CA repeats from nucleosome invasion. Science 2021, 372, eabd5581. [Google Scholar] [CrossRef]
- Baker, S.A.; Chen, L.; Wilkins, A.D.; Yu, P.; Lichtarge, O.; Zoghbi, H.Y. An AT-Hook Domain in MeCP2 Determines the Clinical Course of Rett Syndrome and Related Disorders. Cell 2013, 152, 984–996. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef]
- Nan, X.; Ng, H.-H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef]
- Kimura, H.; Shiota, K. Methyl-CpG-binding Protein, MeCP2, Is a Target Molecule for Maintenance DNA Methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Okuno, H. Regulation and function of immediate-early genes in the brain: Beyond neuronal activity markers. Neurosci. Res. 2011, 69, 175–186. [Google Scholar] [CrossRef]
- Osenberg, S.; Karten, A.; Sun, J.; Li, J.; Charkowick, S.; Felice, C.A.; Kritzer, M.; Nguyen, M.V.C.; Yu, P.; Ballas, N. Activity-dependent aberrations in gene expression and alternative splicing in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E5363–E5372. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Pintchovski, S.A.; Chin, J.; Peebles, C.L.; Mitra, S.; Finkbeiner, S. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat. Neurosci. 2006, 9, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.D.; Rumbaugh, G.; Wu, J.; Chowdhury, S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 Mediates Homeostatic Synaptic Scaling of AMPA Receptors. Neuron 2006, 52, 475–484. [Google Scholar] [CrossRef]
- Siqueira, E.; Obiols-Guardia, A.; Jorge-Torres, O.C.; Oliveira-Mateos, C.; Soler, M.; Ramesh-Kumar, D.; Setién, F.; van Rossum, D.; Pascual-Alonso, A.; Xiol, C.; et al. Analysis of the circRNA and T-UCR populations identifies convergent pathways in mouse and human models of Rett syndrome. Mol. Ther.—Nucleic. Acids 2022, 27, 621–644. [Google Scholar] [CrossRef]
- Pérez-Cadahía, B.; Drobic, B.; Davie, J.R. Activation and function of immediate-early genes in the nervous systemThis paper is one of a selection of papers in a Special Issue entitled 31st Annual International Asilomar Chromatin and Chromosomes Conference, and has undergone the Journal’s usual peer review process. Biochem. Cell Biol. 2011, 89, 61–73. [Google Scholar] [CrossRef]
- Hawk, J.D.; Abel, T. The role of NR4A transcription factors in memory formation. Brain Res. Bull. 2011, 85, 21–29. [Google Scholar] [CrossRef]
- Lin, Y.; Bloodgood, B.L.; Hauser, J.L.; Lapan, A.D.; Koon, A.C.; Kim, T.-K.; Hu, L.S.; Malik, A.N.; Greenberg, M.E. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 2008, 455, 1198–1204. [Google Scholar] [CrossRef]
- Su, D.; Cha, Y.M.; West, A.E. Mutation of MeCP2 alters transcriptional regulation of select immediate-early genes. Epigenetics 2012, 7, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, A.; Calza, A.; Santoru, F.; Grasso, F.; Concas, A.; Sassoè-Pognetto, M.; Giustetto, M. Morphine withdrawal produces ERK-dependent and ERK-independent epigenetic marks in neurons of the nucleus accumbens and lateral septum. Neuropharmacology 2013, 70, 168–179. [Google Scholar] [CrossRef]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 Is Expressed at Near Histone-Octamer Levels and Globally Alters the Chromatin State. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Boxer, L.; Renthal, W.; Greben, A.W.; Whitwam, T.; Silberfeld, A.; Stroud, H.; Li, E.; Yang, M.G.; Kinde, B.; Griffith, E.; et al. MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol. Cell 2020, 77, 294–309.e9. [Google Scholar] [CrossRef]
