
Full-Length Model of SaCas9-sgRNA-DNA Complex in Cleavage State

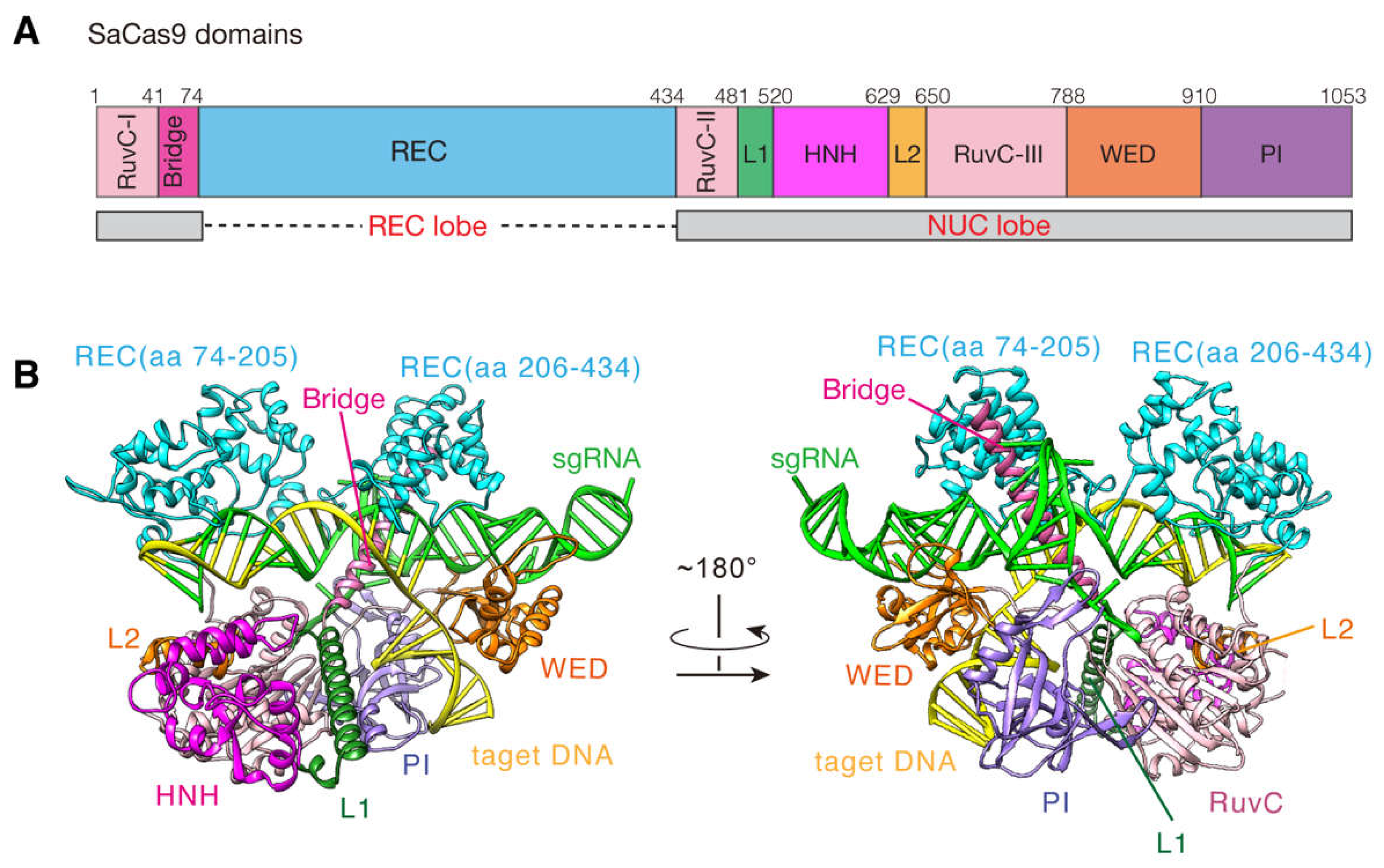{kind=link}
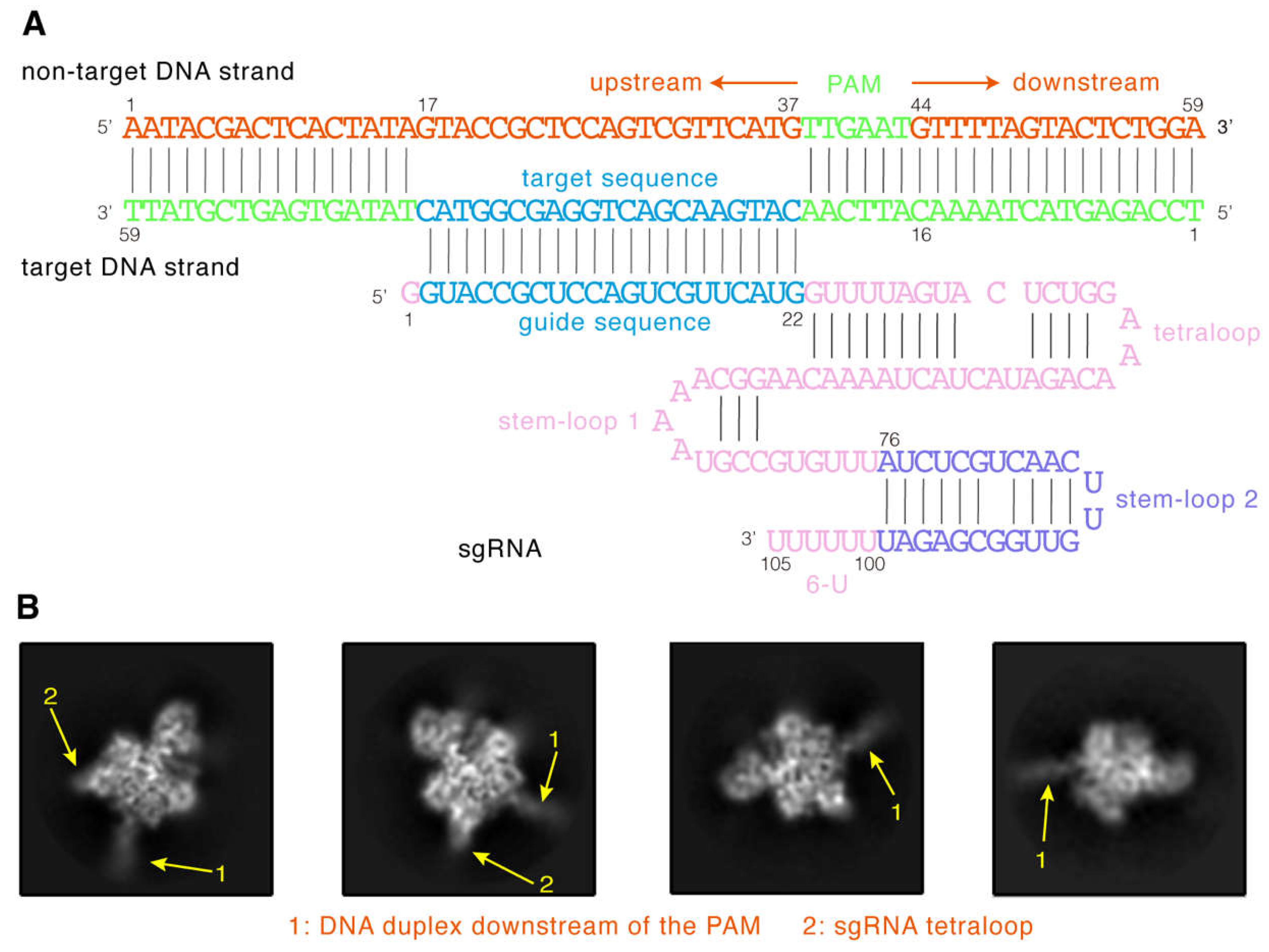{kind=link}
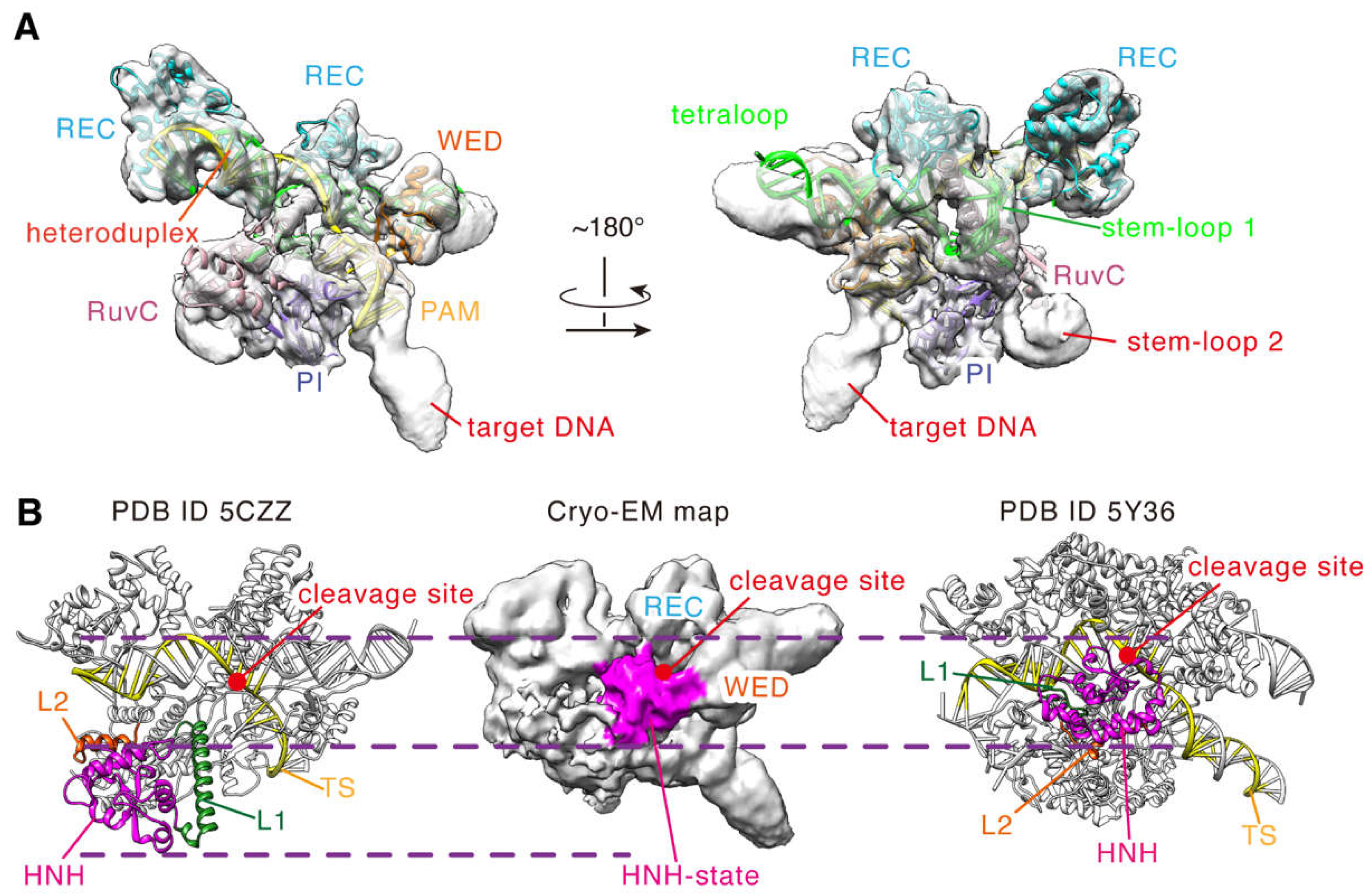{kind=link}
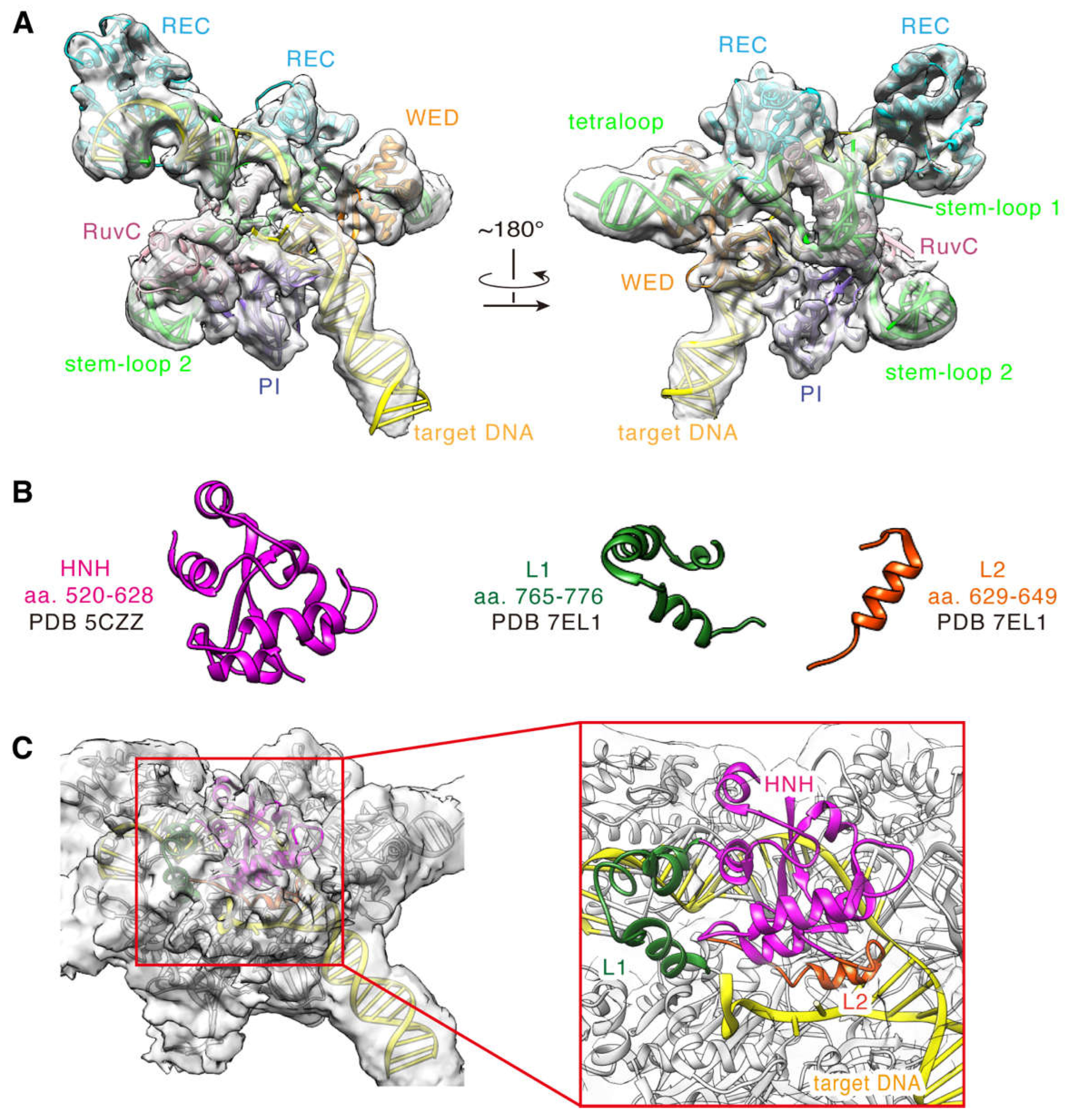{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cryo-EM Map of the SaCas9-sgRNA-DNA Complex
2.2. Conformational State of the Cryo-EM Density Map
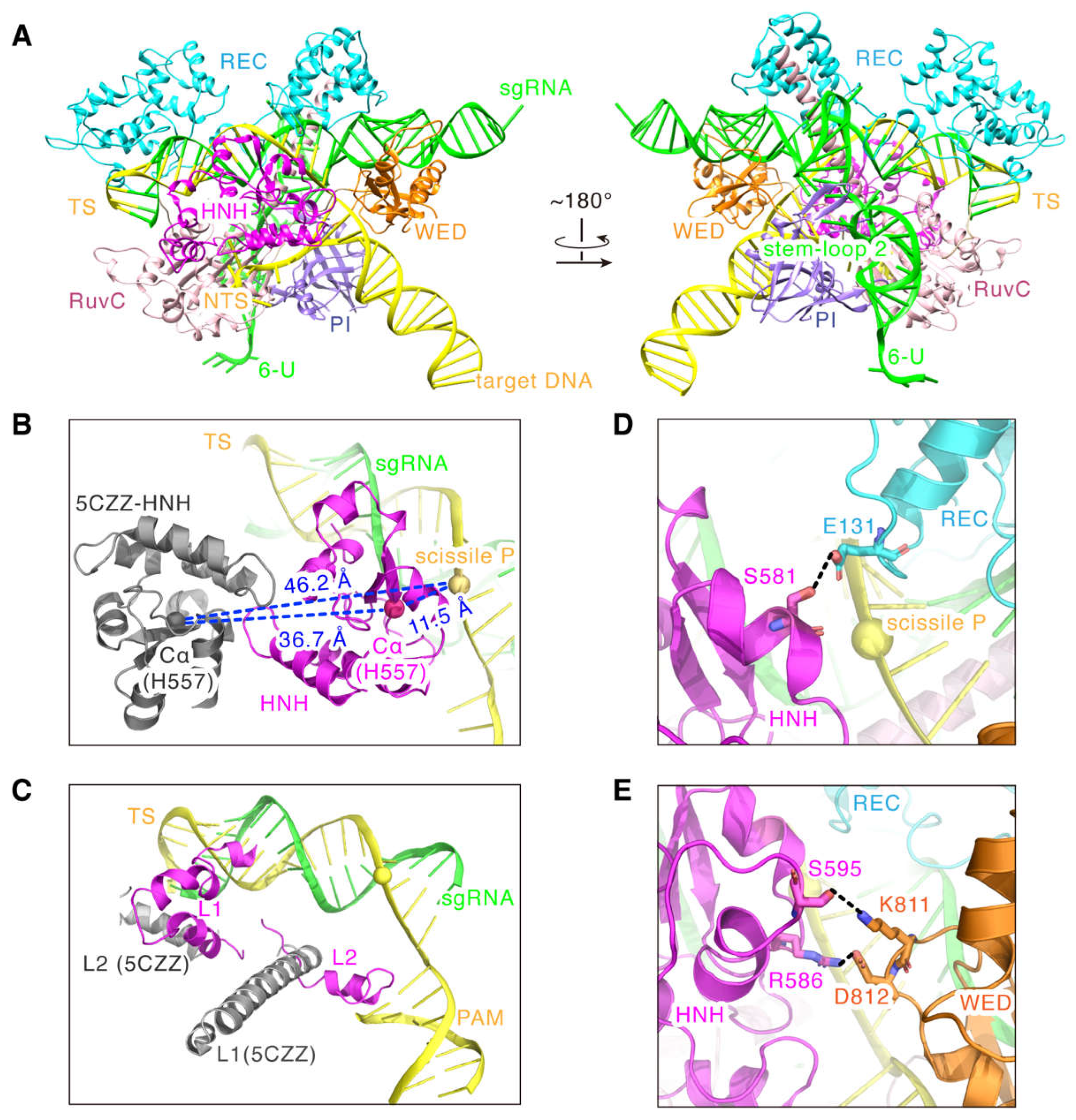
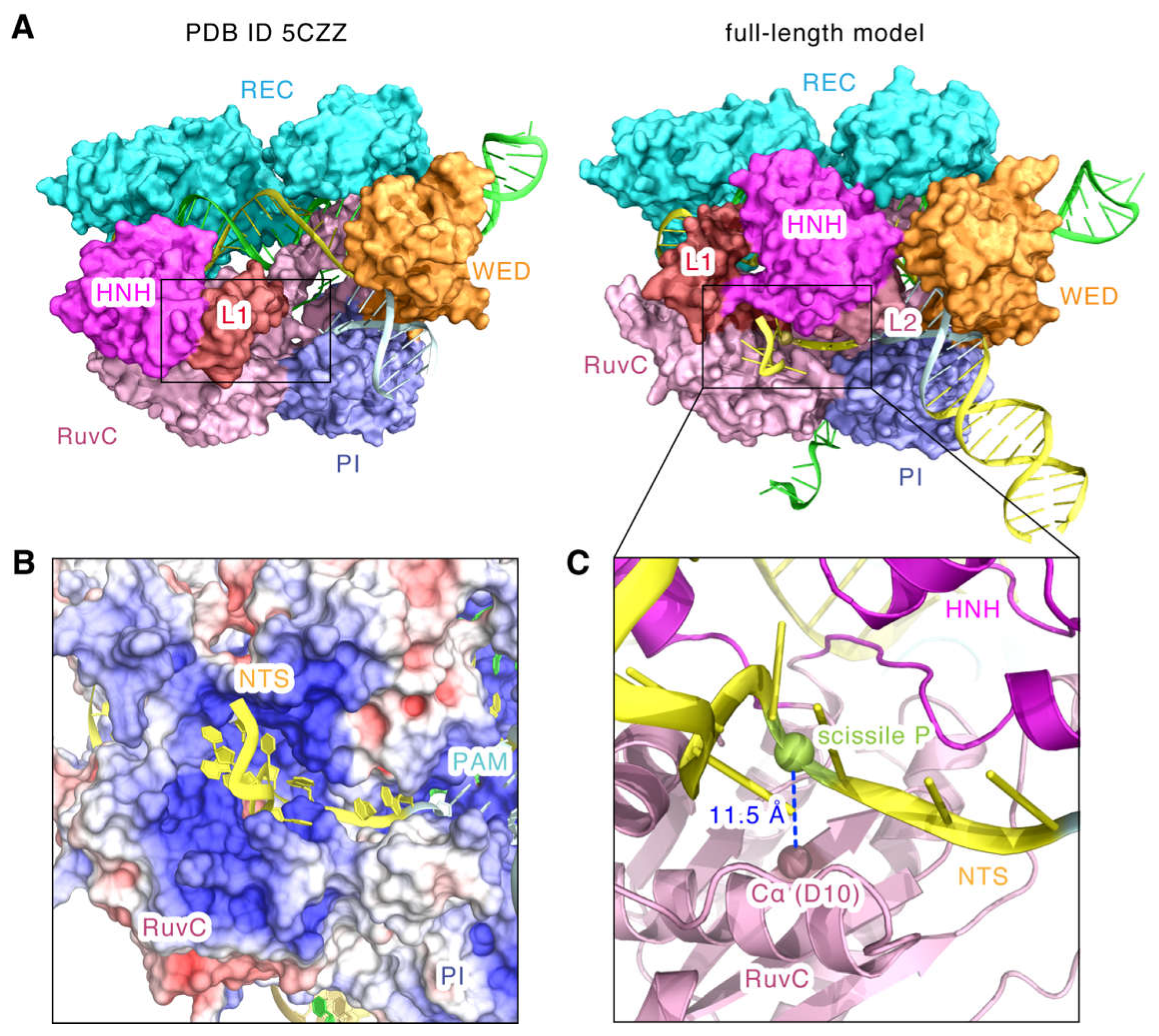
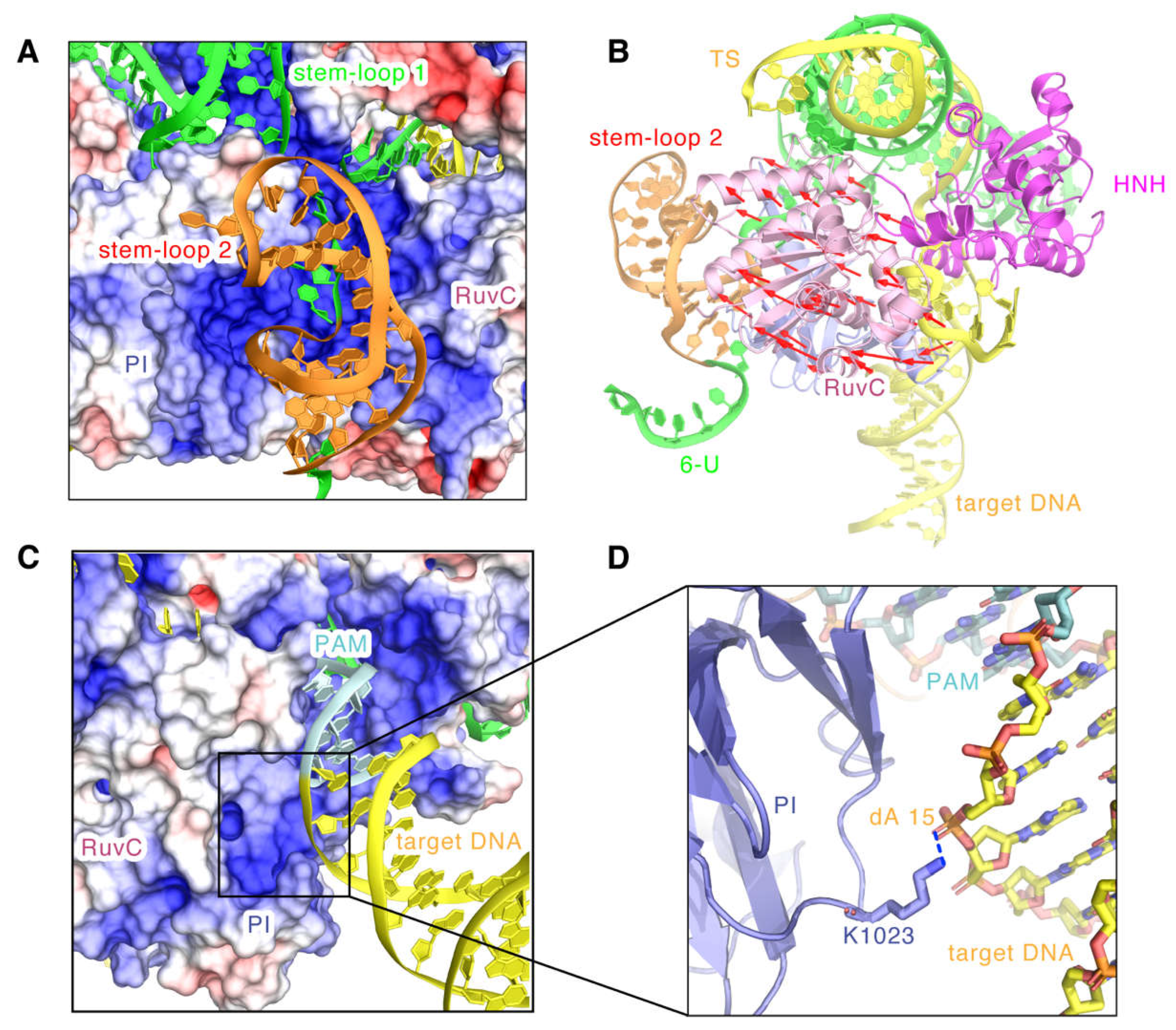
2.3. Model Features of the SaCas9-sgRNA-DNA Complex in Cleavage State
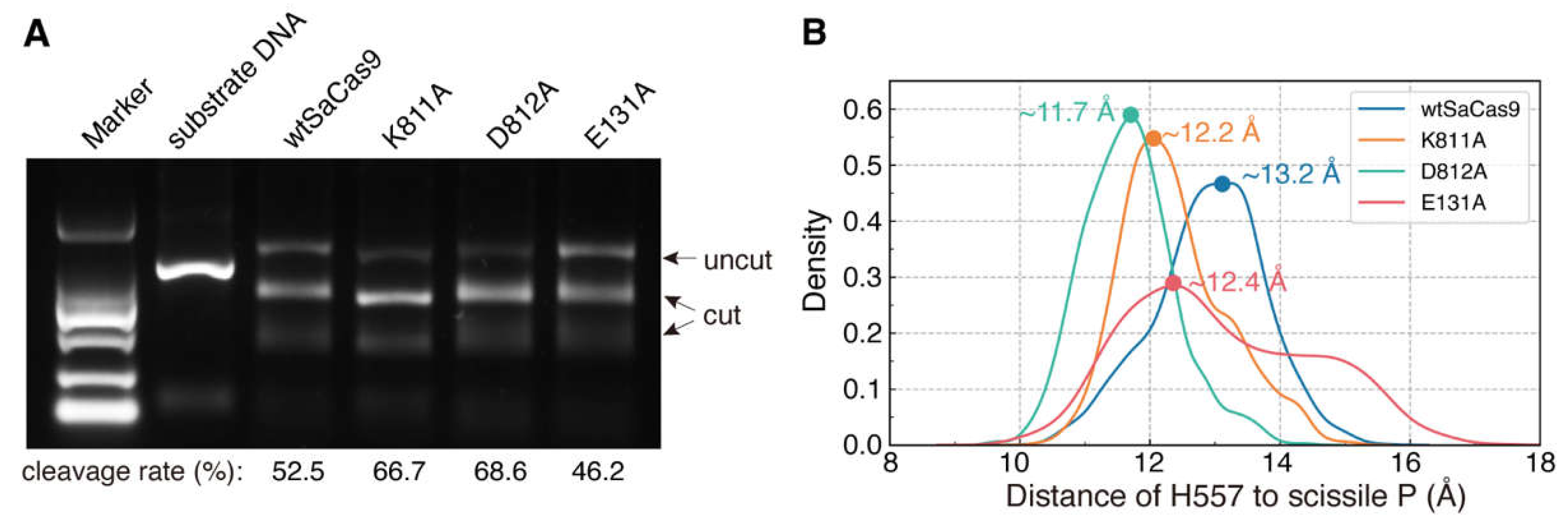
2.4. Site-Directed Mutagenesis and Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Preparation of Target DNA and sgRNA
4.3. Cryo-EM Samples and Data Collection
4.4. Single Particle 3D Reconstruction
4.5. Full-Length Model Building of the Complex
4.6. Activity Detection of SaCas9 Proteins