- Seamon, K.B.; Padgett, W.; Daly, J.W. Forskolin: Unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. USA 1981, 78, 3363–3367. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, I.V.; Lester, H.A.; Davidson, N. Postsynaptic Mechanisms Are Essential for Forskolin-Induced Potentiation of Synaptic Transmission. J. Neurophysiol. 2006, 95, 2570–2579. [Google Scholar] [CrossRef]
- Chavez-Noriega, L.E.; Stevens, C.F. Modulation of synaptic efficacy in field CA1 of the rat hippocampus by forskolin. Brain Res. 1992, 574, 85–92. [Google Scholar] [CrossRef]
- Honkaniemi, J.; Kononen, J.; Kainu, T.; Pyykönen, I.; Pelto-Huikko, M. Induction of multiple immediate early genes in rat hypothalamic paraventricular nucleus after stress. Mol. Brain Res. 1994, 25, 234–241. [Google Scholar] [CrossRef]
- Honkaniemi, J.; Sagar, S.M.; Pyykönen, I.; Hicks, K.J.; Sharp, F.R. Focal brain injury induces multiple immediate early genes encoding zinc finger transcription factors. Mol. Brain Res. 1995, 28, 157–163. [Google Scholar] [CrossRef]
- Näkki, R.; Sharp, F.R.; Sagar, S.M.; Honkaniemi, J. Effects of phencyclidine on immediate early gene expression in the brain. J. Neurosci. Res. 1996, 45, 13–27. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Cossart, R. Kainate, a double agent that generates seizures: Two decades of progress. Trends Neurosci. 2000, 23, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Sperk, G. Kainic acid seizures in the rat. Prog. Neurobiol. 1994, 42, 1–32. [Google Scholar] [CrossRef]
- Liu, W.; Feldman, J.D.; Machado, H.B.; Vician, L.J.; Herschman, H.R. Expression of depolarization-induced immediate early gene proteins in PC12 cells. J. Neurosci. Res. 2003, 72, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Myouzen, K.; Kochi, Y.; Shimane, K.; Fujio, K.; Okamura, T.; Okada, Y.; Suzuki, A.; Atsumi, T.; Ito, S.; Takada, K.; et al. Regulatory polymorphisms in EGR2 are associated with susceptibility to systemic lupus erythematosus. Hum. Mol. Genet. 2010, 19, 2313–2320. [Google Scholar] [CrossRef]
- Cronk, J.C.; Derecki, N.C.; Ji, E.; Xu, Y.; Lampano, A.E.; Smirnov, I.; Baker, W.; Norris, G.T.; Marin, I.; Coddington, N.; et al. Methyl-CpG Binding Protein 2 Regulates Microglia and Macrophage Gene Expression in Response to Inflammatory Stimuli. Immunity 2015, 42, 679–691. [Google Scholar] [CrossRef]
- O’Driscoll, C.M.; Lima, M.P.; Kaufmann, W.E.; Bressler, J.P. Methyl CpG binding protein 2 deficiency enhances expression of inflammatory cytokines by sustaining NF-κB signaling in myeloid derived cells. J. Neuroimmunol. 2015, 283, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, S.; De Felice, C.; Signorini, C.; Zollo, G.; Cortelazzo, A.; Durand, T.; Galano, J.-M.; Guerranti, R.; Rossi, M.; Ciccoli, L.; et al. Cytokine Dysregulation in MECP2- and CDKL5-Related Rett Syndrome: Relationships with Aberrant Redox Homeostasis, Inflammation, and ω-3 PUFAs. Oxidative Med. Cell. Longev. 2015, 2015, 1–18. [Google Scholar] [CrossRef]
- Nguyen, M.V.C.; Felice, C.A.; Du, F.; Covey, M.V.; Robinson, J.K.; Mandel, G.; Ballas, N. Oligodendrocyte Lineage Cells Contribute Unique Features to Rett Syndrome Neuropathology. J. Neurosci. 2013, 33, 18764–18774. [Google Scholar] [CrossRef]
- Vora, P.; Mina, R.; Namaka, M.; Frost, E.E. A novel transcriptional regulator of myelin gene expression: Implications for neurodevelopmental disorders. Neuroreport 2010, 21, 917–921. [Google Scholar] [CrossRef]
- Svaren, J.; Meijer, D. The molecular machinery of myelin gene transcription in Schwann cells. Glia 2008, 56, 1541–1551. [Google Scholar] [CrossRef]
- Le, N.; Nagarajan, R.; Wang, J.Y.T.; Svaren, J.; LaPash, C.; Araki, T.; Schmidt, R.E.; Milbrandt, J. Nab proteins are essential for peripheral nervous system myelination. Nat. Neurosci. 2005, 8, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Sanfeliu, A.; Kaufmann, W.E.; Gill, M.; Guasoni, P.; Tropea, D. Transcriptomic Studies in Mouse Models of Rett Syndrome: A Review. Neuroscience 2019, 413, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Krishnaraj, R.; Haase, F.; Coorey, B.; Luca, E.J.; Wong, I.; Boyling, A.; Ellaway, C.; Christodoulou, J.; Gold, W.A. Genome-wide transcriptomic and proteomic studies of Rett syndrome mouse models identify common signaling pathways and cellular functions as potential therapeutic targets. Hum. Mutat. 2019, 40, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Kron, M.; Howell, C.J.; Adams, I.T.; Ransbottom, M.; Christian, D.; Ogier, M.; Katz, D.M. Brain Activity Mapping in Mecp2 Mutant Mice Reveals Functional Deficits in Forebrain Circuits, Including Key Nodes in the Default Mode Network, that are Reversed with Ketamine Treatment. J. Neurosci. 2012, 32, 13860–13872. [Google Scholar] [CrossRef] [PubMed]
- Swanberg, S.E.; Nagarajan, R.P.; Peddada, S.; Yasui, D.H.; LaSalle, J.M. Reciprocal co-regulation of EGR2 and MECP2 is disrupted in Rett syndrome and autism. Hum. Mol. Genet. 2008, 18, 525–534. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Lovén, J.; Kwok, S.-M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global Transcriptional and Translational Repression in Human-Embryonic-Stem-Cell-Derived Rett Syndrome Neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef]
- Ebert, D.H.; Gabel, H.W.; Robinson, N.D.; Kastan, N.R.; Hu, L.S.; Cohen, S.; Navarro, A.J.; Lyst, M.J.; Ekiert, R.; Bird, A.P.; et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 2013, 499, 341–345. [Google Scholar] [CrossRef]
- Loebrich, S.; Nedivi, E. The Function of Activity-Regulated Genes in the Nervous System. Physiol. Rev. 2009, 89, 1079–1103. [Google Scholar] [CrossRef]
- Zhang, L.; He, J.; Jugloff, D.G.M.; Eubanks, J.H. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus 2007, 18, 294–309. [Google Scholar] [CrossRef]
- DeSteno, D.; Schmauss, C. Induction of early growth response gene 2 expression in the forebrain of mice performing an attention-set-shifting task. Neuroscience 2008, 152, 417–428. [Google Scholar] [CrossRef]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef]
- Young, M.R.; Colburn, N.H. Fra-1 a target for cancer prevention or intervention. Gene 2006, 379, 1–11. [Google Scholar] [CrossRef]
- Nedivi, E.; Hevroni, D.; Naot, D.; Israeli, D.; Citri, Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 1993, 363, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Singh, J.; Pillai, P.P.; Frost, E.E. Involvement of MeCP2 in Regulation of Myelin-Related Gene Expression in Cultured Rat Oligodendrocytes. J. Mol. Neurosci. 2015, 57, 176–184. [Google Scholar] [CrossRef]
- Bahey, N.G.; Gadalla, K.K.; McGonigal, R.; Bailey, M.E.; Edgar, J.M.; Cobb, S.R. Reduced axonal diameter of peripheral nerve fibers in a mouse model of Rett syndrome. Neuroscience 2017, 358, 261–268. [Google Scholar] [CrossRef]