4.7. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduction Targeted Ther. 2020, 5, 1. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Charpentier, E. CRISPR-Cas: Biology, mechanisms and relevance. Phil. Trans. R. Soc. B 2016, 371, 20150496. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Huai, C.; Li, G.; Yao, R.; Zhang, Y.; Cao, M.; Kong, L.; Jia, C.; Yuan, H.; Chen, H.; Lu, D.; et al. Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat. Commun. 2017, 8, 1375. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef]
- Le Rhun, A.; Escalera-Maurer, A.; Bratovic, M.; Charpentier, E. CRISPR-Cas in Streptococcus pyogenes. RNA Biol. 2019, 16, 380–389. [Google Scholar] [CrossRef]
- Nishimasu, H.; Cong, L.; Yan, W.X.; Ran, F.A.; Zetsche, B.; Li, Y.; Kurabayashi, A.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Staphylococcus aureus Cas9. Cell 2015, 162, 1113–1126. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Goncalves, M.A.F.V. Adeno-associated virus: From defective virus to effective vector. Virol. J. 2005, 2, 43. [Google Scholar] [CrossRef]
- Friedland, A.E.; Baral, R.; Singhal, P.; Loveluck, K.; Shen, S.; Sanchez, M.; Marco, E.; Gotta, G.M.; Maeder, M.L.; Kennedy, E.M.; et al. Characterization of Staphylococcus aureus Cas9: A smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015, 16, 257. [Google Scholar] [CrossRef]
- Tang, H.; Yuan, H.; Du, W.; Li, G.; Xue, D.; Huang, Q. Active-site models of Streptococcus pyogenes Cas9 in DNA cleavage state. Front. Mol. Biosci. 2021, 8, 653262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Q.; Hou, X.-M.; Guo, L.; Wang, F.; Bi, L.; Zhang, X.; Li, H.-H.; Wen, F.; Xi, X.-G.; et al. Dynamics of Staphylococcus aureus Cas9 in DNA target Association and Dissociation. EMBO Rep. 2020, 21, e50184. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Y.; Lu, Z.; Huang, Z. Structural basis of Staphylococcus aureus Cas9 inhibition by AcrIIA14. Nucleic Acids Res. 2021, 49, 6587–6595. [Google Scholar] [CrossRef]