- Jin, X.-R.; Chen, X.-S.; Xiao, L. MeCP2 Deficiency in Neuroglia: New Progress in the Pathogenesis of Rett Syndrome. Front. Mol. Neurosci. 2017, 10, 316. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Petazzi, P.; Sandoval, J.; Szczesna, K.; Jorge, O.C.; Roa, L.; Sayols, S.; Gomez, A.; Huertas, D.; Esteller, M. Dysregulation of the long non-coding RNA transcriptome in a Rett syndrome mouse model. RNA Biol. 2013, 10, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, G.; Lee, S.-H.; Singh, D.; Yuan, Y.; Ng, Y.-G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Szczesna, K.; de la Caridad, O.; Petazzi, P.; Soler, M.; Roa, L.; Saez, M.A.; Fourcade, S.; Pujol, A.; Artuch-Iriberri, R.; Molero-Luis, M.; et al. Improvement of the rett syndrome phenotype in a mecp2 mouse model upon treatment with levodopa and a dopa-decarboxylase inhibitor. Neuropsychopharmacology 2014, 39, 2846–2856. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Frontal Cortex | Hippocampus | |||||
|---|---|---|---|---|---|---|
| Gene Symbol | Gene Name | Transcript ID | Fold Change (KO vs. WT) | Adjusted p-Value | Fold Change (KO vs. WT) | Adjusted p-Value |
| Arc | Activity-regulated cytoskeleton-associated protein | uc007wfo.1 | 3.71 | 0 | 2.24 | 2.36 × 10−83 |
| Fos | Proto-oncogene c-Fos | uc007oha.1 | 2.85 | 5.46 × 10−36 | 2.70 | 2.97 × 10−18 |
| Junb | Transcription factor jun-B | uc012ghn.1 | 2.61 | 2.33 × 10−17 | 1.81 | 8.47 × 10−10 |
| Egr2 | Early growth response protein 2 | uc007flx.1 | 2.39 | 5.03 × 10−7 | 4.29 | 2.76 × 10−9 |
| Npas4 | Neuronal PAS domain-containing protein 4 | uc008gbu.2 | 2.30 | 4.51 × 10−14 | 3.60 | 2.64 × 10−21 |
| Nr4a1 | Nuclear receptor subfamily 4 group A member 1 | uc007xsv.2 | 2.29 | 3.78 × 10−63 | 1.84 | 3.96 × 10−25 |
| Fosb | Protein fosB | uc009flk.1 | 1.96 | 4.59 × 10−8 | 1.90 | 2.59 × 10−5 |
| Egr1 | Early growth response protein 1 | uc008elt.1 | 1.70 | 6.69 × 10−95 | 1.60 | 8.24 × 10−40 |
| Nr4a3 | Nuclear receptor subfamily 4 group A member 3 | uc008suw.1 | 1.39 | 4.29 × 10−4 | n.d | n.d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petazzi, P.; Jorge-Torres, O.C.; Gomez, A.; Scognamiglio, I.; Serra-Musach, J.; Merkel, A.; Grases, D.; Xiol, C.; O’Callaghan, M.; Armstrong, J.; et al. Global Impairment of Immediate-Early Genes Expression in Rett Syndrome Models and Patients Linked to Myelination Defects. Int. J. Mol. Sci. 2023, 24, 1453. https://doi.org/10.3390/ijms24021453
Petazzi P, Jorge-Torres OC, Gomez A, Scognamiglio I, Serra-Musach J, Merkel A, Grases D, Xiol C, O’Callaghan M, Armstrong J, et al. Global Impairment of Immediate-Early Genes Expression in Rett Syndrome Models and Patients Linked to Myelination Defects. International Journal of Molecular Sciences. 2023; 24(2):1453. https://doi.org/10.3390/ijms24021453
Chicago/Turabian StylePetazzi, Paolo, Olga Caridad Jorge-Torres, Antonio Gomez, Iolanda Scognamiglio, Jordi Serra-Musach, Angelika Merkel, Daniela Grases, Clara Xiol, Mar O’Callaghan, Judith Armstrong, and et al. 2023. "Global Impairment of Immediate-Early Genes Expression in Rett Syndrome Models and Patients Linked to Myelination Defects" International Journal of Molecular Sciences 24, no. 2: 1453. https://doi.org/10.3390/ijms24021453
APA StylePetazzi, P., Jorge-Torres, O. C., Gomez, A., Scognamiglio, I., Serra-Musach, J., Merkel, A., Grases, D., Xiol, C., O’Callaghan, M., Armstrong, J., Esteller, M., & Guil, S. (2023). Global Impairment of Immediate-Early Genes Expression in Rett Syndrome Models and Patients Linked to Myelination Defects. International Journal of Molecular Sciences, 24(2), 1453. https://doi.org/10.3390/ijms24021453