- Luan, B.; Xu, G.; Feng, M.; Cong, L.; Zhou, R. Combined computational-experimental approach to explore the molecular mechanism of SaCas9 with a broadened DNA targeting range. J. Am. Chem. Soc. 2019, 141, 6545–6552. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chu, A.H.Y.; Bao, S.; Hoang, D.A.; Kebede, F.T.; Xiong, W.; Ji, M.; Shi, J.; Zheng, Z. Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 20969–20976. [Google Scholar] [CrossRef]
- Xie, H.; Ge, X.; Yang, F.; Wang, B.; Li, S.; Duan, J.; Lv, X.; Cheng, C.; Song, Z.; Liu, C.; et al. High-fidelity SaCas9 identified by directional screening in human cells. PLoS Biol. 2020, 18, e3000747. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Qian, J.; Huang, Q. Deep-learning with synthetic data enables automated picking of cryo-EM particle images of biological macromolecules. Bioinformatics 2020, 36, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Beckers, M.; Mann, D.; Sachse, C. Structural interpretation of cryo-EM image reconstructions. Prog. Biophys. Mol. Biol. 2021, 160, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Chen, J.S.; Ricci, C.G.; Rivalta, I.; Jinek, M.; Batista, V.S.; Doudna, J.A.; McCammon, J.A. Key role of the REC lobe during CRISPR-Cas9 activation by ‘sensing’, ‘regulating’, and ‘locking’ the catalytic HNH domain. Q. Rev. Biophys. 2018, 51, e91. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Z.; Wang, F.; Zhang, S.; Chen, H.; Gu, X.; Wen, F.; Jin, J.; Zhang, X.; Huang, X.; et al. Efficient DNA interrogation of SpCas9 governed by its electrostatic interaction with DNA beyond the PAM and protospacer. Nucleic Acids Res. 2021, 49, 12433–12444. [Google Scholar] [CrossRef]
- Lv, J.; Xi, H.; Lv, X.; Zhou, Y.; Wang, J.; Chen, H.; Yan, T.; Jin, J.; Zhao, J.; Gu, F.; et al. Two high-fidelity variants: EfSaCas9 and SaCas9-HF, which one is better? Gene Ther. 2022, 29, 458–463. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef]
- Wang, S.; Tao, C.; Mao, H.; Hou, L.; Wang, Y.; Qi, T.; Yang, Y.; Ong, S.G.; Hu, S.; Chai, R.; et al. Identification of SaCas9 orthologs containing a conserved serine residue that determines simple NNGG PAM recognition. PLoS Biol. 2022, 20, e3001897. [Google Scholar] [CrossRef]
- Nierzwicki, L.; Arantes, P.R.; Saha, A.; Palermo, G. Establishing the allosteric mechanism in CRISPR-Cas9. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1503. [Google Scholar] [CrossRef]
- Saha, A.; Arantes, P.R.; Palermo, G. Dynamics and mechanisms of CRISPR-Cas9 through the lens of computational methods. Curr. Opin. Struct. Biol. 2022, 75, 102400. [Google Scholar] [CrossRef]
- Cui, C.; Wang, D.; Huang, B.; Wang, F.; Chen, Y.; Lv, J.; Zhang, L.; Han, L.; Liu, D.; Chen, Z.Y.; et al. Precise detection of CRISPR-Cas9 editing in hair cells in the treatment of autosomal dominant hearing loss. Mol. Ther. Nucleic Acids 2022, 29, 400–412. [Google Scholar] [CrossRef]
- Li, X.; Mooney, P.; Zheng, S.; Booth, C.R.; Braunfeld, M.B.; Gubbens, S.; Agard, D.A.; Cheng, Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 2013, 10, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H.W. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef]
- Kladwang, W.; VanLang, C.C.; Cordero, P.; Das, R. A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nat. Chem. 2011, 3, 954–962. [Google Scholar] [CrossRef]
- Das, R.; Baker, D. Macromolecular modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Wriggers, W.; Birmanns, S. Using situs for flexible and rigid-body fitting of multiresolution single-molecule data. J. Struct. Biol. 2001, 133, 193–202. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar] [PubMed]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 61–69. [Google Scholar] [CrossRef]
- Lin, Y.N.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Nose, S.; Klein, M.L. Constant pressure molecular-dynamics for molecular-systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, W.; Zhu, H.; Qian, J.; Xue, D.; Zheng, S.; Huang, Q. Full-Length Model of SaCas9-sgRNA-DNA Complex in Cleavage State. Int. J. Mol. Sci. 2023, 24, 1204. https://doi.org/10.3390/ijms24021204
Du W, Zhu H, Qian J, Xue D, Zheng S, Huang Q. Full-Length Model of SaCas9-sgRNA-DNA Complex in Cleavage State. International Journal of Molecular Sciences. 2023; 24(2):1204. https://doi.org/10.3390/ijms24021204
Chicago/Turabian StyleDu, Wenhao, Haixia Zhu, Jiaqiang Qian, Dongmei Xue, Sen Zheng, and Qiang Huang. 2023. "Full-Length Model of SaCas9-sgRNA-DNA Complex in Cleavage State" International Journal of Molecular Sciences 24, no. 2: 1204. https://doi.org/10.3390/ijms24021204
APA StyleDu, W., Zhu, H., Qian, J., Xue, D., Zheng, S., & Huang, Q. (2023). Full-Length Model of SaCas9-sgRNA-DNA Complex in Cleavage State. International Journal of Molecular Sciences, 24(2), 1204. https://doi.org/10.3390/ijms24021204